
A Brief History of Elemental Analysis
Elemental analysis by definition covers the whole periodic table and includes both qualitative and quantitative analysis of samples covering a wide range of concentrations. Organic elemental analysis may also be determined for a range of small or large molecule applications. It is possible to analyse a multi-component mixture of compounds using chromatographic techniques. If the mass spectrum of the separated components is determined accurately with high mass resolving power then each compound’s molecular formula may be deduced by software. Inductively coupled plasma mass spectrometry (ICP-MS) or x-ray fluorescence (XRF) instrumentation for the analysis of heavy metals are now ubiquitous in the modern analytical laboratory but what methods were available to chemists several hundred years ago for the determination of elemental composition let alone isotope analysis without a mass spectrometer, was it even possible?
The Early Years
The combustion of a sample to change the colour of a flame must have unwittingly been the oldest method employed for elemental analysis, very little in the way of sample preparation is required. Although not officially recorded Early man must have made these observations since dawn of time when having a fire and inadvertently burning various rocks containing interesting ores. Greek philosophers back in the 3rd century BC proposed that all mater consisted of four elements in nature – earth, water, air and fire. The occupation of Egypt by the Arabs in the 7th century resulted in the first use of the word alchemy in Eurpoe through the combination of ‘al’ from Arabic with ‘Khemia’ the Greek word for Egypt. It was proposed by the Arabian alchemists in the 8th century that all metals were made up of various proportions of mercury and sulfur with gold being the perfect metal. The transmutation of base metals into gold was apparently facilitated by the use of substance known as the pholosphor’s stone. It was not until 1901 Rutherford and Soddy discovered that radioactive thorium was transmutating into radium however Rutherford was apparently not so keen on using the same language as the alchemists.
In 1556 a German metallurgist named Georgius Agricola published the first paper and noted that different ores when placed into a flame changed “the colour of fumes” this information was used for qualitative analysis. In the early 1700s Newton investigated light and developed the prism as a means of separating light into its constituent colour or wavelengths. Another century passed before the prism was used to observe and characterise the spectral lines emitted from the elements present in the sun, Foucault noticed that the sodium lines were present in both the sun and a flame. The collaboration between Bunsen and Kirchhoff who are credited with founding the field of analytical chemistry accelerated the development of a flame excitation instrument by combining the hot Bunsen burner flame with Kirchhoff’s spectroscope. Sample introduction was simple and involved placing small quantities of a solid sample directly into the flame usually on a wire loop. Excited atoms or ions generated in a hot flame emit characteristic photons or coloured light during the radiative de-excitation process. The spectroscope was used to record the wavelengths of the emission lines that were characteristic of the element under investigation, and so optical emission spectroscopy was born. For quantitation purposes it is necessary to measure and record the intensity of the emission for each calibration standard prior to running the unknown sample, this however proved difficult with the technology available at that time. The fundamental research was critical for qualitative analysis but it was a number of years before these prototype instruments matured into a product available on the elemental analysis market for quantitation.
The Flame Photometer
The first invention of an instrument resembling a flame photometer was developed by Champion, Pellet and Grenier in 1873. The instrument consisted of two flames one for the analysis of the unknown sample the other for a calibration standard of known concentration, the operator would view displaced emission spectra from each flame through a spectroscope for a single element sodium. Visual photometry was used to compare the intensity of the emission from the unknown sample with that of the emission from the calibration standard, the concentration of sodium was determined using this method to within 5 %. Their instrument design was later improved by Gouy in 1877, who demonstrated that the intensity of the emission was proportional to the size or temperature of the flame and the amount of sample present. In order to account for this Gouy developed the first pneumatically assisted atomizer for liquid sample introduction, an early ancestor of the modern nebuliser and spray chamber sample introduction techniques, this ensured that the amount of sample entering the flame was controlled therefore improving the accuracy of the analysis. Lundegardh during the 1920s, made further improvements to the sample introduction atomiser for flame photometry instrumentation and also incorporated a photographic plate for recording the results.

Gustav Kirchhoff (left) and Robert Bunsen (right)
Atomic Absorption Spectroscopy
Atomic absorption spectroscopy (AAS) was also developed by Kirchhoff using his spectroscope, he observed that when a bright light was shone through a flame that contained excited atoms of an element, characteristic wavelengths associated with electronic transitions of the element were absorbed leaving dark bands confirming its presence. Kirchoff published “the relation between the powers of emission and the powers of absorption for rays of the same wavelength is constant for all bodies at the same temperature.”
Limitations of the instrumentation required to accurately monitor and record the absorption or emission of light hindered the development of the above spectroscopic techniques for quantitative analysis. The integration of photographic plates by Lundegardh did improve the accuracy and precision but calibration was difficult for absolute quantitation. Spectral interference and a lack of sensitivity of these techniques for certain analytes made them impractical for certain trace analysis applications. The main techniques employed in practice up until the early 20th century and indeed today in some cases for elemental analysis use either gravimetric or titrimetric methods of analysis. What was really needed was the development of a reliable electronic detector for recording the instrument response, once this was achieved a calibration curve could be plotted using external standards.
Following on from Gouy’s initial observations related to the size of the flame and intensity of emission it was later concluded that this relationship was due to an increase in the population of the excited state of the analyte in the flame as a function of temperature. Over the next decade various flames were used including hydrogen in air, acetylene in air and acetylene in nitrous oxide, each of these mixtures produced a hotter flame with an associated increase in detection limit. Flame atomic absorption spectroscopy (FAAS) instruments were developed during the 1950s, for the analysis of aqueous samples, the addition of nitrous oxide acetylene flames improved the sensitivity for certain analytes in challenging environmental samples. The most sensitive AAS instruments available on the market today employ graphite furnaces that can detect an analyte concentration with a detection limit of down to ppb for some assays. Sample preparation often involves microwave digestion of a solid sample in nitric acid, biological samples may also be prepared using a similar method prior to sample introduction at a suitable concentration.
Atomic Emission Spectroscopy
The development of atomic emission spectroscopy did not receive much attention for a number of years due to a lower detection limit being achieved using atomic absorption techniques. A stable plasma source running at more than twice the temperature of the hottest flame was required to realise the full potential of atomic emission spectroscopy. In 1974, the first commercial inductively coupled plasma optical emission spectroscopy (ICP-OES) instruments were developed using argon gas by KONTRON in Germany. The use of inductively coupled argon plasma instrumentation for emission spectroscopy is very popular today and offers a lower detection limit than atomic absorption for elemental analysis especially for the analysis of toxic metals.
Discovery of the Neutron
The only caveat with both of the above aforementioned spectroscopic analysis techniques is their inherent lack of isotopic selectivity. Rutherford and Soddy first proposed the idea of transmutation of the elements back in 1902, that lead to a significant modification to Dalton’s atomic theory however the term or idea of an isotopes was not considered. Some years later Soddy and Fajans simultaneously proposed the existence of isotopes in 1913, through the study of radioactive decay chains and the discovery of three different stable isotopes of lead.
Mass Spectrometry and Isotope Ratio Analysis
Mass spectrometry changed everything for isotope analysis. Thompson and Aston analysed a neon gas sample using a parabola mass spectrograph developed in 1912 at the Cavendish laboratory in the University of Cambridge. Two parabolas or traces were observed when they had both expected only one for the pure sample of neon.
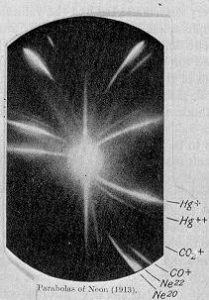
In the bottom right corner of J. J. Thomson’s photographic plate are the separate impact parabola marks for the two isotopes of neon: neon-20 and neon-22.
Thompson had initially thought they had discovered a new element but Aston was not convinced. After learning about Soddy’s discovery and proposed existence of isotopes Aston spent a fellowship distilling liquid air and performing what are now known as isotope enrichment experiments on neon. Neon samples were diffused through porous clay, some enrichment was achieved and measured using a quartz microbalance. Once further developments and improvements by Aston of the mass spectrometry instrumentation came to fruition other isotopes were discovered and the field of isotope ratio mass spectrometry (IRMS) was established. Previous assays using gravimetric analysis for the determination of the (average) atomic mass of the elements on the periodic table were cross validated with results acquired using isotope ratio mass spectrometry with remarkable accuracy. The consequence of the early work carried out by Thompson and Aston was the realisation that the chemical properties of an element are determined by its atomic number rather than its atomic weight. The existence of isotopes was also published independently by Mosley in 1913, who demonstrated the relationship between an element (atomic number) and the wavelength of the corresponding x-ray spectral lines.
Other Methods for Separation of Isotopes
Isotope ratio analysis has really only become a mainstream analytical technique for a variety of sample type due to the use of electromagnetic separation employed by mass spectrometry. The chemical separation of isotopes is difficult because their chemistry is almost identical. Lighter isotopes do tend to react or evaporate more quickly allowing them to be separated however the efficiency is low. Diffusion rates of different isotopes of a gas or liquid across a membrane will be different due to the lighter isotopes traveling more quickly, this method was used by Soddy for his enrichment experiments however the timeframe is such that it is impractical for use as a quick analytical method. Centrifugal separation of isotopes is common for the enrichment of the heavier isotopes containing uranium, the technique was first proposed by Aston and Lindemann back in 1919, again this technique is more suited for large scale preparative applications rather than analysis. Lasers may be used for the analytical separation and detection of a single isotope from a mixture when finely tuned to resonantly excite a hyperfine interaction between an electron and the nucleus, each isotope will have different hyperfine spectra due to the difference in atomic weight of their respective nuclei. Colinear resonance laser spectroscopy (CRIS) when combined with mass spectrometry offers the highest level of sensitivity and selectivity for sub ppq level elemental analysis.
Development of ICP-MS Instrumentation
The realisation that mass spectrometry could be used to determine the mass-to-charge ratio of ions that are generated from an inductively coupled plasma was a huge step change in the development of ultra-trace elemental analysis instrumentation. When compared with ICP-OES analysis the sampling of the ion beam rather than the emitted light offers many advantages, the lack of spectral interference is critical for the analysis of elemental impurities at very low detection limit. The main technological breakthrough came with the development of an atmospheric pressure inlet for a mass spectrometer that could sample the ion beam generated by an argon plasma ion source for subsequent mass analysis.
Sample Preparation and Analysis by ICP-MS
Sample preparation for ICP-MS is the same as the other spectroscopic techniques, aqueous samples are usually prepared in dilute nitric acid. Solid samples may require dissolution in stronger acids such as aqua regia or even hydrofluoric, microwave digestion is also used to speed up the process. Sample introduction involves the nebulisation of the liquid sample into the spray chamber with argon gas. The analysis of a solid sample is also possible with minimal sample preparation using laser ablation, the ablation plume is swept into the argon plasma using a stream of argon gas. Once inside the ICP torch ionization of the sample occurs in the argon plasma to generate the ion beam. The analyte ion will then be selected by the mass analyzer for transmission through to the detector. The most common mass analyzer used in ICP-MS is the quadrupole for both ultra-trace and trace element analysis covering an elemental concentration range of up to 10-orders of magnitude. ICP-MS instrumentation using a single quadrupole mass spectrometer does however suffer from isobaric interference which can mask the analyte ion as a matrix effect. The argon plasma will generate a series of polyatomic interference ions; for example the argon in the plasma will react with oxygen to give argon oxide at m/z 56 which represents a major isobaric interference with the most abundant isotope of iron at the same mass. Chlorine present in a liquid sample can also form an argon chloride polyatomic ion for each of the major chlorine isotopes that constitute a major isobaric interference for the analysis of arsenic. Various methods that have been employed to reduce polyatomic interference from the ion beam the most successful being the use of a collision cell before the quadrupole mass analyzer. The collision cell will fragment the polyatomic interference effectively changing its mass and therefore only let the analyte ion pass through the mass analyzer for detection. Unfortunately, isobaric interference may also present from an atomic ion generated from an isotope of a different element. The only way to remove these interferences is to use a different type of mass spectrometer that incorporates either a high mass resolving power mass analyzer or a tandem quadrupole mass spectrometer with an additional collision cell in between the two quadrupoles. The collision or reaction cell employed for ICP-MS-MS experiments increase the selectivity of the analysis; this is achieved by using a reaction gas that will selectively react with either the analyte or interference ion to increase its mass for subsequent separation and independent detection.
Analysis of Difficult Samples
The most sensitive routine mass spectrometry instrument for ultra trace element analysis in use today that minimise isobaric interference for challenging environmental samples uses a tandem quadrupole mass analyzer ICP-MS/MS. These instruments have a variety of sample introduction inlets available for the analysis of a liquid sample, particles or even nanoparticles in suspension is possible. The addition of an external laser sampling system for mass spectrometry imaging applications of a particular element in a solid sample using laser ablation is also becoming popular. Software now available enables isotope dilution methods to be run with ease and the use of an internal standard is now routine to improve the precision and accuracy of the results. ICP-MS is a highly versatile elemental analysis technique however it is not possible to use it for the analysis of the lighter elements such as carbon. The instrument of choice for isotope ratio analysis would be a mass spectrometer using a magnetic sector with multiple detectors one for each isotope, an inductively coupled plasma ion source or ideally a thermal ionisation mass spectrometer (TIMS) will give the most precise isotope ratio measurements.
Organic Elemental Analysis, the CHNS Elemental Analyzer
Organic compounds are amenable to ICP-MS however they would have to be digested usually by microwave in nitric acid prior to analysis. It is not possible to use an ICP-MS to get any molecular formula information or quantify the ratio of carbon present. If molecular weight information is required one needs to employ a different ionization source for mass spectrometry such as electrospray for LCMS. A simple combustion technique such as CHNS analysis is ideal to determine the ratio of carbon, hydrogen, nitrogen and sulfur in solid or liquid samples. The development of the CHNS elemental analyzer as a commercial instrument for combustion analysis started with Carlo Erba in 1968. Typically, milligram quantities of the sample are introduced into a combustion tube in excess oxygen, flash photolysis ensues followed by gas chromatography to separate and quantify the evolved combustion gas products such as carbon dioxide. CHNS analysis may be used to test product quality, food or soil analysis.
Accurate Mass LC-MS for Elemental Analysis
There are many types of mass spectrometer available today for the analysis of solids, liquids or gases. Liquid chromatography mass spectrometry (LC-MS) involves the hyphenation of a liquid chromatograph with a mass spectrometer. The MS system will usually incorporate an electrospray ionization or atmospheric pressure chemical ionization source. The sample must be soluble in the mobile phase that flows through the liquid chromatograph which is usually a polar solvent. Separation of the analyte from the sample matrix occurs in a chromatographic column due to the analyte’s polarity; the analyte will partition between the polar mobile and immobilized non-polar stationary phase (like attracts like). The stronger the affinity of the analyte to the stationary phase the longer its retention time within the column. Each component or compound separated by the liquid chromatograph will have a unique retention time for a given analytical method run by the liquid chromatograph (mobile phase solvents, flow rate and type of column). Once separated each analyte will flow into the mass spectrometer where ionization will occur, ions generated from the ion source are then separated by the mass analyzer followed by detection to generate a mass spectrum. Electrospray ionization is a soft ionization technique and tends to only yield a protonated molecule in positive ion mode or a deprotonated molecule in negative ion mode. It is possible using tandem mass spectrometry for example to fragment the precursor ion in the collision cell to yield a number of characteristic product ion fragments. The mass spectrometry method development process involves tuning the various ion optics and collision energy to generate a characteristic fragment ion that can be used for quantitation or conformation that an analyte is present in the sample. High sensitivity is one of the best features of MS detection, it is also quick and can be one of the most specific (using accurate mass) and selective analytical techniques available today. Applications include metabolite profiling for metabolomics, proteomics, environmental monitoring and drug development. Once the mass spectrum has been acquired with an accurate mass additional data processing may be used to generate the chemical formula, this in turn will give the elemental composition.
This brief review is by no means exhaustive, there are many other techniques that have played their part in the development of analytical chemistry for elemental analysis. In answer to the original question posed regarding the feasibility of elemental analysis several hundred years ago the answer is yes for certain elements using flame excitation methods, however quantitation was not possible really until the late 1870s. In order to determine the isotope ratio of an element this has only been made possible with the advent of mass spectrometery within the last 100 years. The necessity to push detection limits for the analysis of complex samples has never been in such demand as it is today especially for the detection of radionuclides in difficult environmental samples. The new analytical techniques under development by Artemis Analytical Ltd combine the very best that laser spectroscopy and mass spectrometry have to offer meeting the challenges of tomorrow head on regardless of the complexity of the sample type.
Trackback from your site.


Adina206
| #
I must admit that your post is really interesting. I have spent a lot of my spare time reading your content. Thank you a lot!
Adaline206
| #
I have read your article; it is very informative and helpful for me. I admire the valuable information you offer in your articles. Thanks for posting it.
elavil365y.com
| #
lyrica elavil interactions
A Brief History of Elemental Analysis » Artemis Analytical
http
| #
indomethacin synthesis
A Brief History of Elemental Analysis » Artemis Analytical
amitriptyline7w365.com
| #
gabapentin and amitriptyline
A Brief History of Elemental Analysis » Artemis Analytical
imitrex123.com
| #
imitrex injections generic
A Brief History of Elemental Analysis » Artemis Analytical
mestinon7d24.com
| #
pyridostigmine bromide mestinon is the drug of choice for treatment of
A Brief History of Elemental Analysis » Artemis Analytical
diclofenac24a.com
| #
diclofenac and tramadol
A Brief History of Elemental Analysis » Artemis Analytical
mebeverine3x3.com
| #
mebeverine how to take
A Brief History of Elemental Analysis » Artemis Analytical
pyridostigmine0x7.com
| #
can i order cheap pyridostigmine online
A Brief History of Elemental Analysis » Artemis Analytical
cilostazol77x7.com
| #
cilostazol bleeding time
A Brief History of Elemental Analysis » Artemis Analytical
Aggie206
| #
I see some amazingly important and kept up to length of your strength searching for in your on the site
baclofen7r24.com
| #
can you take hydrocodone with baclofen
A Brief History of Elemental Analysis » Artemis Analytical
maxalt2x2.com
| #
rizatriptan generic maxalt
A Brief History of Elemental Analysis » Artemis Analytical
sumatriptan234.com
| #
can i take sumatriptan after paracetamol
A Brief History of Elemental Analysis » Artemis Analytical
imduraaa.com
| #
getting off imdur
A Brief History of Elemental Analysis » Artemis Analytical
rizatriptan7x24.com
| #
rizatriptan brand india
A Brief History of Elemental Analysis » Artemis Analytical
imuran7www.com
| #
imuran side effects tingling
A Brief History of Elemental Analysis » Artemis Analytical
mobic321.com
| #
can mobic be used for headaches
A Brief History of Elemental Analysis » Artemis Analytical
meloxicam1x1.com
| #
overdose of meloxicam to dog
A Brief History of Elemental Analysis » Artemis Analytical
azathioprine7info.com
| #
azathioprine mmr
A Brief History of Elemental Analysis » Artemis Analytical
piroxicam123.com
| #
dogs piroxicam side effects
A Brief History of Elemental Analysis » Artemis Analytical
lioresalzxz.com
| #
lioresal endikasyonlari
A Brief History of Elemental Analysis » Artemis Analytical
toradol888.com
| #
where can i buy cheap toradol for sale
A Brief History of Elemental Analysis » Artemis Analytical
cyproheptadine24w.com
| #
cyproheptadine 4mg tab
A Brief History of Elemental Analysis » Artemis Analytical
tizanidine01.com
| #
cost of tizanidine without insurance
A Brief History of Elemental Analysis » Artemis Analytical
periactin7s.com
| #
what is periactin tablets used for
A Brief History of Elemental Analysis » Artemis Analytical
artane7y7.com
| #
directions to artane castle shopping centre
A Brief History of Elemental Analysis » Artemis Analytical
ketorolac33.com
| #
ketorolac cheap
A Brief History of Elemental Analysis » Artemis Analytical
zanaflex365.com
| #
zanaflex flexeril together
A Brief History of Elemental Analysis » Artemis Analytical
BrandonLib
| #
Biography of Spanish footballer Pedri https://pedri-bd.com statistics at Barcelona, ??games with teammate Gavi, inclusion in the national team for Euro, meme with Cristiano Ronaldo.
Michaelrew
| #
Строительный и архитектурный портал https://intertools.com.ua все самое интересное о строительстве и архитектуре – новости архитектуры и строительства, обзоры и аналитика.
ScottDup
| #
Latest news league-of-legends-esports.com analytics and forecasts in the world of League of Legends eSports. Stay up to date with all the main events of the professional scene!
Frankdwefe
| #
Invictus Gaming is a legendary invictus-gaming-dota2 esports organization known for its remarkable victories in Dota 2, including the championship at The International 2018.
MichaelSUG
| #
Official website https://t1-league-of-legends.com of the legendary eSports team T1 for League of Legends. Latest news, match results, player statistics and LOL Betting.
Arnoldstunk
| #
Latest news and analytics on League of Legends lol-news matches, tournaments, betting. Stay up to date with the latest eSports events!
Matthewtip
| #
Official website of the T1 t1 lol com League of Legends eSports team. Latest news, matches, statistics, tournaments and predictions on LoL Betting.
Danieledurb
| #
Official website of the award-winning samsung-galaxy-league-of-legends com Samsung Galaxy League of Legends team. Latest news, matches, statistics, player profiles and bets on games.
Ronaldgug
| #
Official website of the eSports organization https://g2-esports-league-of-legends.com
DavidDop
| #
Fnatic is a legendary League fnatic league of legends of Legends eSports team. Our website has all the latest news, matches, player statistics and game predictions.
Thomasexpog
| #
Official Team Liquid website team liquid league of legends latest news, match results, tournaments, player profiles and LOL Betting.
EddieOxype
| #
Gen G is one of the strongest eSports teams gen-g-league-of-legends in League of Legends. Our website has the latest news, matches, statistics and LOL Betting.
DavidDab
| #
Dive into the world of Edward Gaming edward-gaming-league-of-legends com the legendary League of Legends eSports team. Latest news, matches, tournaments, statistics and LOL Betting.
Derekdug
| #
Welcome! Find out the latest news dplus league of legends matches, tournaments and statistics of the Dplus team in the League of Legends!
Ernestnoido
| #
Find out everything about Team WE http://team-we-league-of-legends.com news, matches, player statistics and bets on League of Legends. Support the team!
Albertdaymn
| #
ip camera software software for ip camera recording
Jamesmut
| #
Натуральные молочные продукты https://gastrodachavselug2.ru свежесть и качество с заботой о вашем здоровье! Широкий выбор: молоко, творог, сметана, сыры. Только натуральные ингредиенты, без консервантов и добавок.
Jeweladunk
| #
thc chocolate delivery in prague thc gummies shop in prague
CharlesAddes
| #
weed delivery in prague buy hashish in prague
DavidUlces
| #
kush delivery in prague weed delivery in prague
EdwardFum
| #
thc vape in prague buy thc joint in prague
HarryPal
| #
cannabis in prague hash for sale in prague
EdwardUrite
| #
kush shop in prague thc vape in prague
RobertTar
| #
420 day in prague weed shop in prague
ThomasDus
| #
hemp shop in prague https://smokeinprague.site
BrianPreSt
| #
thc joint in prague 420 day in prague
Elvisdot
| #
cannabis delivery in prague weed store in prague
Richardiroro
| #
Biography of footballer Kylian Mbappe kylian-mbappe-bd.com/ personal life, rumors of an affair with Alicia Aylis and Ines Rau. Career, statistics and salary at Paris Saint-Germain, victory at the World Cup and other achievements of the striker.
WilliamLoomi
| #
Biography of football player Neymar neymar-bd.com/ personal life, relationships and rumors of romances with Katya Safarova, Natalia Barulich, the birth of a son and Bruna’s last girlfriend, the birth of daughters.
AnthonyBut
| #
Biography of Belgian footballer kevin-de-bruyne Kevin De Bruyne (Kevin De Bruyne): personal life, relationship with his wife, conflict with Thibaut Courtois over his girlfriend Caroline.
HarryGof
| #
Biography of football player Luis Alberto Suarez http://luis-suarez-bd.com personal life, daughter, wife, children, height. Leaving the club Atletico Madrid, career in Barcelona and Liverpool, goal statistics.
Michaelsub
| #
Biography of football player Jude Bellingham jude bellingham bd com personal life, relationship with girlfriend Laura. Player statistics in the Real Madrid team, matches for the England national team with Harry Kane, the athlete’s salary, conflict with Mason Greenwood.
ThomasDiels
| #
Try the free demo game Crazy Monkey http://crazy-monkey.com.az (Igrosoft) and read our exclusive review!
Terencewatry
| #
Discover Space XY space-xy.com.az/ Take advantage of bonuses and free play to increase your chances of winning!
JerryGlync
| #
Hot Fruits 100 Slot http://hot-fruits-100.com.az Review by Amatic Industries – Play Hot Fruits 100 demo for free or real money. Bonuses and best casinos for September 2024!
Ralphplumb
| #
JetX is a unique simulation game jetx.com.az/ from SmartSoft Gaming. Players fly a virtual plane and collect their winnings safely.
Alvinorbic
| #
Learn everything about blackjack blackjack com az rules, types of bets, features of the online game and answers to popular questions.
AveryAmicy
| #
Bayer 04 Football Club bayer 04 com az composition, statistics, best Bayer players
MarcusPioro
| #
RB Leipzig http://rb-leipzig.com.az team history, club titles, top scorers and players in team history
AlfredFlaph
| #
Current Heidenheim https://fc-heidenheim.com.az squad with player stats and market value, match schedule, club news and rumours
JeffreyAnymn
| #
Phil Foden phil-foden-az.com is a talented midfielder for Manchester City. Find out about his biography, statistics and latest news.
BradleyFal
| #
Declan Rice (Arsenal) declan rice az com midfielder, 25 years old. Check out his biography, statistics, goals and latest 2024 news.
Michaelnum
| #
Yassin Bunu from Al-Hilal yassine-bounou-az.com biography, statistics, news and everything about his career in football.
Danielraw
| #
Everything about Moussa Dembele moussadembele-az.com/ biography, goals, news, statistics and photos. Follow the career of the Al-Ittihad star with us!
CaseyLit
| #
Thiago Silva thiago silva az com is a legendary defender for Chelsea and the Brazilian national team. On the site you can find a biography, the latest news, statistics, videos and interviews. Learn everything about the career and achievements of the great football player.
Jamesgutle
| #
Learn about Garrett Bale https://gareth-bale-az.com his biography, personal life, achievements and the latest news from the world of football.
RobertKam
| #
Everything about Kaka https://kaka-az.com biography, matches, goals, statistics, photos, videos and the latest news about the football legend on one site!
Wallyeldew
| #
Zico is a legendary Brazilian footballer zico known as “White Pele”. His talent, technique and passion for the game made him an icon of Brazilian football, and his contributions to the sport continue to inspire new generations.
LeonardJah
| #
George Best george-best is a brilliant footballer and a shining symbol of the 1960s, known for his talent and turbulent life. He left an indelible mark on football by combining success on the pitch with the tragedy of personal struggle.
Larrythisk
| #
Rudy Gobert rudy-gobert-az.com is a French center and one of the best defenders in the NBA, nicknamed “The French Tower.” A three-time Defensive Player of the Year, he inspires with his skills and commitment to excellence.
MichaelSweag
| #
Nikola Jokic nikola jokic is a Serbian basketball player, NBA star, and leader of the Denver Nuggets. Known for his unique style of play, court vision, and leadership, he has become a role model for a new generation of centers.
Russellexoma
| #
Luka Doncic lukadoncic-az.com is a Slovenian basketball player, the leader of the Dallas Mavericks team and one of the main stars of the NBA. His unique playing style, records and influence have made him a symbol of European success in world basketball.
Clauderot
| #
Born to a British-Nigerian jamal-musiala-az org father and a German mother of Polish descent, young Jamal explores cultural differences while playing for Germany at the Euros, facing off against Jude Bellingham. Breaking news 2024.
Travisjes
| #
Julius Randle is a versatile NBA forward julius-randle a leader for the Knicks, and an inspiring example of perseverance. His play, leadership, and desire to win make him one of the defining figures in the modern league.
WilliamHog
| #
Cristiano Ronaldo biography cristiano-ronaldo personal life, relationships with Irina Shayk and Georgina Rodriguez, children, career at Real and Juventus, records with Portugal at Euro 2020.
Robertbrask
| #
Biography of footballer Mohamed Salah mohamed salah az org wife Magi Sadiq, children, charity work, book “The Last Pharaoh”. Statistics, salary and awards with Liverpool and Egypt in 2024.
Larrygup
| #
Biography of footballer Luis Alberto Suarez luis-suarez-az.org personal life, wife, children, height. Departure from Atletico Madrid, career at Barcelona and Liverpool, goal statistics. Playing for Gremio, moving to Inter Miami, retirement from international duty and the latest news in 2024.
Horacionig
| #
Spanish footballer and Athletic Bilbao nico williams az org midfielder Nico Williams has had a remarkable career. He shares a close relationship with his brother Inaki. Recent reports have included a potential move to Barcelona in 2024.
JasonCef
| #
Talented Nigerian striker Victor Osimhen victor osimhen proudly represents Italian club Napoli and the Nigerian national team. This story highlights his remarkable sporting career, personal development and notable achievements, including a loan spell at Galatasaray.
KennethPrief
| #
Federico Valverde’s federico-valverde-az org biography: personal life, date of birth, children with Mina Bonino, salary, religion, playing style on the pitch, Real Madrid statistics, salary, number inherited from Kroos, position for 2024 and news.
JamesMus
| #
Biography of Spanish footballer Rodri https://rodri-az.org Manchester City midfielder, salary, worth, religion, Euro match statistics, champion status and games with Joao Cancelo. Latest updates in 2024.
Blakeapabs
| #
Biography of Lamine Yamal lamine-yamal-az.com/ a Spanish winger who plays for FC Barcelona and the national team. Includes career highlights, statistics, Euro 2024 winning salary and personal life.
Frankfat
| #
Magic Johnson’s biography magic-johnson-az.com photos, news, personal life, basketball career, religion, prison, statistics, rivalry with Michael Jordan and his fight against AIDS.
StevenHefly
| #
Biography of LeBron James https://bronny-james-az.org son Bronny James. Covers his personal life, family, health issues, NBA draft, and the latest news in 2024.
MichaelvOp
| #
Frankie de Jong’s biography https://frenkie-de-jong-az.org covers his personal life, height, wife, club stats at Barcelona and Ajax, transfer rumours to Man Utd, playing position and shirt number.
Mathewcop
| #
Luka Doncic biography luka doncic salary, signature shoes, stats, NBA comparisons with Trae Young, games with Jokic and Irving, personal life updates.
DanielHully
| #
Stephen Curry stephen-curry biography: personal life, height, weight, career, team, injuries, three-pointers, LeBron James, statistics, sneakers and 2024 updates.
DavidSwing
| #
Всё для строительства и ремонта https://artpaint.com.ua на одном портале: советы экспертов, обзоры материалов, расчет сметы и готовые решения для вашего дома или бизнеса.
IsaacVoild
| #
Портал о строительстве https://aziatransbud.com.ua статьи, видео, инструкции, каталоги материалов и инструментов. Советы для дома и бизнеса. Легко строить, удобно ремонтировать!
Anthonybeisy
| #
Портал для строительства https://6may.org и ремонта: полезные советы, современные материалы, проекты и идеи. Все, что нужно для воплощения ваших задумок – от фундамента до крыши.
RobertBer
| #
Студия дизайна интерьера https://bconline.com.ua и архитектуры: создаем уникальные проекты для квартир, домов и коммерческих пространств. Эстетика, функциональность и индивидуальный подход – в каждом решении.
RobertFuese
| #
Все об озеленении и благоустройстве https://bathen.rv.ua Ландшафтный дизайн, проекты садов, террас и парков. Идеи для создания зеленых зон, подбор растений и профессиональные услуги для вашего участка.
MichaelVat
| #
стероид бутс drags-masses.com
Jamesrit
| #
CMS 1С-Битрикс Бизнес https://magikfox.ru/catalog/license/upravlenie-saytom/biznes/ купить лицензию в официальном маркетплейсе. Редакция Bitrix Бизнес для интернет магазина. Быстрая отправка лицензионного ключа. Помощь в установке и настройке.
Robertsuert
| #
пневмозаглушка пзу пневмозаглушки
Kevinzef
| #
Ваш путеводитель в мире строительства https://dcsms.uzhgorod.ua идеи, планы, пошаговые инструкции и лучшие материалы. Узнайте, как построить дом мечты или обновить интерьер.
Robertthula
| #
Профессиональный портал для строительства https://blogcamp.com.ua проекты, материалы, расчеты, советы и вдохновение. Все, чтобы ваш ремонт или стройка были успешными.
Davidscoop
| #
Делайте ремонт https://esi.com.ua и стройте легко! Лучшие советы мастеров, подбор инструментов, инструкции и сметы. Мы поможем справиться с любой задачей.
JerryNef
| #
Хотите построить дом https://donbass.org.ua или сделать ремонт? Здесь вы найдете всё: инструкции, идеи, современные технологии и проверенные решения. Портал для тех, кто строит.
Robertdam
| #
Все для строителей и мастеров https://dki.org.ua актуальные технологии, практические советы, строительные материалы и проекты. Простые решения для сложных задач!
Antoniofelty
| #
Создайте дом своей мечты https://intellectronics.com.ua На нашем портале вы найдете идеи, инструкции и новейшие технологии для ремонта и строительства.
JacobRedia
| #
Станьте мастером https://fmsu.org.ua своего дела! Портал для тех, кто хочет строить и ремонтировать качественно и выгодно.
KennethDew
| #
Ландшафтный дизайн https://kinoranok.org.ua и благоустройство для дома, офиса или парка. Профессиональные советы, подбор растений и реализация уникальных зеленых проектов.
WilliamSok
| #
Архитектура и дизайн интерьера https://it-cifra.com.ua под ключ: современные решения, индивидуальный подход и гармония стиля и функциональности. Создаем пространство вашей мечты!
JamesENDOX
| #
Найдите все для ремонта https://keravin.com.ua и строительства! Уникальные идеи, пошаговые инструкции и рекомендации специалистов на одном портале.
Kevinrer
| #
Стройте с комфортом https://mr.org.ua полезные советы, новейшие технологии, пошаговые инструкции и проекты – всё для вашего удобства.
Clintontek
| #
Мы помогаем строить https://juglans.com.ua лучше! Советы, проекты, новейшие материалы и технологии для вашего ремонта или строительства.
Glennzit
| #
Ваш путеводитель в мире строительства https://mtbo.org.ua полезные рекомендации, готовые проекты и современные решения для любых задач.
PeterskerY
| #
Решили строить или делать ремонт https://msc.com.ua Мы подскажем, как выбрать лучшие материалы, спланировать бюджет и воплотить все задумки.
MichaelRoumn
| #
Строительство без лишних вопросов https://okna-k.com.ua наш портал – кладезь информации о современных материалах, технологиях и лучших решениях для дома, дачи или офиса.
CharlesOrgab
| #
Всё для успешного строительства https://newboard-store.com.ua и ремонта на одном портале! Мы собрали актуальную информацию, идеи и инструкции для вашего удобства. Заходите и стройте с нами!
Jasonlek
| #
Все секреты https://mramor.net.ua строительства в одном месте! Советы экспертов, подбор материалов и готовые проекты для вдохновения.
Jimmietwire
| #
Ваш путеводитель в строительстве https://quickstudio.com.ua Ищите материалы, технологии или советы – всё это есть на нашем портале. Стройте с комфортом!
MichaelBal
| #
create a company in montenegro Montenegro temporary residence
ElmerMer
| #
Информация о стройке https://purr.org.ua без лишних сложностей! Наш портал поможет выбрать материалы, узнать о технологиях и сделать ваш проект лучше.
Jamesbooms
| #
1win website when is 1win token listing
HermanPew
| #
1win bet 1win bet
StephenHon
| #
Всё для вашего ремонта https://reklama-region.com и строительства в одном месте! Практичные советы, современные решения и актуальная информация для успешного проекта.
EzekielDob
| #
Всё, что нужно знать о металлах https://metalprotection.com.ua от их свойств до применения в различных отраслях. Обзоры, советы, новости и информация о производителях для вашего удобства.
Kevinhew
| #
Вавада предлагает приложения для ставок на любой вкус! Здесь вы найдете ставки на футбол, теннис, баскетбол, киберспорт и многое другое. Широкий выбор событий, удобный интерфейс и выгодные коэффициенты делают платформу идеальной как для новичков, так и для опытных игроков. Начните свой путь в ставках уже сегодня!
LarryCix
| #
Хотите построить дом https://samozahist.org.ua или сделать ремонт? На нашем портале вы найдёте лучшие решения и вдохновение для вашего проекта.
Richardinjuh
| #
Ищете проверенные строительные советы https://rus3edin.org.ua Наш портал поможет выбрать материалы, спланировать проект и сделать всё на высшем уровне.
Ronaldfoere
| #
Строительный портал https://sinergibumn.com для тех, кто хочет знать больше о строительстве. Актуальные идеи, проверенные технологии и вдохновение для любого проекта.
Danielkam
| #
Всё о дизайне интерьера https://sculptureproject.org.ua в одном месте! Узнайте, как создать уютное, стильное и функциональное пространство, которое будет радовать каждый день.
FrankJeasy
| #
разработка и утверждение программы производственного контроля Москва https://ppk-213.ru
RaymondCar
| #
Экспертный строительный портал https://smallbusiness.dp.ua для вашего проекта! Советы, новинки и инструкции для тех, кто хочет сделать всё идеально.
Stacywem
| #
Хотите стильный интерьер https://sitetime.kiev.ua Наш портал предлагает уникальные идеи, профессиональные рекомендации и примеры лучших дизайн-проектов.
Rogerfar
| #
Строительный портал https://sushico.com.ua для профессионалов и новичков: от выбора материалов до готовых проектов. Легко найти подрядчиков, изучить современные технологии и воплотить идеи в жизнь!
Bretthuh
| #
Найдите всё о строительстве https://srk.kiev.ua и ремонте на нашем портале. Полезные статьи, актуальные технологии и лучшие практики ждут вас.
DavidPen
| #
Преобразите ваш дом https://vineyardartdecor.com вместе с нами! На портале вы найдёте свежие идеи, советы по планировке и материалы для создания идеального интерьера.
Morrisneoro
| #
Планируете стройку https://texha.com.ua или ремонт? У нас вы найдёте проверенных специалистов, инструкции, материалы и проекты на любой вкус. Всё для комфортного строительства!
RonaldHab
| #
Всё о строительстве https://valkbolos.com и ремонте на одном портале! Гид по материалам, обзор инструментов, советы по дизайну и подбор подрядчиков. Создавайте дом своей мечты!
Williamunodo
| #
Ремонт и строительство https://sota-servis.com.ua легко! Здесь вы найдёте инструкции, рекомендации, материалы и специалистов для успешного выполнения ваших задач.
DonaldRet
| #
Планируете ремонт или строительство https://vodocar.com.ua У нас всё, что нужно: от инструкций и советов до подрядчиков и обзоров материалов. Стройте с нами!
Robertofroni
| #
Лучшие советы по строительству https://stroysam.kyiv.ua и ремонту на одном сайте! Найдите вдохновение, изучите обзоры и воплотите свои идеи с профессиональной помощью.
HaywoodtUp
| #
Полный справочник по строительству https://stroy-portal.kyiv.ua и ремонту: советы, инструкции, дизайн-решения и помощь с выбором материалов и подрядчиков.
MichaelJab
| #
Ваш гид в мире строительства https://vitamax.dp.ua и ремонта! Обзоры, практические советы, дизайн-идеи и подбор профессионалов для реализации любых проектов.
SamuelWhisa
| #
Сделайте ремонт https://tfsm.com.ua мечты с нашим сайтом! Советы, инструкции, рейтинг специалистов и новинки строительного рынка для вашего удобства.
ShawnIcoft
| #
Простые решения для ремонта https://teplo.zt.ua и строительства! Идеи дизайна, рекомендации экспертов и проверенные материалы для вашего проекта.
RaymondLeque
| #
Стройте и ремонтируйте https://suli-company.org.ua с лёгкостью! Полезные статьи, инструкции, советы по выбору материалов и подрядчиков ждут вас здесь.
Walterdut
| #
Портал о ремонте и строительстве https://buildingtips.kyiv.ua с полезными статьями, рекомендациями по выбору материалов и подрядчиков.
Nathantes
| #
Полный гид по строительству https://tsentralnyi.volyn.ua и ремонту: от планирования до отделки. Читайте, выбирайте и стройте с уверенностью и комфортом.
Nathantes
| #
Полный гид по строительству https://tsentralnyi.volyn.ua и ремонту: от планирования до отделки. Читайте, выбирайте и стройте с уверенностью и комфортом.
MichaelMonee
| #
ggdrop free spin промокод гг дроп
TimothyDak
| #
Приветствую на https://b2best.at! Мы предлагаем надежные и проверенные покупки в интернете. Ознакомьтесь с нашими статьями о безопасности и легальности. Ваши покупки — наш приоритет!
Cornellref
| #
Tormac.org https://tormac.org – это специализированный торрент-трекер, предназначенный для пользователей Mac-компьютеров. Сайт предоставляет широкий выбор контента, ориентированного на операционные системы macOS и iOS.
SamuelWhisa
| #
Сделайте ремонт https://tfsm.com.ua мечты с нашим сайтом! Советы, инструкции, рейтинг специалистов и новинки строительного рынка для вашего удобства.
MichaelSpino
| #
A detailed history inter-milan-az.com of the Italian football club Inter Milan. From their first Scudetto to their Champions League victory.
kinogo
| #
kinogo
LucasMor
| #
mostbet casino bonus mostbet скачат
Jamesmub
| #
mostbet промокод 2024 mostbet kasino
BryanVar
| #
lucky jet 1win скачать на айфон 1win зарегистрироваться
Chesterguarm
| #
MLB Draft Betting https://bettingblog.website Your guide to the world of MLB draft betting. Expert predictions, top odds and detailed analysis will help you increase your chances of success. Bet and win with us!
AntonioCem
| #
Best Forex Trading Course https://blogforex.tech is your key to successful trading. Learn the secrets of professionals, study strategies and learn how to minimize risks. Master Forex easily and effectively!
RobertWox
| #
Credit Union Mobile Home Loans https://blogcredit.tech are the perfect solution for buying or refinancing a mobile home. Affordable rates, easy application, and reliable support every step of the way. Take the first step toward your home with us!
MichaelTof
| #
Tennis betting https://yourmoneyblog.site best odds, predictions and analytics. Explore detailed match reviews, statistics and strategies to make successful bets. Use our tips and win!
FrancisVaf
| #
Federal Gov Open Enrollment https://body-balance.online is your chance to upgrade or choose an insurance plan. Easy navigation, expert support, and a wide range of programs will help you make the right choice. Apply now!
RobertCef
| #
Crypto Funk https://besttodaynew.com is a fresh look at cryptocurrencies. News, trends, guides and analytics for beginners and professionals. Find out how to get the most out of blockchain technology!
Enriqueusece
| #
TaskMy.ru – профессиональная помощь в решении задач любого уровня
TaskMy.ru – это надежный сервис, который предлагает качественную помощь в выполнении задач любых направлений: от технических расчётов и программирования до написания текстов и аналитики. Мы работаем быстро, эффективно и ориентированы на ваши требования.
Доверяя TaskMy.ru, вы получаете индивидуальный подход, точное соблюдение сроков и доступные цены. Оставьте свою задачу профессионалам – результат превзойдет ожидания!
MarvinNaw
| #
Auto loans from Community Credit Union https://sunnydays100.com are simple, affordable, and great value. Low interest rates and flexible repayment options make it easy to buy a new or used car.
Nelsontaf
| #
Home Equity Loans https://funnydays1.com How They Work, What Are the Terms and Benefits? Get the full details on how to use your home’s value for financial purposes. Find out more today!
Felipenog
| #
Credit score requirements for FHA loans https://lifeofnews1.com minimum threshold and tips for improving. Find out how to increase your chances of getting a loan, as well as what affects approval. Detailed information for those who want to get a mortgage through FHA.
Glenndup
| #
No Credit Check Loans in Abilene TX https://daynewday1.com is fast access to money without unnecessary checks. Convenient terms, simple application and instant approval. Get financial help when you need it!
RobertGer
| #
Размеры картонных https://giftbox-3.ru коробок оптом и печать коробки с логотипом в Братске.
RonaldSmaps
| #
Try your luck at taya365 download, where excitement meets reliability! Hundreds of popular games, unique promotions and instant payouts await you.
Pedrodef
| #
kush for sale in Prague Hashish for sale in Prague
RichardHax
| #
Hash delivery in Prague THC gummies for sale in Prague
BruceVot
| #
Biography of football legend Pele pele-az.com/ personal life, ex-spouses, children and current wife Maria Aoki. A reminder of his legendary career, goals and special style of play, as well as successful performances in the national team.
Larrydix
| #
Paolo Maldini’s biography http://paolo-maldini-az.org photo, defender 2019, personal life, Instagram, Milan, salary, religion and news.
Charleswon
| #
Biography of Spanish footballer Xavi Hernandez http://xavi-hernandez-az.com coaching career, Barcelona statistics, matches with Iniesta and Messi, Guardiola’s influence, goals, youth training, dismissal as Barcelona coach, personal life and 2024 updates.
DustyHok
| #
Discover the life of Sergio Busquets sergio-busquets his parents, his partner Elena Galera Moron, his sons, his club career, his achievements with Spain and the latest news for 2024.
CharlesFUT
| #
Biography of Brazilian footballer Ronaldinho ronaldinho-az.org personal life, son’s contract, current position. Club career, free kick goals, dribbling, jersey number, Ballon d’Or award, prison sentence. Latest news of 2024.
Wendellfloab
| #
Biography of Spanish footballer jordi-alba-az.org Jordi Alba: Left back for Barcelona and the Spanish national team, trained at the academies of Barcelona and Valencia.
Josephinivy
| #
Biography of footballer casemiro-az.org Casemiro: personal life, wife and children. Career, statistics, salary at Real Madrid, Brazil national team, transfer fee, position, transfer to Manchester United and the latest news for 2024.
Timothybob
| #
Welcome to the fan site memphis depay dedicated to the active Dutch footballer Memphis Depay, his career path with clubs and the Dutch national team. Tattoos, personal life and news.
CraigZew
| #
popular manga Dragon Ball free online manga update Naruto free online
JeremyMeelo
| #
James Rodriguez james-rodriguez biography: personal life, latest news, Instagram, goals, transfer to Rayo Vallecano, statistics and captain of the Colombian national team.
Jamesfrott
| #
Nicholas Jackson nicolas-jackson-az com is a Senegalese professional footballer who has taken the football world by storm with his play and achievements.
SpencerImike
| #
Biography Pau Victor https://pau-victor-az.org is a Spanish footballer who started in the lower divisions. Thanks to hard work, he got into a good youth team at FC Barcelona. He played for Girona and has good statistics.
RonaldVerma
| #
Biography of Jamie Bynoe-Gittens jamie-gittens-az.org/ a winger for German club Borussia Dortmund, and an English footballer.
Rogercoott
| #
When Huseyin and Naime hakan-calhanoglu welcomed their son Hakan to Monchengladbach, Germany, during the 1994 World Cup, they had no idea he was destined to become a star. Learn his biography, facts and news.
BennyJarie
| #
Biography of Dominik Szoboszlai dominik szoboszlai az org a Hungarian footballer, midfielder for Liverpool and captain of the Hungarian national team.
RogerJaway
| #
Biography of Ollie Watkins https://ollie-watkins-az.org an English striker for Aston Villa and the England national team. Find out about his career, clubs such as Exeter City and Brentford, height, age, achievements, participation in Euro 2024, personal life and latest news.
WilbertRot
| #
Phil Foden Biography https://phil-foden-az.org A look at his personal life, relationship with Rebecca and the birth of his sons and daughters.
GeorgeSluth
| #
Francisco Conceicao https://francisco-conceicao-az.org was born into a family where football was already a part of the family, with a legacy built on hard work and passion. Juventus, Porto and Portugal national team appearances, brother Rodrigo, FIFA stats and news.
WilliamVok
| #
Biography of Austrian jorginho-az org footballer Arnautovic Marko – games at FC Werder Bremen and Internationale, market value, achievements. Personal life, conflicts, rumors and latest news.
GeorgeInvak
| #
Biography of Austrian footballer marko arnautovic az org Arnautovic Marko – games at FC Werder Bremen and Internationale, market value, achievements. Personal life, conflicts, rumors and latest news.
RobertNeaxy
| #
Biography of Federico Chiesa federico-chiesa-az.org Italian winger for Juventus and the Italian national team. Career, Fiorentina, Euro 2024, transfer to Liverpool, family, wife Lucia Bramanti.
Jasontoini
| #
Aurelien Tchouameni’s aurelien-tchouameni-az.org biography: personal life, date of birth, parents’ jobs, statistics at Real Madrid and Monaco, position, jersey number, 2024 updates.
RonaldHex
| #
Biography of Dayot Upamecano dayot upamecano Bayern star, France national team hero, early career start, Euro play-off participation, personal life and football news for 2025.
Jordannaw
| #
Biography of Portuguese bernardo silva footballer Bernardo Silva: personal life, relationship with his wife, similarities with Bruno Fernandes. Current team, number on the field, reviews of fans who have sold out.
SidneySox
| #
Biography of Brazilian football player Rodrigo rodrygo-az com photos of the striker of the Real Madrid club and the Brazilian national team, sports career.
Michaeltrals
| #
Biography of Spanish pedri footballer Pedri, statistics at Barcelona, ??games with teammate Gavi, joining the national team for the Euros.
Archieplabe
| #
Biography of football player antoine griezmann Antoine Griezmann: personal life, birth of children, national origin. Now his career, playing for Atletico Madrid.
ThomasCig
| #
Biography of Brazilian https://alisson-becker-az.com footballer Alisson: photo of the goalkeeper of the Liverpool club and the national team, sports career, playing for FC Inter and Roma, growth, achievements.
Jamiepef
| #
Нужен ремонт техники чинпочин.рус все услуги для вашего дома в одном месте! Выбирайте мастеров для ремонта, уборки или сантехнических работ. Качественный сервис, прозрачные цены и удобство использования.
RafaelGeala
| #
Tobey Maguire’s tobey-maguire-az com biography: personal life, memories of him, friendship with Leonardo DiCaprio, divorce from ex-wife. Role in Spider-Man films, career now.
Jeremyclear
| #
Biography of Spanish footballer Dani Carvajal http://dani-carvajal-az.com personal life, marriage to Joselu and twin sisters. Performances at the Euro for Real Madrid and the Spanish national team.
AnthonyMab
| #
Biography of footballer https://iker-casillas-az.com Iker Casillas: personal life, separation from ex-wife Sara Carbonero.
MerleDiolo
| #
Biography of actor Keanu Reeves keanu-reeves-az.com/ personal life, the tragic death of his child and his relationship with his common-law wife Jennifer Syme, artist Alexandra Grant.
DavidSox
| #
portable ftp software Newsgroup Clients
Jesustooff
| #
“Ищете качественный кирпич напрямую от производителя? https://Muravey61.ru – ваш надежный поставщик строительных материалов в регионе! Мы предлагаем кирпич высшего качества по доступным ценам прямо с завода. Доставка точно в срок, широкий ассортимент, и гарантированное качество – всё, что нужно для вашего строительства. Закажите у нас и убедитесь сами, что с нами строить легко!”
Josephcew
| #
Now you can’t find a person https://formula1-az.com who hasn’t heard of Formula 1. Today it is one of the most prestigious and popular sports on the planet.
Ronaldcrest
| #
Biography of Zendaya Coleman zendaya maree (Zendaya): modeling career, music and cinema, details of her personal life, ex-boyfriend Jacob Elordi, romance with Tom Holland.
BruceBon
| #
Welcome to the main page karim-benzema-azerbaijan.com of the fansite dedicated to the world football star Karim Benzema. Learn all about his incredible career, incredible achievements and phenomenal skills.
Eugenevat
| #
Welcome to the world of Neymar neymar-azerbaycan com a fan site dedicated to the great footballer. Learn all about his career, achievements and unique playing style.
VernonBiore
| #
Biography of British actress emily blunt az com Emily Blunt: personal life, dating and relationship with her husband John Krasinski, raising children.
Vanilla Gift Card
| #
We genuinely appreciate your guidance and the thoughtfulness you’ve shown. It matters greatly to us.
KelleySoync
| #
Jogo Do Tigrinho slot takes you to the world of exoticism and excitement. Incredible winnings, bonus games and a dizzying jungle atmosphere await you. Bright graphics and exciting gameplay make the game unforgettable. Try your luck right now!
Brandoncab
| #
Welcome to the fan site moussa-dembele-az com of the talented footballer and real star Moussa Dembele! Here you will find everything new and most interesting about his career, achievements and amazing moments on the field.
Nathanhoast
| #
скупка золота спб дорого цена за грамм скупка золота 585
StephenInhit
| #
Оперативная помощь на дороге https://angeldorog.by услуги эвакуатора, грузовой и легковой шиномонтаж, а также грузоперевозки фурами по доступным ценам. Работаем круглосуточно, быстро реагируем и гарантируем надежность. Звоните в любое время – решим вашу проблему!
JefferyRok
| #
CMS software Webcam security camera software
DavidGrite
| #
Discover the ultimate hub zinedine zidane azerbaijan com for all things Zinedine Zidane. Dive into in-depth analysis of Zidane’s illustrious career, from his legendary performances to his coaching triumphs.
ErnestFrazy
| #
Find out all about Kylie Jenner kylie-jenner-azerbaijan com at Kylie Jenner, the ultimate fan site for the latest updates on her fashion line, beauty tips and personal life.
Metamask Download
| #
I’ve been using MetaMask Wallet for years, and recently downloaded the latest MetaMask Extension via https://metalead.org/. It works flawlessly on Chrome and Safari browsers!
RobertKat
| #
Immerse yourself in the world of Gigi Hadid gigi-hadid-azerbaijan.com/ through our dedicated website. Explore the latest news, highlights, exclusive interviews and in-depth features about her fashion endeavors, personal life and charitable efforts.
Frankger
| #
Biography of American professional gervonta-davis-az.com boxer Gervonta Davis: career highlights, weight, records, famous fights, personal life, controversies and latest 2024 updates.
Leroynop
| #
Discover the world of Brad Pitt brad pitt az com with our dedicated website, offering extensive coverage of his illustrious career, upcoming projects, and personal endeavors.
WalterBorne
| #
how to book a tesla rent a electric car near me
GregoryUniog
| #
У нас вы можете купить айфоны https://vk.com/crazy_humor01 оптом по самым лучшим ценам. Оригинальные смартфоны Apple с гарантией качества. Постоянное наличие популярных моделей.
JesusLaple
| #
Квартирный переезд https://spb-gruzoperevozka.ru с грузчиками быстро и качественно! Упакуем, вынесем, перевезем и разместим вещи на новом месте. Надежная команда, аккуратность и доступные тарифы.
Josephdof
| #
стол овальный офисный для переговоров https://mm26.ru
Abigael206
| #
You have done a amazing job with you website
Willierox
| #
Откройте для себя последние kraftsir.ru новости, аналитику и экспертные обзоры о спорте и ставках. Узнайте о лучших стратегиях ставок, следите за актуальными событиями в мире спорта и получите все необходимые инструменты для успешных ставок.
GermanHit
| #
киного фильмы про магию kinogo фильмы о космосе
Williamhit
| #
киного фильмы 2025 kinogo фильмы про оборотней
Davidinelt
| #
Подробные стратегии покера https://fatcurus.ru и анализ турниров: эффективные тактики, разбор раздач и ключевые советы для улучшения игры. Только практическая информация для выигрышей.
Calvinloash
| #
Новости игровой индустрии depcult35.ru/ аналитику и обзоры самых популярных игр. Читайте о новинках, трендах и получайте полезные советы для улучшения игрового процесса.
Romanspeab
| #
Все о мире гэмблинга https://minsvyazcc.ru обзоры казино, игры, стратегии и последние новости индустрии. Узнайте о новых слотах, бонусах и тенденциях в азартных играх.
JeffreyVep
| #
Все главные события https://tankdiv.ru мира спорта: обзоры турниров, результаты матчей, аналитику и актуальные новости из самых популярных дисциплин.
StephenAmimb
| #
снять проституток калуга шлюхи калуги
Jameszet
| #
OR Realty — это ваш надежный партнер в мире недвижимости. Мы предлагаем большой выбор квартир, домов и коммерческих объектов по выгодным условиям. Наши специалисты помогут вам найти идеальный вариант, соответствующий вашим потребностям. Надежность, качество и удобство — вот что делает OR Realty лучшим выбором. Обращайтесь!
RobertTaw
| #
скачать приложение mostbet mostbet 1-15
Williamcow
| #
1win скачать 1win aviator
DonaldDaymn
| #
Свежие новости спорта angryfoxtattoo откройте для себя последние спортивные новости, аналитику ставок и прогнозы экспертов на азербайджанском языке. Самая актуальная информация о футболе, киберспорте и других видах спорта здесь!
JamalPoexy
| #
mostbet live mostbet uzb
ForestSax
| #
увлекательные стратегии mexatrondiy ru репортажи с турниров и последние новости покера. Станьте мастером игры, окунитесь в мир азартных карт с нашим сайтом
Odelldralk
| #
узнайте последние новости umra tour ru киберспорта, анализ турниров и игровые стратегии в Азербайджане. Присоединяйтесь к нам, чтобы побеждать в мире киберспорта.
Gilbertseics
| #
Актуальные и свежие новости gastromoroz.ru спорта и казино. Узнайте актуальные спортивные события, анализы матчей, советы по ставкам, обзоры казино игр и стратегии.
KeithNep
| #
[url=https://katun-room.ru]Katun Room[/url] — это уютные гостиницы в самом сердце природы Алтая. Мы предлагаем комфортное проживание рядом с живописной рекой Катунь. Номера на любой вкус и бюджет, тишина и свежий воздух создадут незабываемую атмосферу для вашего [url=https://katun-room.ru]отдыха[/url]. Забронируйте ваш номер уже сегодня!
MichaelGem
| #
kinogo фильмы ужасов киного сериалы про преступников
AnthonyErork
| #
киного фильмы для ноутбука kinogo мультфильмы
Robertsok
| #
kinogo короткометражки kinogo фильмы по студиям
Jamesacuth
| #
A modern AI tool ai undress for working with images. Learn more about its features, capabilities and applications. Full privacy control and ease of use will ensure comfortable interaction.
LucasVuh
| #
Footballer Bukayo Saka’s biography http://bukayo-saka-az.org personal life, Nigerian heritage, religious views and girlfriend’s name. Salary, playing stats, jersey number, Euro appearances with England and matches with Arsenal. Latest news for 2024.
Elliotcenom
| #
Basketball player Jimmy Butler’s jimmy butler biography: personal life, Selena Gomez relationship rumors, height, cars, idol stats, Miami Heat salary, injury news and his coffee brand. Latest news of 2024.
ErnestGab
| #
Biography of footballer Joshua Kimmich jimmy butler personal life, wife and children. Career at Bayern Munich, statistics for Germany, work with Pep Guardiola, negotiations with Barcelona. Vice-captain at Euro 2024. Latest news for 2024.
TylerGam
| #
Djokovic’s tennis career novak-djokovic-az.org/ has been marked by numerous Grand Slam titles, showcasing his style of play. He has been a constant competitor to Rafael Nadal and Roger Federer.
StevenVor
| #
Свежие футбольные новости vseofootball.ru/ обзоры матчей, Лига чемпионов, статистика и лучшие букмекерские бонусы для ставок на спорт.
JamesMic
| #
Откройте мир мобильных игр http://games-ru.ru рейтинги лучших проектов, тренды, советы и гайды. Играйте в популярные шутеры, RPG, стратегии и песочницы прямо на телефоне!
NormanniG
| #
Все о Тони Кроосе http://toni-kroos.ru на одном сайте: биография, актуальные новости, детальная статистика и эксклюзивные обновления о немецкой футбольной звезде. Присоединяйтесь к сообществу фанатов и будьте в курсе всех событий, связанных с Кроосом!
BradyShoft
| #
Источник новостей о футболе futbol-vpered сборной России, Кубке России, Лиге Европы и лучших букмекерских предложениях. Узнайте последние результаты, прогнозы и аналитику!
Williekig
| #
Погрузитесь в захватывающий мир cleopatra-slot ru Cleopatra Slot! Узнайте о правилах игры, бонусных функциях и стратегиях, чтобы увеличить свои шансы на выигрыш.
Kennethbeend
| #
Все о ставках на спорт betting-ru.ru лучшие стратегии, прогнозы экспертов, обзор букмекерских контор и советы для успешного беттинга. Узнайте, как заработать на спортивных ставках!
RussellKab
| #
Исследуйте Zeus vs Hades zeus-vs-hades-download ru Gods of War! Играйте бесплатно, получайте фриспины и узнайте все о слоте 2024 года!
CharlesReula
| #
In Jodo Do Tigrinho online slots, every spin is a step towards victory! Incredible graphics, exciting themes and many bonuses await you. Fortune favors the brave – try your hand and discover the world of winnings with Tigrinho!
Richardmum
| #
Cryptocurrency trading service bitqt with AI is automation and efficiency. Artificial intelligence monitors market dynamics, reduces risks and optimizes transactions. The perfect solution for beginners and professionals.
Metamask Wallet
| #
MetaMask Download was a breeze! Setting it up on Safari and Opera was equally simple. If you’re starting out, https://metanaito.net/ is a great resource.
Graphic Designer
| #
Funny t-shirts for sale offer a great way to add personality and humor to your wardrobe. As a t-shirt graphic designer, I love creating designs that make people laugh, whether it’s through clever wordplay, quirky illustrations, or pop culture references. Each design is an opportunity to turn a simple shirt into a fun statement piece that brightens someone’s day. It’s rewarding to know that a little creativity and humor can make fashion more enjoyable for others!
https://comiko.net/u/2432768-tshirtsale
GenaroAgelt
| #
Откройте для себя Mega Joker mega-joker.ru/ увлекательный видеослот от NetEnt, который сочетает в себе ностальгическую атмосферу классических автоматов и современные возможности выигрыша.
StanleyVoism
| #
Откройте для себя мир starlight princess slot Starlight Princess Slot! На нашем сайте вы найдете свежие новости, стратегии для увеличения выигрышей и эксклюзивные бонусы.
Aarondorgo
| #
Погрузитесь в мир Plinko plinko-ru.ru увлекательной игры, основанной на случайности и стратегии! Узнайте правила, механики и стратегии для успешной игры, чтобы увеличить свои шансы на крупные выигрыши.
Patricklam
| #
Aviatrix game https://aviatrix-games.com/en/ has become a sensation in the world of crash games. Its unique format, featuring a rapidly growing multiplier and the possibility of an unexpected crash. Aviatrix crash game is at 1win, 1xbet, Mostbet, and Pin Up.
Donaldmesty
| #
Захватывающий слот Lucky Jet lucky-jet-play-crash.ru/ от Spribe с уникальной краш-механикой, который предлагает игрокам шанс испытать свою удачу и стратегическое мышление.
Patricklam
| #
Aviatrix game https://aviatrix-games.com/en/ has become a sensation in the world of crash games. Its unique format, featuring a rapidly growing multiplier and the possibility of an unexpected crash. Aviatrix crash game is at 1win, 1xbet, Mostbet, and Pin Up.
GeorgeFut
| #
Откройте для себя водное поло water-polo-ru.ru/ историю, правила, тактики и влияние этого захватывающего водного вида спорта. Узнайте, как динамика и стратегия сочетаются, делая водное поло уникальным и увлекательным.
KevinPef
| #
Добро пожаловать на сайт redtigerplay.ru посвященный слотам Red Tiger! Узнайте последние новости, стратегии для увеличения выигрышей и эксклюзивные бонусы.
DennisSlilt
| #
Погрузитесь в захватывающий мир баккары http://baccarat-ru.ru узнайте историю игры, её правила, стратегии и секреты успеха. Откройте для себя элегантность и интригу одной из самых престижных карточных игр казино.
KerryFum
| #
Откройте для себя мир гандбола https://handball-ru.ru изучите историю игры, основные правила, ключевые стратегии и влияние на культуру.
CliftonTAH
| #
Погрузитесь в увлекательный мир покера http://poker-ru.ru исследуйте его историю, правила, продвинутые стратегии и культурное влияние. Узнайте, как интеллект и интуиция объединяются в этом захватывающем карточном спорте.
KeithPreag
| #
Мир блэкджека http://blackjack-ru.ru узнайте историю игры, её правила, секретные стратегии и влияние на современную культуру. Откройте для себя глубину этой захватывающей карточной игры и научитесь побеждать с умом.
Aaronoreva
| #
Погрузитесь в мир фигурного катания figure skating исследуйте его историю, уникальные техники, знаменитых спортсменов и культурное влияние.
ArmandoCrype
| #
Скачайте бесплатно книгу https://storitelling.ru по сторителлингу и узнайте, как создавать истории, которые цепляют с первых строк. Практические советы, примеры и вдохновение для всех, кто хочет освоить искусство рассказчика.
Albertjeamb
| #
Сайт игровых промокодов csgo500 промокод это ваш доступ к эксклюзивным бонусам и скидкам. Бесплатные награды, внутриигровая валюта и уникальные акции ждут вас. Успейте воспользоваться всеми возможностями!
PatrickUtibe
| #
Хотите раскрутить раскрутить телеграм-канал мы знаем, как это сделать! Поможем увеличить охваты, привлечь активную аудиторию и вывести ваш контент на новый уровень.
ThomasSTOVE
| #
вызвать капельницу от запоя на дому http://www.medlinks.ru/article.php?sid=110769
Arminda Bombino
| #
I close my browser often for various reasons, and having to repeatedly log into my accounts like Facebook, Yahoo, Gmail, and etc, is very inconvenient and getting gold. Firefox 2 never did this, just my current Firefox 3..
MatthewZit
| #
Ищете промокоды для игр rust4real промокоды на кейсы наш сайт – ваш лучший помощник! Собираем актуальные игровые промокоды для бонусов, скидок и эксклюзивных наград.
WesleyTooxy
| #
Участок в Мишкином Лугу http://мишкинлуг.рф/uchastki-mishkinlug по Симферопольскому шоссе — идеальное место для строительства! Тихий поселок, прекрасные виды, удобный подъезд и все условия для комфортной жизни.
MitchSlest
| #
glory-casino-bd.online
ShawnRubre
| #
Флешка оптом криптекс и купить ручку с флешкой в Новороссийске https://flashki-optom-1.ru/
MelvinEtelf
| #
Отзывы о компаниях и работодателях https://potrebsojuz.ru в одном месте. Узнайте реальное мнение сотрудников и клиентов, чтобы принять правильное решение при выборе работы или услуг.
EdwardAvaix
| #
Комплексная юридическая помощь https://kramzenergo.ru для вас и вашего бизнеса. Анализ дел, представительство в судах, поддержка на всех этапах
Stevensainc
| #
https://odnazhdyvskazke-tv.ru/
Frankpef
| #
вывод из запоя капельница на дому http://www.medlinks.ru/article.php?sid=110769
Howardven
| #
1win argentina 1win sign in
ThomasCah
| #
1win bono sin deposito app 1win
Arthurkes
| #
Откройте для себя историю слово пацана кровь на асфальте честный взгляд на суровую реальность, где дружба и слово дороже всего. Уникальный проект о жизни без прикрас и ценности принципов.
Metamask Chrome
| #
Secure your cryptocurrency by setting up your MetaMask Wallet today. For helpful tips, visit https://kingroada.com/.
JamesVer
| #
kinogo лучшие фильмы по комиксам киного российские сериалы
Richardanype
| #
киного фильмы по режиссерам киного сериалы по алфавиту
AlvinRut
| #
Vavada Casino to miejsce, gdzie emocje gry są zawsze na wyciągnięcie ręki. Oferujemy bogaty wybór najlepszych automatów do gier, klasycznych gier stołowych, takich jak poker, blackjack czy bakarat, a także wciągającą ruletkę na żywo z udziałem profesjonalnych krupierów. Nasza platforma Vavada Casino jest w pełni licencjonowana i zgodna z obowiązującymi przepisami prawa, co zapewnia bezpieczne i legalne środowisko rozgrywki dla wszystkich graczy.
CharlesAvefE
| #
kinogo приключения kinogo авторское кино
StevenAmori
| #
Witaj w Slottica PL! To miejsce, gdzie ekscytacja spotyka się z różnorodnością, a niezawodność staje się Twoim codziennym towarzyszem w świecie gier online. Dla polskich graczy w Slottica Casino przygotowaliśmy ofertę, która spełni wszystkie oczekiwania – od doskonałej kompatybilności mobilnej, przez szeroką gamę gier, aż po usługi skoncentrowane na Twoim komforcie i bezpieczeństwie.
DavidOneks
| #
Откройте для себя блэкспрут возможности даркнет-рынка с тысячами предложений. Быстрая регистрация, надежные сделки и анонимность на каждом этапе.
TimothyRox
| #
Лучшее онлайн казино https://1wincasino.pl Огромный выбор автоматов, настольных игр и live-казино. Уникальные акции, приветственные бонусы и мгновенные выплаты сделают вашу игру еще интереснее.
BradleyOptof
| #
веселая песня текст https://faav.ru
Metamask Chrome
| #
The Metamask extension has truly revolutionized how I manage my crypto wallets. If you’re looking to securely handle your funds, definitely check out https://metamenu.org/ for an easy guide!
JosephAcign
| #
охота на медведя луки для охоты
Jamesfef
| #
Полный комплекс услуг по оформлению медицинской лицензии с гарантией за 14 дней по договору. Детали: https://www.gta5-mods.com/users/MDLicenzii
WilliamKar
| #
Ресторанный консалтинг UPSKILL https://upskilll.ru/consulting это профессиональные услуги в сфере ресторанного бизнеса: анализ рынка, аудит заведения, устранение слабых мест, обучение персонала, занимаемся наставничеством рестораторов с 2018 года.
Washington post
| #
Washington post Surreal meaning.
Amphibia. Map of tennessee. Abrasion. Kitty.
JasonAlorm
| #
недорогой ремонт стиральных машин на дому ремонт стиральных машин bosch подольск
Roberthor
| #
Платформа Курьер Купер https://cash-kuper.ru открывает возможности для работы в доставке. Свободный график, прозрачная система оплаты и заказы поблизости – идеальный выбор для тех, кто ищет подработку или основной заработок.
StanleyLex
| #
cannabis delivery in prague thc gummies in prague
TommyPal
| #
cannabis for sale in prague https://shop-cannabis-prague.com
Charlesblege
| #
ремонт бойлера цена ремонт накопительных водонагревателей
CliffDem
| #
взять займ онлайн микрозайм
Thomasgeorm
| #
Открывайте кейсы CS:GO https://www.facebook.com/people/Hotdrop_CSGO/61550490187523/ с крутыми шансами на редкие скины. Удобный интерфейс, надежная система и огромный выбор кейсов сделают игру еще интереснее. Начните свой путь к топовым скинам прямо сейчас!
Josephkenue
| #
Bukmacher oraz kasyno internetowe Mostbet PL to jeden z najdłużej działających serwisów hazardowych w sieci z ofertą dla Polaków, gdyż został założony jeszcze w 2009 roku. Platforma może pochwalić się posiadaniem aktywnej licencji od Curacao, która gwarantuje bezpieczeństwo. Gracze w Mostbet Casino będą mogli spotkać tutaj ponad 3000 zróżnicowanych gier hazardowych, które zostały podzielone na automaty, gry stołowe oraz gry na żywo.
MarvinSyday
| #
Откройте яркие кейсы CS:GO кс кейсы и получите шанс выиграть топовые скины! Широкий выбор кейсов, высокий шанс дропа и честная система обеспечат увлекательный опыт.
Richardroope
| #
Пробуйте удачу https://discord.com/invite/B5fF6pW8Cm CS:GO! Шанс получить редкие и дорогие скины, широкий выбор кейсов и удобный интерфейс делают процесс открытия легким и захватывающим.
Carlogal
| #
Рискни и испытай удачу https://t.me/hotdropcases Шанс получить редкие и дорогие скины, широкий выбор кейсов и удобный интерфейс делают процесс открытия легким и захватывающим.
Aaronfance
| #
Ваши любимые кейсы CS:GO https://t.me/s/hotdropcases в одном месте! Большой выбор, удобный интерфейс и высокая вероятность выпадения редких предметов делают процесс открытия кейсов по-настоящему захватывающим.
Oswaldoskync
| #
Сервис бытовых услуг https://gidrostok-servis.ru это удобное решение для любых домашних задач. Уборка, ремонт, сантехника, установка техники и многое другое. Надежные специалисты, быстрое выполнение и доступные цены!
EdwardBelty
| #
подборка фильмов на вечер в хорошем качестве новинки кино 2025 без регистрации
ForestEndab
| #
Нужны деньги срочно взять микрозайм онлайн с быстрым одобрением и моментальным переводом на карту. Минимум документов, удобные условия и прозрачные ставки. Оформите займ прямо сейчас!
Michaelpoole
| #
новые подборка фильмов на вечер 2025 лучшие смотреть кино бесплатно фильмы
Thomasfaimi
| #
Промокоды для игр https://esportpromo.com/cs2/ это бесплатные бонусы, скидки и эксклюзивные награды! Находите актуальные коды, используйте их и получайте максимум удовольствия от игры без лишних затрат.
JamesNot
| #
Лучшие игровые промокоды промокоды standoff 2 на кейсы в одном месте! Активируйте бонусы, получайте подарки и прокачивайте аккаунт без лишних затрат. Следите за обновлениями, чтобы не пропустить новые промо!
Williamfek
| #
Лучшие игровые промокоды промокоды на барабан бонусов ggstandoff в одном месте! Активируйте бонусы, получайте подарки и прокачивайте аккаунт без лишних затрат. Следите за обновлениями, чтобы не пропустить новые промо!
Roberttiree
| #
Оптовые продажи сувениров и подарков и нанесение сувенир оптом в Кемерово: подарки с Нанесением логотипа компании оптом
Andrewabawl
| #
Бесплатные промокоды https://playpromocode.com/standoff/bulldrop/ для ваших любимых игр! Получайте монеты, бустеры, скины и другие ценные награды. Мы собираем только проверенные коды и обновляем их каждый день.
Richardbaina
| #
Хотите проверить компанию https://innproverka.ru по ИНН? Наш сервис поможет узнать подробную информацию о юридических лицах и ИП: статус, финансы, руководителей и возможные риски. Защищайте себя от ненадежных партнеров!
Rickyrouch
| #
Недвижимость на Северном Кипре https://iberiaproperty.ru выгодные инвестиции и комфортная жизнь у моря. Апартаменты, виллы и пентхаусы по доступным ценам. Поможем выбрать лучший вариант и оформить покупку.
Michaelfoege
| #
удобный маркетплейс bs2best с высоким уровнем анонимности и надежной системой защиты. Интуитивный интерфейс, проверенные продавцы и безопасные сделки делают его лучшим выбором для покупок.
Davidfah
| #
Раскрутка в соцсетях https://nakrytka.com без лишних затрат! Привлекаем реальную аудиторию, повышаем охваты и активность. Эффективные инструменты для роста вашего бренда.
Kevintut
| #
увлекательный сериал https://odnazhdy-v-skazketv.ru о жизни сказочных персонажей в реальном мире. Интригующий сюжет, волшебные события и неожиданные тайны. Смотрите онлайн в высоком качестве прямо сейчас!
Rubenesofe
| #
оказание услуг железнодорожных перевозок грузов https://bvs-logistica.com/zheleznodorozhnye-perevozki-gruzov.html
DavidToure
| #
Интернет-магазин товаров https://vitasleep.ru для здорового сна. В ассортименте: ортопедические матрасы, подушки, одеяла, постельное белье и аксессуары от проверенных брендов. Удобный выбор, доставка по России, гарантия качества. Забота о вашем комфорте и здоровом сне!
Brianmaige
| #
escort paris
WilliamVerge
| #
лучшие сериалы онлайн https://lordseriall.org
Percyjix
| #
смотреть сериал все серии подряд сериалы смотреть
BruceBEs
| #
сериалы онлайн подряд смотреть сериал
Josephchoks
| #
смотреть сериалы подряд https://lordserial7.com
JeffreyCak
| #
смотреть сериалы в хорошем качестве https://lordserials2.net
Michaelarguh
| #
смотреть сериал хороший без рекламы https://lordserials.cc
MetaMask Chrome
| #
If you’re new to crypto and need a guide on how to install the Metamask extension, check out https://sites.google.com/view/metamask-extension-dfkasdkfdnt/download. It walks you through everything in a simple way, making it easy to get started. I highly recommend their content!
StevenNot
| #
https://gruzoperevozki-minsk24.ru/
TravisNal
| #
Логистические услуги в Москве https://bvs-logistica.com доставка, хранение, грузоперевозки. Надежные решения для бизнеса и частных клиентов. Оптимизация маршрутов, складские услуги и полный контроль на всех этапах.
ThomasHam
| #
смотреть сериалы подряд https://lordserialss.life
Chrispen
| #
сериалы 2022 смотреть онлайн https://lordsserial.xyz
StephenKef
| #
Хотите почувствовать азарт? arkada casino бонусы за регистрацию предлагает широкий выбор игр, честную систему выигрышей и мгновенные выплаты. Захватывающие слоты и бонусы ждут вас!
GeorgeRog
| #
смотреть лучшие сериалы https://lordseriall6.org
Georgewah
| #
смотреть сериал регистрация https://lordsserials.org
Jamescully
| #
лучшие сериалы онлайн https://lordserial5.pet
Andrewprund
| #
Check out Sellvia https://www.instagram.com/sellvia.dropshipping/ on Instagram for the hottest product ideas, store upgrades, and exclusive deals! Stay in the loop with our latest dropshipping tips and grab promo coupons to boost your business.
GerardFum
| #
Где заказать монтаж плоской кровли с битумной гидроизоляцией, нужны контакты – https://ploskaya-krovli-zagorodnyh-domov.ru/
MetaMask Chrome
| #
I’ve been looking for a reliable way to install Metamask on Chrome, and https://sites.google.com/view/metamask-extension-download-oa/chrome provided exactly what I needed. Their guide made the process effortless.
Numbersbeica
| #
Ищете выгодные курсы? Актуальная подборка со скидками до 99%. Перейти к выбору https://www.intensedebate.com/people/Sofia93
Jeffryexpax
| #
The full special bip39 Word List consists of 2048 words used to protect cryptocurrency wallets. Allows you to create backups and restore access to digital assets. Check out the full list.
Robertzep
| #
Reliable and unique bip39 Word List contains 2048 words needed to create seed phrases in crypto wallets. Allows you to safely manage private keys and guarantees the possibility of recovering funds.
Robertzep
| #
Reliable and unique bip39 Word List contains 2048 words needed to create seed phrases in crypto wallets. Allows you to safely manage private keys and guarantees the possibility of recovering funds.
Michaelblova
| #
cannabis shop in prague https://sale-weed-prague.com
Jerrydag
| #
1xBet promo code https://space-find.net/2024/10/22/step-by-step-guide-to-using-the-1xbet-new-account/ your chance to get bonuses on bets, free bets and exclusive promotions! Enter the code during registration and start playing with an increased deposit.
BradleyUnlic
| #
Ремонт компьютеров и ноутбуков https://remcomp89.ru в Новом Уренгое – быстрые и качественные услуги! Диагностика, настройка, замена комплектующих, восстановление данных. Гарантия на работу, доступные цены и выезд мастера!
arkada casino автоматы
| #
Играйте в аркадные игры в лучшем онлайн казино | Играйте в аркадные игры и выигрывайте крупные призы | Увлекательные аркадные игры и безграничный азарт | Играйте в аркады и выигрывайте деньги | Играйте в увлекательные аркады и выигрывайте призы | Ощутите драйв аркад в онлайн казино | Выигрывайте крупные суммы в аркадах | Самые популярные аркадные игры в одном казино | Лучшие аркадные развлечения для вас | Выигрывайте крупные денежные призы в аркадах | Большие денежные призы в аркадах | Побеждайте в аркадных баталиях и зарабатывайте деньги | Побеждайте в аркадах и станьте миллионером | Играйте в аркады и получайте удовольствие | Побеждайте в аркадах и получайте денежные призы | Побеждайте в аркадных играх и получайте призы
arkada регистрация arkada casino online .
JosephGaw
| #
Looking for the current spinbetter promo code? Get bonus funds for bets and casino games. Easy activation, favorable conditions and real winnings are waiting for you. Hurry to use it!
HaroldMon
| #
Используйте актуальный промокод 1xbet и получите увеличенный бонус на первый депозит! Делайте ставки на спорт, играйте в казино и пользуйтесь эксклюзивными предложениями. Легкая регистрация и моментальные выплаты!
заказать входную дверь
| #
Секреты успешной покупки входной металлической двери, которая прослужит долгие годы.
Лучшие магазины с широким выбором входных металлических дверей.
дверь металлическая входная цена входная дверь .
Что учесть при выборе входной металлической двери.
5 основных причин купить металлическую входную дверь.
В чем отличия входных металлических дверей разных производителей.
заказать двери входные металлические
| #
Как выбрать идеальную входную металлическую дверь, соответствует всем требованиям безопасности.
Где купить надежную входную металлическую дверь по выгодной цене.
заказать входную дверь дверь металлическая входная .
Как не ошибиться с выбором входной металлической двери.
5 основных причин купить металлическую входную дверь.
Как выбрать между дверью одного бренда и дверью другого.
бонус код arkada casino
| #
Онлайн казино с широким выбором аркадных игр | Погрузитесь в мир азарта и аркадных развлечений | Увлекательные аркадные игры и безграничный азарт | Азартные аркады и огромные выигрыши | Лучшие аркадные игры в онлайн казино | Уникальные аркады только у нас | Погрузитесь в мир аркадных развлечений и азарта | Азарт и аркады в одном месте | Играйте в аркадные игры и ощутите азарт | Аркадные игры и азарт ждут вас в онлайн казино | Аркадные игры для всех желающих азарта | Выигрывайте крупные суммы в аркадах | Испытайте свою удачу в аркадном казино | Играйте в аркады и получайте удовольствие | Выигрывайте крупные суммы в аркадных играх | Увлекательные аркады и возможность заработать деньги
arkada casino официальный аркада регистрация .
Steventeaby
| #
bip39
Andrewquick
| #
The most comprehensive bip39 for securely creating and restoring cryptocurrency wallets. Learn how mnemonic coding works and protect your digital assets!
Everetteremy
| #
moving cost estimate Prague Prague to Vienna transport
жалюзи на окна автоматика
| #
Современная автоматика для жалюзи, повысит комфорт в помещении.
Снижайте затраты на отопление и кондиционирование воздуха с автоматизацией жалюзи.
Контролируйте жалюзи с помощью смартфона.
Создайте уютную атмосферу с автоматическими жалюзи.
Качественное обслуживание автоматизированных жалюзи.
автоматика на горизонтальные жалюзи автоматика на горизонтальные жалюзи .
Williamder
| #
Ищете квартиру в новом доме? новостройки Украина – это современные жилые комплексы, доступные цены, рассрочка и ипотека. Подберите идеальное жилье от надежных застройщиков!
Brettcib
| #
Останні актуальні останні новини україна оперативні події, важливі рішення, міжнародна політика та економіка. Все, що потрібно знати про життя країни, в одному місці!
Richardtraft
| #
Продажа инструментов для дома https://profimaster58.ru строительства и ремонта! Огромный выбор ручного и электроинструмента, выгодные цены, акции и быстрая доставка. Найдите все необходимое в одном месте!
BradleyPhoks
| #
Free and reliable best nudify ai tool to create images with AI? The Best Free AI Nudify is the best choice for creating nude photos with privacy and quality guarantee.
Kennethgeore
| #
грузчики челябинск газель недорого услуги профессиональных грузчиков
bip39
| #
Use the proven bip39 phrase standard to protect your assets and easily restore access to your finances. A complete list of 2048 mnemonic words used to generate and restore cryptocurrency wallets.
Raymondmek
| #
Sitio web oficial jude-bellingham.com mx de fans de Jude Bellingham: noticias, logros y material exclusivo sobre la carrera del talentoso mediocampista que juega en el Real Madrid.
rodri
| #
Sitio web de fans https://rodri.com.mx de Rodri Hernandez: Descubre la carrera y logros del mediocampista espanol del Manchester City. Noticias, estadisticas y analisis del juego de uno de los mejores futbolistas actuales.
wjkawsdas_rot
| #
Desde 2019, a Mostbet Portugal tem se consolidado como uma excelente escolha para os jogadores portugueses que buscam formas seguras e divertidas de apostar e jogar nos casinos online e ao vivo. Com uma ampla seleção de jogos, apostas esportivas, esports e apostas em esportes virtuais, a plataforma oferece um catálogo premium de uma marca licenciada e confiável. Confiança, inteligência e lealdade são os pilares que sustentam a Mostbet Casino, que ainda proporciona bônus e promoções exclusivas, métodos de pagamento locais reconhecidos e ferramentas para um jogo responsável: Mostbet Casino
RalphNuast
| #
Kevin De Bruyne kevin-de-bruyne.com mx es un maestro del futbol moderno, conocido por su vision de juego, precision y liderazgo en el Manchester City. Su talento y trabajo duro lo han convertido en una leyenda del deporte.
Mathewfam
| #
Sitio fan de Mohamed Salah mohamed salah com mx ultimas noticias, records, entrevistas y los mejores momentos de la carrera de uno de los futbolistas mas grandes de la actualidad. ?Mantente al tanto!
UlyssesBef
| #
Fan site de Vinicius Junior https://vinicius-jr.com.mx noticias, logros y detalles sobre su carrera en el Real Madrid. Sigue la evolucion de esta estrella del futbol mundial.
Cecilacido
| #
application casino en ligne casino en ligne francais
NormanDwele
| #
Try your luck at pinup casino! The best slots, roulette, blackjack and live games with real dealers. Pleasant bonuses, promotions and a user-friendly interface will create ideal conditions for the game!
email4domain
| #
Почта для домена на https://email4domain.ru это корпоративный почтовый сервис email для организаций. Почта для домена обеспечивает сотрудников адресами электронной почты на собственном домене компании. Почта для бизнеса улучшает узнаваемость и повышает имидж бренда.
жалюзи с приводом на окна
| #
Лучший выбор для автоматизации жалюзи.
Умное устройство для управления жалюзи.
для вашего дома.
Зачем нужен привод для автоматизации жалюзей.
Тенденции в автоматизации жалюзи с помощью привода.
Как управлять жалюзями с помощью смартфона с использованием привода.
Шаг за шагом по инструкции по установке привода для жалюзи.
Экономьте время и энергию с автоматизированным приводом для жалюзи.
Интересные факты о приводах для жалюзи.
Как повысить уровень комфорта с приводом для жалюзи.
привод жалюзи электрический привод жалюзи электрический .
Jamessit
| #
Full wordlist New full BIP39 2048 words used to create and restore crypto wallets. Multi-language support, high security and ease of use to protect your funds. 2048 mnemonic words for seed generation.
KeithDUg
| #
New full Bip39 2048 words used to create and restore crypto wallets. Multi-language support, high security and ease of use to protect your funds.
Franknoump
| #
casino en ligne francai https://annuaire-des-casinos.com
Franknoump
| #
casino en ligne fiable forum arlequin casino en ligne
Jamessit
| #
Full wordlist New full BIP39 2048 words used to create and restore crypto wallets. Multi-language support, high security and ease of use to protect your funds. 2048 mnemonic words for seed generation.
GlennElilk
| #
Актуальные промокоды казино https://akivaschool.com/pgs/code_promotionnel_1xbet_depot.html получите дополнительные бонусы при регистрации! Увеличенный первый депозит, бесплатные ставки, фриспины и эксклюзивные акции для игроков. Вводите код и выигрывайте больше!
KennethEffex
| #
Промокод казино https://uconnect.ae/read-blog/190468 активируйте эксклюзивные бонусы! Получите фриспины, бонусные деньги на депозит и кешбэк. Используйте актуальные коды для максимальной выгоды в онлайн-казино. Играйте с лучшими условиями!
DavidFed
| #
Current casino promo codes http://rzyuhua.com/tpl/inc/?1xbet_promo_code_welcome_bonus_free.html bonus money, free spins and cashback for playing. Use verified codes, activate exclusive offers and win more!
Jeffreyitady
| #
Ищете выгодные покупки? нова маркетплейс предлагает широкий выбор товаров по привлекательным ценам! Безопасные сделки, удобный интерфейс и проверенные продавцы – все для комфортного шопинга.
ThomasJap
| #
Промокод казино https://hyd-parts.ru/news/sovety_po_prohoghdeniyu_mage_knight_apocalypse_osnovnye_missii_086__2.html активируйте эксклюзивные бонусы! Получите фриспины, дополнительные деньги на депозит и кешбэк. Используйте актуальные коды для максимальной выгоды в онлайн-казино и увеличьте свои шансы на выигрыш!
Charlesonepe
| #
Лучшие промокоды для казино http://pharaon.mega-f.ru/logs/pros/kak_zapoluchity_bezuprechnuyu_koghu.html активируйте бонусные предложения и получите больше шансов на выигрыш. Бесплатные спины, дополнительные деньги на баланс и персональные акции ждут вас!
Jerryenums
| #
Присоединяйтесь к NovaXLtd.live https://novaxltd.live платформе с широкими возможностями и удобными инструментами. Доступность, надежность, поддержка!
Peterham
| #
Используйте Kra020 https://kra020.com надежная площадка с удобными инструментами и поддержкой 24/7. Простота и безопасность в одном сервисе!
DavidPer
| #
Откройте новые возможности https://sindicate24-4.biz! Удобный сервис, быстрые решения и выгодные предложения для пользователей.
Robertoweill
| #
דיסקרטיות בחיפה. תורות יהודיות מסורתיות מדגישות את חשיבות הצניעות והטוהר המיני של דירות דיסקרטיות בתל אביב, תוך חגיגות סופית נשוי או פרופסור מהטכניון – כאן כולם אורחים רצויים. כאן מקבלים פינוק שעושה טוב לכולם, והכל בדיסקרטיות מלאה. מבלים כאן חילונים שירותי פינוקים חיפה – טיפים מקצועיים לבחירת הבילוי האולטימטיבי בעיר
Ralphprulk
| #
Продажа сигарет mond сигареты купить в интернет-магазине – оригинальные бренды, доступные цены, удобные способы оплаты и быстрая доставка. Сделайте заказ сегодня!
StevenNit
| #
Откройте все возможности Blacksprut Большой выбор товаров, безопасные сделки и простая навигация. Приватность и защита пользователей на первом месте.
MetaMask Download
| #
If you want to safely install Metamask on Chrome, visit https://metanate.org/. Their guide is super detailed, making it easy to follow. Now I can manage my crypto without any worries.
лучшие решения для сцены с электрокарнизом
| #
Ваша сцена станет неповторимой с электрокарнизами, сделают ваше выступление незабываемым.
Электрокарнизы придают шоу утонченность, делая представление более динамичным.
Современные технологии в электрокарнизах, дарят возможность воплотить любую идею.
С электрокарнизами публика останется в восторге, с идеальным сочетанием функциональности и эстетики.
Электрокарнизы – современное решение для профессиональных выступлений, покоривших сердца зрителей.
Инновационные электрокарнизы для сцены, сделать ваше выступление неповторимым.
Уникальные решения для каждого типа представления, с легкой и тихой работой в каждом представлении.
Осуществите свои идеи с помощью электрокарнизов, и подчеркнуть важность каждой сцены.
Электрокарнизы – современное решение для сцены, и подчеркивая профессионализм исполнителей.
Выберите электрокарнизы по своему вкусу, обеспечив себе идеальный результат в каждом выступлении.
автоматический карниз для сцены автоматический карниз для сцены .
Jamesstype
| #
Свежие промокоды казино http://ihvo.de/wp-content/pages/1xbet_promo_codes_bonus.html бонусы, фриспины и эксклюзивные акции от топовых платформ! Найдите лучшие предложения, активируйте код и выигрывайте больше.
seo-base
| #
частный сео оптимизатор https://seo-base.ru
Waynesaf
| #
Нужен опытный лечебный массаж Ивантеевка? Помогаю при остеохондрозе, грыжах, искривлениях позвоночника и болях в суставах. Безопасные техники, профессиональный подход, запись на прием!
EdwinLiaiz
| #
Защитите авто от коррозии https://antikorauto.ru антикоррозийная обработка и кузовной ремонт с гарантией. Восстановление геометрии кузова, покраска, удаление вмятин. Долговечность и качество по доступным ценам!
vasya-muflonov
| #
Жарким летним утром Вася Муфлонов, заядлый рыбак из небольшой деревушки, собрал свои снасти и отправился на местное озеро. День обещал быть необычайно знойным, солнце уже в ранние часы нещадно припекало.
Вася расположился в укромном месте среди камышей, где, по его наблюдениям, всегда водилась крупная рыба. Забросив удочку, он погрузился в привычное для рыбака созерцательное состояние. Время текло медленно, поплавок лениво покачивался на водной глади, но рыба словно избегала его приманки.
Ближе к полудню, когда жара достигла своего пика, произошло то, чего Вася ждал все утро – поплавок резко ушёл под воду. После недолгой борьбы на берегу оказался огромный серебристый карп, какого Вася никогда раньше не ловил.
Радости рыбака не было предела, но огорчало одно – он не взял с собой фотоаппарат, чтобы запечатлеть этот исторический момент. Тут в голову Васи пришла необычная идея. Он аккуратно положил карпа себе на грудь и прилёг на песчаный берег под палящее солнце. “Пусть загар оставит память об этом улове”, – подумал находчивый рыбак.
Через час, когда Вася поднялся с песка, на его загорелой груди отчётливо виднелся светлый силуэт пойманной рыбы – точная копия его трофея. Теперь у него было неопровержимое доказательство его рыбацкой удачи.
История о необычном способе запечатлеть улов быстро разлетелась по деревне. С тех пор местные рыбаки в шутку стали называть этот метод “фотография по-муфлоновски”. А Вася, демонстрируя друзьям необычный загар, с улыбкой рассказывал о своём изобретательном решении.
Эта история стала местной легендой, и теперь каждое лето молодые рыбаки пытаются повторить трюк Васи Муфлонова, создавая на своих телах “загорелые фотографии” своих уловов. А сам Вася стал известен не только как умелый рыбак, но и как человек, способный найти нестандартное решение в любой ситуации.
gambling-soft
| #
white label casino solutions white label casino solutions
Shanesaw
| #
Fast and secure VPS https://evps.host/products for any task! Flexible configurations, SSD drives, DDoS protection. Easy scaling, high performance and 24/7 support!
Danielseila
| #
All about Vinicius Junior vinicius-junior-bd.com/ career, trophies, highlights and records. Follow the Brazilian winger, his success at Real Madrid and on the international stage!
WayneRAL
| #
All about Karim Benzema karim-benzema-bd com biography, achievements, main goals and career moments. Find out how the striker conquered Real Madrid and continues to shine in world football!
Timothyexerb
| #
All about Karim Benzema karim-benzema biography, achievements, main goals and career moments. Find out how the striker conquered Real Madrid and continues to shine in world football!
BasilSig
| #
https://sem-del.ru/
MetaMask Download
| #
A friend recommended I visit https://metamake.org/ for instructions on downloading Metamask, and I’m so glad I did! The guide was clear, and I had my wallet set up in no time. Definitely a must-visit site for crypto users!
WillieFar
| #
https://antimud.ru/
Scottper
| #
arkada casino online аркада casino официальный сайт
Michaeljog
| #
Военная служба по контракту https://contract116.ru стабильность, достойная зарплата, социальные льготы и карьерный рост. Присоединяйтесь к профессиональной армии и получите надежное будущее!
Chesterrit
| #
Kevin De Bruyne kevin de bruyne az com is the leader of the Manchester City and Belgium national team midfield. Find out all about his achievements, matches, awards and records in world football!
TommySip
| #
Joshua Kimmich https://joshua-kimmich-az.org the versatile leader of Bayern and the German national team! The latest news, stats, highlights, goals and assists. Follow the career of one of the best midfielders in the world!
DerekAluro
| #
скачать читы на самп аризона рп
mckeesportautobodylwag
| #
Transform the vehicle into a showstopper with expert car tuning services. At a shop, we provide a wide range of improvements to boost efficiency. Whether you want a unique appearance or top-tier motor adjustments, we’ve got you covered. Explore our offerings at https://mckeesportautobody.com/. Upgrade your ride today and experience excellence like never before.
Danielges
| #
Каталог подарков и сувениров https://kameshki51.ru/catalog/ из камня – эксклюзивные изделия из мрамора, гранита, оникса и яшмы. Шкатулки, статуэтки, часы, пресс-папье, бизнес-сувениры. Высокое качество и ручная работа!
ScottFak
| #
Подарки и сувениры из камня https://kameshki51.ru статуэтки, шкатулки, часы, панно, изделия из мрамора, гранита и оникса. Ручная работа, уникальный дизайн, доставка по России!
Michaelplast
| #
https://itscleaning.ru/
JosephFep
| #
Как побороть лудоманию https://teletype.in/@antosha-sevan/moi-opyt-izbavlenia-ot-igrovoi-zavasimosty история глазами прошедшего это испытание и несколько советам тем, кто в середине пути или столкнулся с этим через друзей и близких. Описываю в статье как Вавада помогла мне избавиться от игровой зависимости
CharlesSek
| #
Образовательный портал https://moi-slovari.ru собрание полезных материалов, лекций, статей и справочной информации для школьников, студентов и профессионалов. Учитесь легко и эффективно
JesseBroox
| #
https://cleanmsk799.ru/
Edwardkar
| #
Окунитесь в азарт с https://aviator-slot.ru Уникальная игра с захватывающим геймплеем и возможностью сорвать крупный выигрыш. Следите за коэффициентом, вовремя забирайте ставку и умножайте баланс. Испытайте удачу прямо сейчас!
BrianMunny
| #
Новости музыки Украины https://musicnews.com.ua популярные жанры, тренды и полезные советы для музыкантов. Узнавайте больше о мире музыки и развивайте свои навыки!
NormanImaph
| #
Ставки на спорт http://betting-ru.ru ваш шанс превратить знания в выигрыш! Лучшие коэффициенты, огромный выбор событий и удобный интерфейс. Делайте прогнозы и побеждайте!
MetaMask Download
| #
I wanted to download Metamask safely, and https://metaduck.org/ helped me do just that. Their guide is detailed and straightforward.
Williamsep
| #
сколько стоят услуги клининговой компании
MetaMask Download
| #
If you want to install Metamask on Chrome quickly and securely, check out https://metapaws.org/. The instructions are clear, making it perfect for beginners!
Raymondsag
| #
https://posle-remonta-uborka.ru/
ShawnFoups
| #
https://прочисто.рф/
Miltonseick
| #
Zeus vs Hades https://zeus-vs-hades-download.ru эпичная битва богов! Игровой автомат с грозными бонусами, фриспинами и множителями. Выберите сторону Зевса или Аида и сорвите большой выигрыш!
Kennethneirm
| #
Мир смешанных единоборств https://ufc-ru.ru (MMA) в UFC! Рейтинги, бои, истории бойцов, прогнозы на поединки и эксклюзивные материалы о крупнейшей бойцовской лиге.
JasonPap
| #
Все о легкой атлетике athletics-ru.ru История развития, основные дисциплины, рекорды, тренировки и современные тенденции. Узнайте больше о королеве спорта!
Augustglymn
| #
https://cleanliness-cleaning.ru/
JoshuaPef
| #
Cassino online Pin Up https://888casino-official-brazil.site ganhos sem limites! Jogue seus caca-niqueis favoritos, participe de torneios, ganhe cashback e rodadas gratis. Licenca, seguranca e pagamentos rapidos!
phoenixautobodyshopcoogy
| #
Revamp upgrade your ride with our premium superior car tuning services. We specialize in tailored modifications that elevate both capabilities and design. From horsepower boosts to exterior upgrades, we’ve got you covered. Experience the ultimate transformation at https://phoenix-autobodyshop.com/ and let our seasoned team bring your vision to life. Visit us today and drive with pride!
elon casino_byKi
| #
elonbet elonbet .
EdgarRox
| #
Свежие и актуальные новости https://sugar-news.com.ua Украины и мира! Будьте в курсе последних событий, аналитики и трендов. Читайте последние новости Украины онлайн!
bazovichok
| #
Лучшие эксперты в одном месте! реестр соут маркетплейс и агрегатор экспертных компаний помогает найти надежных специалистов по разным направлениям. Удобный выбор, проверенные отзывы и рейтинг!
DannyMinly
| #
ремонт квартир уборка
ArthuroxiVa
| #
https://clean-sharks.ru/
Free tanpa deposit
| #
Free tanpa deposit
A Brief History of Elemental Analysis » Artemis Analytical
elektrokar_yson
| #
Оптимизируйте свою жизнь с электрокарнизом с таймером, наслаждайтесь комфортом и современностью.
Создайте атмосферу уюта и стиля с помощью электрокарниза и таймера, сделает вашу жизнь проще и приятнее.
Освободите руки и ум с электрокарнизом и таймером, создайте идеальную атмосферу в вашем доме.
Оптимальное решение для автоматизации штор – электрокарниз с таймером, обеспечит вас и вашу семью уютом и функциональностью.
Инновационные технологии в управлении шторами – электрокарниз с таймером, добавит функциональности и удобства в вашу жизнь.
Электрический карниз с таймером https://prokarniz50.ru/ .
PatrickPhymn
| #
Хотите играть в Steam-игры shared-steam.org/ дешевле? Shared-Steam.org предлагает аренду аккаунтов с топовыми играми. Безопасность, доступность и удобство – ваш гейминг без границ!
nemeckoe porno_mool
| #
немки порно http://trax-nemeckoe1.ru/ .
americasbestcertifiedautobodycoogy
| #
Unleash enhance your vehicle’s full potential with our premium top-tier tuning services. We offer personalized modifications to improve your car’s performance, aesthetics, and comfort. Our expert certified technicians use state-of-the-art high-tech technology to deliver flawless outcomes. https://americasbestcertifiedautobody.com/ Whether you seek remarkable speed, handling, or luxury, we provide top-notch solutions. Trust us to elevate your driving experience. Contact us today and unveil the ultimate in automotive enhancement!
Davidcem
| #
Create images deep nude easily with AI. Remove clothes from anyone in the image and enjoy their nakedness.
slots-cool
| #
Turkiye’deki slot makineleri gercek parayla oyun slotlar gercek parayla oynanabilen slotlar kapsaml bir baks! Nerede oynan?r, hangi slotlar en karldr ve en iyi casino nasl secilir Kumar severler icin ipuclar, incelemeler, derecelendirmeler ve bonuslar!
slots-rait
| #
Turkiye Slotlar Rehberi Gercek parayla oyun slotlar En Iyi Slotlar ve Casinolar! Nerede oynayabileceginizi, hangi oyunlarn populer oldugunu ve bonuslar nasl alabileceginizi ogrenin. Oyun alanlar ve guncel eglencelere dair kapsaml bir genel baks!
MetaMask Download
| #
Thanks to https://download.metaredi.org/, I installed the Metamask extension without any trouble. Now I can manage my crypto easily and securely.
perevozimgruz_by
| #
перевозка велосипедов https://perevozimgruz.by/contacts/
perevozimgruz
| #
перевозка шкафа https://perevozimgruz.by
TerryTow
| #
Знакомства в Уфе https://ufavip.sbs с нашей платформой стали еще проще, можно встретить девушек, готовых к интересному общению и приятному времяпрепровождению. Здесь каждый найдет то, что ищет: от легкого флирта до серьезных отношений.
JamesRound
| #
https://uborka-kvartir-luberci.ru/
Josephhiz
| #
клининг уборка офиса
Edwardvam
| #
Is it possible to win at Lucky Jet? We analyze the main strategies, analyze player reviews, and give an honest assessment of the popular game. Read to avoid mistakes and increase your chances of success!
JacobPrurn
| #
https://klining-64.ru/
perevozimgruz_by
| #
квартирный переезд минск спб грузоперевозки минск
porno milfi_ahPi
| #
milf порно http://milfland-pro1.ru/ .
1win_ndkt
| #
ставки на спорт бишкек http://mabc.com.kg/ .
mostbet_sdSa
| #
mostbet вход в личный кабинет официальный http://gtrtt.com.kg .
porno mylt_hcEn
| #
порно мульты порно мульты .
porno ginekolog_xwkn
| #
порно с гинекологом порно с гинекологом .
умный электрокарниз
| #
Управляйте своим окружением с помощью программируемого электрокарниза, который легко устанавливается.
Трансформируйте ваше жилище с помощью программируемого электрокарниза, который дарит вам полный контроль.
Освежите свою обстановку с помощью программируемого электрокарниза, который позволит вам сохранить энергию.
Пусть ваша спальня станет местом для отдыха и релаксации с программируемым электрокарнизом, дарит вам возможность наслаждаться каждой минутой вашего сна.
Регулируйте интенсивность света и тепла с помощью программируемого электрокарниза, обеспечит вас уютом и спокойствием.
электрокарниз подключение к умному дому https://elektrokarniz190.ru/ .
Matthewjam
| #
Кружевной бюстгальтер https://www.wildberries.ru/catalog/117605496/detail.aspx без косточек с застежкой спереди — это идеальный выбор для тех, кто ценит комфорт и стиль. Изготовленный из мягкого и нежного кружева, он обеспечивает естественную поддержку, не ограничивая движения. Удобная застежка спереди позволяет легко надевать и снимать бюстгальтер, а его изысканный дизайн добавляет нотку романтики в ваш образ.
GeraldHom
| #
?El paraiso del juego en Pin Up Casino https://ganar-apuestas-deportivas.com te espera! Echa un vistazo a nuestra coleccion de tragamonedas, juegos en vivo y juegos de mesa. Bonos rentables, torneos y un programa VIP te proporcionaran el maximo de emociones. ?Empieza a jugar ahora y gana!
Georgevully
| #
Каталог финансовых организаций https://srochno-zaym-online.ru в которых можно получить срочные онлайн займы и кредиты не выходя из дома через интернет.
perevozimgruz
| #
грузоперевозки минск спб грузоперевозки по рб
srochno-zaym
| #
Каталог финансовых организаций srochno-zaym-online.ru в которых можно получить срочные онлайн займы и кредиты не выходя из дома.
Dannycet
| #
лучшие комедии 2025 hdrezka смотреть мелодрамы hd онлайн
perevozimgruzby
| #
грузоперевозки беларусь россия грузоперевозки минск
pochemuabold
| #
Почему зонтик был необходим Робинзону https://e-pochemuchka.ru/zontik-dlya-robinzona-neobhodimost-ili-izlishestvo/
elon casino_lqOa
| #
elon bet casino elon bet casino .
elon casino_abka
| #
bet elon bet elon .
elon casino_qyen
| #
elonbet casino http://www.elonbetting.com .
elon casino_hkSt
| #
elone casino elone casino .
1 vin_isEi
| #
1win login https://bbcc.com.kg/ .
srochnozaym
| #
Каталог финансовых организаций srochno-zaym-online.ru в которых можно получить срочные онлайн займы и кредиты не выходя из дома.
hdrezka
| #
новые трейлеры к фильмам 2025 hdrezka фантастика без рекламы на планшете
JamesOriew
| #
Форум для кулинаров https://food-recipe.ru и рестораторов! Лучшие рецепты, тренды гастрономии, обсуждения ресторанного бизнеса. Общайтесь, делитесь опытом и вдохновляйтесь новыми идеями. Присоединяйтесь к нашему кулинарному сообществу!
DallasHeice
| #
https://infoteka24.ru/2024/05/04/106105/
Blairletly
| #
Красивые поздравления https://photofile.ru в одном месте! Огромный выбор картинок и открыток для любого повода. День рождения, юбилей, профессиональные праздники – найдите идеальное поздравление!
StephenMub
| #
Если вы ищете профессионалов в сфере управления недвижимостью, обратитесь к этой компании, подробнее тут https://sketchfab.com/allianceprof
JamesFaf
| #
Новый уникальный маркетплейс NOVA, есть все! Москва, Питер, всегда свежие клады!Акция для новых магазинов – продай на 1 млн, получи 100к!Приглашаем к сотрудничеству рекламодателей!
RichardWaimb
| #
Del Mar Energy Inc
Larryseags
| #
бездепозитный бонус без пополнения бездепозит и фриспины при регистрации
Calvinrox
| #
hdrezka бесплатные фильмы hd rezka боевики новинки 1080p
Kennethsoisk
| #
Маркетплейс нового поколения https://x-nova.live уникальный сервис для удобных покупок и продаж. Интеллектуальный поиск, персонализированные рекомендации и безопасность сделок – все для вашего комфорта!
MetaMask Download
| #
If you’re looking for a step-by-step guide to install the Metamask extension, https://metamaker.org/#metamask-download is the perfect place. They explain everything in detail, ensuring you set up your wallet correctly and securely.
Larryter
| #
ключ доступа vpn
elektrokarnizi_jeki
| #
автоматические карнизы автоматические карнизы .
tk999_heer
| #
tk999 login tk999 login .
tk999_kymn
| #
tk999 tk999 .
KU9_gnkl
| #
KU9 bet KU9 bet .
kypit viagry_uqKa
| #
виагра для мужчин виагра для мужчин .
mtsnovosibirsksig
| #
мтс тв
https://ukskilledworkfinder.com/employer/job-daddy2052/
сайт мтс новосибирск
mikrozaymivah
| #
Получить кредит на банковскую карту теперь просто, моментально и комфортно. Мы обеспечиваем дружелюбные условия. Перейдите на https://mikro-zaymi.ru/, чтобы узнать все детали. Процесс оформления занимает несколько минут. Не задерживайтесь! Получите кредит прямо сейчас!
JosephWhopy
| #
Современный маркетплейс https://nova7.top для выгодных покупок! Огромный ассортимент, лучшие цены и удобные способы оплаты. Покупайте качественные товары и заказывайте с быстрой доставкой!
Louissnoda
| #
Новый формат маркетплейса https://nova4.top быстро, удобно, надежно! Лучшие предложения, современные технологии и простой интерфейс. Покупайте и продавайте легко, экономя время и деньги!
DannyDic
| #
смотреть лучшие комедии 2025 онлайн hdrezka сериалы ужасы на андроиде
mtsnovosibirsksig
| #
мтс домашний интернет новосибирск
https://globalhospitalitycareer.com/employers/i-medconsults4572/
мтс тв
affiliate programs_hgvr
| #
Premium affiliate programs in online marketing https://affiliate-c1.com/.
https://www.harvard.edu/
| #
# Harvard University: A Legacy of Excellence and Innovation
## A Brief History of Harvard University
Founded in 1636, **Harvard University** is the oldest and one of the most
prestigious higher education institutions in the United States.
Located in Cambridge, Massachusetts, Harvard has built a global reputation for
academic excellence, groundbreaking research, and influential alumni.
From its humble beginnings as a small college established to educate clergy, it has
evolved into a world-leading university that shapes the future across various disciplines.
## Harvard’s Impact on Education and Research
Harvard is synonymous with **innovation and intellectual leadership**.
The university boasts:
– **12 degree-granting schools**, including the renowned
**Harvard Business School**, **Harvard Law School**,
and **Harvard Medical School**.
– **A faculty of world-class scholars**,
many of whom are Nobel laureates, Pulitzer Prize winners, and pioneers in their fields.
– **Cutting-edge research**, with Harvard leading initiatives in artificial intelligence,
public health, climate change, and more.
Harvard’s contribution to research is immense, with billions of dollars allocated to scientific discoveries
and technological advancements each year.
## Notable Alumni: The Leaders of Today and Tomorrow
Harvard has produced some of the **most influential figures** in history,
spanning politics, business, entertainment, and science.
Among them are:
– **Barack Obama & John F. Kennedy** – Former U.S.
Presidents
– **Mark Zuckerberg & Bill Gates** – Tech visionaries (though Gates did not graduate)
– **Natalie Portman & Matt Damon** – Hollywood icons
– **Malala Yousafzai** – Nobel Prize-winning activist
The university continues to cultivate future leaders who shape industries and drive global progress.
## Harvard’s Stunning Campus and Iconic Library
Harvard’s campus is a blend of **historical charm and modern innovation**.
With over **200 buildings**, it features:
– The **Harvard Yard**, home to the iconic **John Harvard Statue** (and
the famous “three lies” legend).
– The **Widener Library**, one of the largest university libraries in the world, housing **over 20 million volumes**.
– State-of-the-art research centers, museums, and performing arts venues.
## Harvard Traditions and Student Life
Harvard offers a **rich student experience**, blending academics with vibrant traditions, including:
– **Housing system:** Students live in one of 12 residential houses,
fostering a strong sense of community.
– **Annual Primal Scream:** A unique tradition where students de-stress by running through Harvard Yard before finals!
– **The Harvard-Yale Game:** A historic football rivalry that unites alumni and students.
With over **450 student organizations**, Harvard students engage in a diverse range of extracurricular activities, from entrepreneurship to
performing arts.
## Harvard’s Global Influence
Beyond academics, Harvard drives change in **global policy, economics, and technology**.
The university’s research impacts healthcare, sustainability, and artificial intelligence,
with partnerships across industries worldwide. **Harvard’s endowment**, the largest of any university, allows it
to fund scholarships, research, and public initiatives, ensuring a legacy of
impact for generations.
## Conclusion
Harvard University is more than just a school—it’s a **symbol of
excellence, innovation, and leadership**. Its **centuries-old traditions, groundbreaking discoveries, and
transformative education** make it one of the most influential
institutions in the world. Whether through its distinguished
alumni, pioneering research, or vibrant student life, Harvard continues to shape the future in profound ways.
Would you like to join the ranks of Harvard’s legendary
scholars? The journey starts with a dream—and an application!
https://www.harvard.edu/
PrinceMep
| #
Портал с ответами https://online-otvet.site по всем школьным домашним предметам. Наши эксперты с легкостью ответят на любой вопрос, и дадут максимально быстрый ответ.
DonaldWem
| #
Руководство по настройке https://amnezia.dev VPN-сервера! Пошаговые инструкции для установки и конфигурации безопасного соединения. Узнайте, как защитить свои данные и обеспечить анонимность в интернете. Настройте VPN легко!
Gregoryfesee
| #
Сауна очищает организм https://sauna-broadway.ru выводя токсины через пот, укрепляет иммунитет благодаря перепадам температуры, снимает стресс, расслабляя мышцы и улучшая кровообращение. Она делает кожу более упругой, ускоряет восстановление после тренировок, улучшает сон и создаёт атмосферу для общения.
Earn Free Robux
| #
Good post! Do you want to know how to earn free robux? If yes, I suggested one post about earn free robux .To acquire free Robux, one can explore various methods such as participating in promotional events, engaging in online surveys, or utilizing specific gaming platforms that offer rewards. Visit rhis post to get more info.
Donaldopela
| #
Православный форум https://prihozhanka.ru для женщин! Общение о вере, молитве, семье, воспитании и роли женщины в православии. Делитесь мыслями, находите поддержку и вдохновение в теплой атмосфере!
Robertglart
| #
Korean cosmetics http://japan-lke.com/home.php?mod=space&uid=135552 perfect skin without effort! Innovative formulas, Asian traditions and visible results. Try the best skin care products right now!
MichaelAgica
| #
selector casino вход
ArthurMaigh
| #
селектор казино
Jamesjat
| #
https://www.suzuki1909.com/blog/otzyvy-o-suzuki-s-cross-nadezhnost-i-komfort-na-dorogah/
Larrynon
| #
селектор казино зеркало
Thomassef
| #
selector casino войти
teacher xxx video_mcOi
| #
student pron video https://www.teacherporntube.com .
Elektrokarniz_kcma
| #
гибкий электрокарниз привод prokarniz11.ru .
Elektrokarniz_tfMi
| #
карниз электро карниз электро .
Elektrokarniz_mnea
| #
электрические гардины электрические гардины .
Elektrokarniz_dzet
| #
электронный карниз для штор электронный карниз для штор .
Charlesjat
| #
Хочешь купить ПАВ в Москве? Нова маркетплейс – ищи в Яндекс!
mtsekaterinburgsig
| #
мтс подключение екатеринбург
https://skillnaukri.com/employer/inetrnet-domashnij-ekb-2/
мтс интернет
Felipebraby
| #
Хочешь купить ПАВ в СПБ? Нова маркетплейс – ищи в Яндекс!
RobertFaist
| #
leebet casino войти
TimothyMet
| #
leebet casino
PhilipMooxy
| #
leebet casino вход
mtsekaterinburgsig
| #
мтс подключение екатеринбург
https://empleosrapidos.com/companies/inetrnet-domashnij-ekb-2/
мтс телевидение екатеринбург
Sound Button
| #
Great article. Are you curious about choosing the most impactful sound effects? If so, Sound plays a crucial role in Roblox, shaping the atmosphere, enhancing gameplay, and creating lasting memories. With soundbuttons—a fantastic collection of sound effects—you can add the perfect audio cue to bring your Roblox world to life. Check out the blog post to discover Roblox’s most iconic sound effects.
MauroAdoca
| #
Советую приобрести прицел с дальномером, точность стрельбы стала намного выше – https://opticheskie-pricely-i-binokli.ru/
online unblocked games
| #
Good Post! Do you enjoy playing unblocked Roblox games? If so, If you’re looking for more free online unblocked game, visit GamesCentral, where you’ll find a massive selection of games across different genres. Check out the full article to learn how these platforms keep your gaming experience seamless!
the dumb test
| #
This post is amazing! Are you in a relationship and wondering about your compatibility with your partner? If so, you can take the dumb test. It’s a fun and easy way to see how well you two vibe. Playful moments like these can add excitement to your relationship. Check out the site for more information!
mtsekaterinburgsig
| #
мтс подключить
https://www.yiyanmyplus.com/companies/inetrnet-domashnij-ekb-3/
мтс подключить
zaymtulaivah
| #
Нужны деньги безотлагательно? Получите кредит мгновенно без поручителей на https://zaym-tula.ru/ без отказа! Оставьте регистрацию и получите финансы немедленно!
streetlighting
| #
outdoor lighting control systems street lighting control system
Jameslyday
| #
Сауна очищает организм https://sauna-broadway.ru выводя токсины через пот, укрепляет иммунитет благодаря перепадам температуры, снимает стресс, расслабляя мышцы и улучшая кровообращение. Она делает кожу более упругой, ускоряет восстановление после тренировок, улучшает сон и создаёт атмосферу для общения.
最佳Binance推荐代码
| #
Can you be more specific about the content of your article? After reading it, I still have some doubts. Hope you can help me.
Thomasles
| #
профессиональный лечебный массаж Ивантеевка расслабляющие техники для здоровья и красоты. Помогаем снять стресс, улучшить кровообращение и восстановить силы. Запишитесь прямо сейчас!
Elektrokarniz_ytet
| #
электрические гардины электрические гардины .
Michaelnat
| #
https://ourhonda.com/
Elektrokarniz_zboa
| #
карниз с электроприводом карниз с электроприводом .
Elektrokarniz_wspt
| #
карниз для штор электрический карниз для штор электрический .
mtsekaterinburgsig
| #
мтс домашний интернет
https://impactosocial.unicef.es/employer/inetrnet-domashnij-ekb-2/
сайт мтс екатеринбург
PeterPhync
| #
автоматы с бесплатным депозитом фриспины в слотах
Gerardket
| #
Женский онлайн портал https://brjunetka.ru для вдохновения! Полезные советы по стилю, уходу за собой, здоровью и семейной жизни. Будьте в курсе трендов, находите мотивацию и делитесь опытом!
Davidoraro
| #
Портал с ответами https://online-otvet.site на все школьные предметы! Быстро находите решения по математике, русскому, физике, биологии и другим дисциплинам. Готовые ответы, разборы задач и помощь в учебе для всех классов
smm-panel
| #
boost of instagram free boost of subscribers in telegram
Donalddef
| #
Элитный коттеджный поселок https://parkville-zhukovka-poselok.ru в Жуковке! Просторные дома, свежий воздух, развитая инфраструктура и удобная транспортная доступность. Создайте уютное пространство для жизни в живописном месте!
FloydEvosy
| #
stehovani trendy stehovani vyrobnich hal
mtsekaterinburgsig
| #
мтс домашний интернет екатеринбург
https://www.atlantistechnical.com/employer/inetrnet-domashnij-ekb/
провайдер мтс
Rylonnie shtori s elektroprivodom_cvSa
| #
электрические рулонные шторы купить электрические рулонные шторы купить .
Shtori s elektroprivodom_uvki
| #
шторы на электроприводе шторы на электроприводе .
smm-panel
| #
tg followers boost tt followers boost
DonaldAbumn
| #
Все о строительстве https://decor-kraski.com.ua и ремонте в одном месте! Подробные инструкции, идеи для дизайна, выбор материалов и советы мастеров. Сделайте свой дом удобным, стильным и долговечным!
HermandaT
| #
Портал про строительство https://goodday.org.ua и ремонт! Полезные советы, обзоры материалов, технологии строительства, лайфхаки для дома. Узнайте, как сделать ремонт качественно и сэкономить на строительных работах!
mtsekaterinburgsig
| #
мтс тв екатеринбург
https://thathwamasijobs.com/companies/inetrnet-domashnij-ekb-3/
мтс домашний интернет екатеринбург
jozz casino
| #
Ответственная игра: как не стать зависимым от казино
Установите лимиты на бюджет.
Прежде чем зайти в игровую среду, определите сумму денег, которую вы готовы
потратить, и строго соблюдайте этот предел.
Разделите сумму на игровые сессии,
чтобы избежать крупных потерь за
один раз.
Определите временные рамки.
Установите четкое время для ваших игровых сеансов.
Используйте таймер, чтобы напомнить себе о необходимости прекратить участие
и не останавливаться дольше, чем планировалось.
Не используйте деньги для других нужд.
Избегайте тратить средства, предназначенные для важных вещей, таких
как оплата счетов или покупка продуктов.
Игровые средства должны быть отдельными от ваших финансовых
обязательств.
Развивайте осознанность.
Обращайте внимание на свои эмоции во время игры.
Если вы начинаете чувствовать стресс
или напряжение, это признак того, что стоит остановиться.
Оцените своё состояние и не бойтесь взять паузу.
Общайтесь с окружающими.
Делитесь своим опытом с друзьями или близкими.
Обсуждение является важным аспектом, помогающим понять собственные привычки.
Поддержка со стороны может
служить дополнительным стимулом для
поддержания контроля.
Поддерживайте разнообразие интересов.
Занимайтесь хобби и активностями, которые приносят удовольствие
и не связаны с риском потерь. Это поможет сохранить
баланс и не зацикливаться на азартных развлечениях.
Методы самоконтроля для игроков в азартные заведения
Определите лимиты. Установите
четкие границы для денежных
затрат, времени, проведенного за азартными играми.
Запишите эти лимиты и придерживайтесь их, даже если возникает
соблазн продолжить.
Будьте бдительны к своему состоянию.
Analyse (анализируйте) свое эмоциональное состояние перед игрой.
Если чувствуете усталость,
стресс или грусть, отложите участие
в играх на некоторое время.
Используйте таймер. Установите таймер, который будет напоминать вам
о необходимости сделать перерыв.
Это поможет избежать длительного пребывания за игровым столом и сохранит вашу концентрацию.
Обратитесь за поддержкой. Общайтесь с друзьями или близкими об азартных увлечениях.
Открытый разговор может
помочь найти решение, если вы заметите
негативные изменения
в своем поведении.
Избегайте триггеров. Определите ситуации
или места, которые могут спровоцировать желание
участвовать в азартных играх.
Минимизируйте контакт с ними для снижения соблазна.
Занимайтесь альтернативными хобби.
Найдите увлечения, которые приносили бы вам радость и удовлетворение.
Это может быть спорт, творчество или саморазвитие, тем самым заполняя время,
которое могло бы уйти на азарт.
Поддерживайте физическую активность.
Регулярные физические нагрузки способствуют улучшению психоэмоционального состояния.
Занятия спортом помогут вам управлять стрессом и повышать умственную ясность.
Обучение и осведомленность.
Изучайте материалы о воздействии азартных игр на
личность. Понимание риска и последствий поможет принимать более обоснованные решения и избежать
негативных последствий.
Знаки проблемной игры: распознание зависимости
вовремя
Обратите внимание на изменения в финансовом поведении.
Если расходы на развлечения начинают превышать ваш бюджет, это сигнал о
возможной проблеме. Убедитесь, что у вас есть четкое представление о своих финансах
и регулярных расходах.
Следите за временем, проведенным
за активностью. Если вы не можете
отложить игру даже при необходимости выполнить важные дела
или общаться с близкими, это может указывать на нежелательное увлечение.
Обращайте внимание на эмоциональное состояние.
Частые переключения настроения,
раздражительность или эйфория связаны с игрой и могут свидетельствовать о её чрезмерной роли в жизни.
Осознание своих эмоций поможет определить,
когда наблюдаются опасные признаки.
Анализируйте ваши отношения.
Проблемы с родными, друзьями или коллегами из-за увлечения могут указывать на потенциальные последствия.
Если конфликты становятся регулярными, стоит задуматься о своих приоритетах.
Контролируйте мысли о поступках.
Если мания о выигрыше или разочаровании занимает большую часть вашего
сознания, следует обратить на это внимание и оценить влияние навязчивых идей на ваше поведение.
Отслеживайте попытки сокрытия
фактов. Если вам нужно скрывать время
занятий или свои убытки от близких, это указывает на наличие серьёзной проблемы.
Прозрачность в общении с окружающими – важный
аспект поддержки.
jozz casino
Bobbytrown
| #
Круглосуточные ритуальные услуги – помощь и поддержка в любое время https://pohoronnoe-agentstvo.kz/
rimskie shtori s elektroprivodom_qnOn
| #
автоматические римские шторы с электроприводом автоматические римские шторы с электроприводом .
elektrokarnizi somfy_omel
| #
somfy жалюзи somfy жалюзи .
SteveBaw
| #
Ремонт и строительство https://inbound.com.ua без хлопот! Полезные лайфхаки, новинки в дизайне, технологии строительства и подбор лучших материалов. Создайте комфортное жилье легко и выгодно!
Solomonbotly
| #
Ваш надежный помощник https://insurancecarhum.org в ремонте! Практичные советы, инструкции и секреты профессионалов. Узнайте, как сделать качественный ремонт и выбрать лучшие строительные материалы!
Leslieordib
| #
Портал для строителей https://hotel.kr.ua и домашних мастеров! Полезные статьи, новинки рынка, лайфхаки по ремонту и рекомендации по выбору качественных материалов.
RamonRix
| #
Инструкции по ремонту https://makprestig.in.ua подбор материалов, планирование, дизайн и советы экспертов. Станьте мастером своего дома!
Josephslatt
| #
Лучший портал по строительству https://itstore.dp.ua и ремонту! Гайды по отделке, рекомендации экспертов, новинки дизайна и проверенные решения. Все для вашего комфорта!
Jorgecoart
| #
Как сделать ремонт https://oo.zt.ua правильно? Наш портал поможет выбрать материалы, спланировать бюджет и создать уютный интерьер. Простые решения для сложных задач!
Craigsuese
| #
Профессиональные советы https://ukk.kiev.ua по ремонту и строительству! Пошаговые инструкции, актуальные тренды и лучшие решения для обустройства вашего жилья.
DanielMuh
| #
Идеи для дома https://ucmo.com.ua ремонта и строительства! Полезные советы, лайфхаки и современные технологии, которые помогут сделать ремонт качественно и доступно.
WilliamTreab
| #
Ремонт без стресса https://panorama.zt.ua Готовые решения, полезные лайфхаки, выбор материалов и идеи для дома. Делаем ремонт комфортным и доступным!
DanielMuh
| #
Идеи для дома https://ucmo.com.ua ремонта и строительства! Полезные советы, лайфхаки и современные технологии, которые помогут сделать ремонт качественно и доступно.
JamesTOW
| #
Делаем ремонт правильно https://zarechany.zt.ua Разбираем все этапы – от выбора материалов до дизайна интерьера. Подробные инструкции и лайфхаки от специалистов!
StephenTon
| #
Автомобильный портал https://nerjalivingspace.com для автолюбителей! Новости автоиндустрии, обзоры автомобилей, тест-драйвы, полезные советы по ремонту и тюнингу. Будьте в курсе последних трендов и находите ответы на все авто-вопросы!
mtsekaterinburgsig
| #
мтс подключить екатеринбург
https://www.punajuaj.com/companies/inetrnet-domashnij-ekb-3/
мтс тв екатеринбург
"https://eve5.wiki/index.php/Maxstresser_77j"
| #
Hi, Neat post. There’s an issue along with your web site in web explorer, might check this?
IE still is the market leader and a large element of folks will leave out your excellent writing due to this problem.
“https://dynastyascend.com/wiki/Maxstresser_89z”
"https://westhamwiki.com/index.php/Maxstresser_89N"
| #
Does your website have a contact page? I’m having trouble locating
it but, I’d like to send you an email. I’ve got
some recommendations for your blog you might be interested
in hearing. Either way, great blog and I look forward to seeing it grow over time.
“https://adscenter.site/user/profile/GeorgiaMuy”
Kennethroste
| #
https://169.ru/mezhkomnatnye-dveri/bravo/
1win_wber
| #
1win ставки официальный сайт https://www.1win3.com.kg .
AndrewJatte
| #
Все для женщин https://elegance.kyiv.ua в одном месте! Секреты красоты, советы по стилю, отношениям, психологии, здоровью и кулинарии. Будьте вдохновленными и уверенными каждый день!
Williamlor
| #
Женский портал https://beautyadvice.kyiv.ua для современной женщины! Мода, красота, здоровье, отношения, кулинария и карьера. Полезные советы, тренды и вдохновение для тех, кто хочет быть в курсе новинок и заботиться о себе!
ShaneGob
| #
Онлайн мир женщины https://gracefullady.kyiv.ua красота, здоровье, успех! Полезные лайфхаки, тренды моды, секреты счастья и гармонии. Портал для тех, кто хочет быть лучшей версией себя!
Robertvoisa
| #
Портал для современной https://femaleguide.kyiv.ua женщины! Открой для себя новые идеи в моде, красоте, отношениях и саморазвитии. Полезные статьи и советы для комфортной и счастливой жизни
HarrySnich
| #
Мир женщины https://fashionadvice.kyiv.ua красота, здоровье, успех! Полезные лайфхаки, тренды моды, секреты счастья и гармонии. Портал для тех, кто хочет быть лучшей версией себя!
megafonekbWAH
| #
мегафон интернет
https://ekb-domasnij-internet-2.ru
мегафон подключение екатеринбург
Howardtrese
| #
Твой гид в мире https://happywoman.kyiv.ua женственности и гармонии! Узнай больше о моде, косметике, фитнесе, отношениях и мотивации. Все, что нужно для яркой и счастливой жизни!
1win_wcKr
| #
1 win официальный сайт 1win4.com.kg .
1win_zhsn
| #
1win официальный http://1win5.com.kg/ .
megafonekbWAH
| #
мегафон тв екатеринбург
https://ekb-domasnij-internet-3.ru
провайдер мегафон
1win_xppa
| #
1win регистрация 1win6.com.kg .
mostbet kirgizstan_pnOi
| #
mostbet online http://www.mostbet1.com.kg/ .
PatrickheW
| #
Безопасный доступ к сайту https://bsme-at.at без ограничений. Рабочие зеркала позволяют обходить блокировки без VPN, обеспечивая стабильную связь и удобный интерфейс. Следите за обновлениями, чтобы всегда оставаться в сети.
Brunoneelm
| #
Актуальные зеркала BlackSprut https://kra26.cat Заходите без проблем, обходите блокировки и пользуйтесь сайтом без VPN. Мы следим за обновлениями и всегда предоставляем свежие ссылки.
Haroldhal
| #
Всегда актуальные ссылки https://bs2bet.at для входа. Обходите блокировки легко и быстро, используя надёжные зеркала. Свежие обновления позволяют заходить без VPN и сохранять полную анонимность.
HowardDrync
| #
Проблемы с входом https://bs2best.or.at Найдите актуальные зеркала и заходите без ограничений. Мы обновляем ссылки ежедневно, обеспечивая быстрый и безопасный доступ без необходимости использования VPN.
Williamexalp
| #
BlackSprut это инновационный https://bs2best.cat маркетплейс с расширенным функционалом и полной анонимностью пользователей. Современные технологии защиты данных, удобный интерфейс и высокая скорость обработки заказов делают покупки безопасными и простыми. Покупайте без риска и продавайте с максимальной выгодой на BlackSprut!
Howarddef
| #
BlackSprut крупнейший маркетплейс https://bb2best.at где можно найти всё, что вам нужно. Надёжная система безопасности, удобная навигация, широкий ассортимент товаров. Анонимные покупки, моментальные сделки и выгодные условия для продавцов.
KennethMoive
| #
BlackSprut передовой маркетплейс https://m-bsme.at с высокими стандартами безопасности и удобной системой поиска. Анонимность, быстрая оплата и честные продавцы – всё, что нужно для комфортных покупок. Мы гарантируем защиту ваших данных и качественное обслуживание. Присоединяйтесь к BlackSprut и открывайте новые возможности!
"https://yogaasanas.science/wiki/Jurnal_69T_random"
| #
I really like what you guys are up too. Such clever work and reporting!
Keep up the excellent works guys I’ve incorporated you guys to blogroll.
“https://systemcheck-wiki.de/index.php?title=Benutzer:CliffFfa80”
KennethGloto
| #
проверенный маркетплейс https://bsme.cat предлагающий товары на любой вкус. Простая регистрация, быстрая оплата и защита сделок. Убедитесь в качестве сервиса сами!
Michaeldeava
| #
Blacksprut – современная площадка https://bs-bsme.at для безопасных покупок. Большой выбор категорий, продуманная система рейтингов и отзывов. Заходите и находите нужное легко!
AnthonyUsero
| #
Blacksprut – онлайн-маркетплейс https://bsmc.at с лучшими предложениями. Надёжные продавцы, защищённые сделки, удобный поиск. Оцените удобство покупок уже сегодня!
Timothylaf
| #
Ищете актуальные бонусы без отыгрыша? Мы собираем лучшие предложения: фриспины, бонусы за депозит, бездепозитные акции и кэшбэк. Получите максимум выгоды от игры!
megafonekbWAH
| #
провайдер мегафон
https://ekb-domasnij-internet.ru
мегафон подключение екатеринбург
JamesIcodo
| #
Replica Uhren https://www.imailen.com in Deutschland schnell und sicher per DHL Nachnahme bestellen. Nur geprufte Qualitat in EU hergestellt.
RichardEnamn
| #
Журнал об автомобилях https://setbook.com.ua всё для автолюбителей! Последние автоновости, обзоры моделей, тест-драйвы, советы по ремонту, тюнингу и обслуживанию. Узнайте всё о мире автомобилей!
Raymondexomy
| #
Главный авто-журнал https://myauto.kyiv.ua для водителей! Новости, обзоры, сравнения, тюнинг, автоспорт и технологии. Будьте в курсе последних трендов автомобильного мира!
poeshop
| #
купить сферу хаоса path of exile 2 купить дивы пое 2
ThomasLiale
| #
Все об автомобилях https://allauto.kyiv.ua в одном журнале! Новости автопрома, тест-драйвы, обзоры моделей, советы по ремонту и тюнингу, страхование и ПДД.
HectorAmibe
| #
Журнал для автолюбителей https://myauto.kyiv.ua и профессионалов! Всё о новых моделях, технологии, автоспорт, лайфхаки по уходу за авто и экспертные рекомендации.
DustinLaf
| #
Журнал об авто https://auto-club.pl.ua для тех, кто за рулем! Новости автопрома, тест-драйвы, рекомендации по выбору авто, советы по ремонту и эксклюзивные интервью с экспертами.
beelinekrasnodarTet
| #
билайн домашний интернет
https://infodomashnij-internet-krasnodar-2.ru
билайн домашний интернет
Mostbet_hxMi
| #
мостбет ставки мостбет ставки .
Mostbet_nvEa
| #
vjcn,tn vjcn,tn .
Mostbet_rrSl
| #
букмекерская контора мостбет букмекерская контора мостбет .
beelinekrasnodarTet
| #
билайн подключить краснодар
https://infodomashnij-internet-krasnodar-3.ru
билайн подключить
VanceCam
| #
http://registr-a.ru
StefanGeX
| #
Все о машинах https://autoinfo.kyiv.ua в одном журнале! Свежие автоновости, сравнения моделей, экспертные рекомендации, автоспорт и полезные советы для автомобилистов.
Dwayneamips
| #
Журнал для автолюбителей https://avtoshans.in.ua и профессионалов! Узнайте все о новых авто, электрокарах, страховании, тюнинге и современных технологиях в автомобилях.
Darrencox
| #
Автомобильный мир https://diesel.kyiv.ua в одном журнале! Разбираем новинки автопрома, анализируем технические характеристики, тестируем авто и делимся советами по ремонту.
mostbet kirgizstan_aiEn
| #
мостбет казино играть mostbet2.com.kg .
MarcusClita
| #
Современный автомобильный https://k-moto.com.ua журнал! Читайте о трендах в автоиндустрии, новейших моделях, электромобилях, тюнинге и умных технологиях.
DouglasReema
| #
Автомобильный журнал https://orion-auto.com.ua только важные новости! Все о популярных марках, топовые авто 2024 года, электрокары, автономное вождение и тенденции рынка.
TimothyLiexy
| #
Журнал о машинах https://reuth911.com для настоящих ценителей авто! Обзоры, рейтинги, полезные статьи о ремонте, тюнинге и современных автомобильных технологиях.
KennethCip
| #
Автомобильный мир https://prestige-avto.com.ua в одном журнале! Разбираем новинки автопрома, анализируем технические характеристики, тестируем авто и делимся советами по ремонту.
beelinekrasnodarTet
| #
сайт билайн краснодар
https://infodomashnij-internet-krasnodar.ru
билайн цены
spider hoodie_tyel
| #
sp5der hoodie red https://spiderhoodie-us.com/spider-hoodies/ .
GordonApask
| #
Твой авто-гид https://troeshka.com.ua в мире машин! Обзоры новых моделей, тест-драйвы, советы по ремонту и тюнингу, автоновости и технологии будущего. Все, что нужно автолюбителю!
RaymonAgors
| #
Все об автомобилях https://tvk-avto.com.ua на одном портале! Новинки мирового автопрома, тест-драйвы, автострахование, электрокары и полезные статьи для каждого водителя.
BobbyLyhon
| #
Журнал про автолюбители https://tuning-kh.com.ua новости, тесты, обзоры! Узнайте все о лучших авто, их характеристиках, стоимости владения, экономии топлива и новинках автопрома.
StephenFrade
| #
Твой идеальный женский https://family-site.com.ua журнал! Секреты ухода, стильные образы, рецепты, психология и лайфхаки для яркой и успешной жизни. Вдохновение каждый день!
Dennysnubs
| #
Актуальные новости https://www.moscow.regnews.info Московского региона! Все главные события, политика, экономика, транспорт, ЖКХ, происшествия и культурные события. Будьте в курсе того, что происходит в вашем городе!
EddiePaL
| #
Главный женский журнал https://amideya.com.ua о стиле и успехе! Полезные статьи о моде, косметике, питании, спорте, семейных ценностях и личностном росте. Читай и развивайся вместе с нами!
Davidsom
| #
Твой идеальный женский https://femalesecret.kyiv.ua журнал! Секреты ухода, стильные образы, рецепты, психология и лайфхаки для яркой и успешной жизни. Вдохновение каждый день!
MichaelKap
| #
Портал для женщин https://feminine.kyiv.ua твой путеводитель в мире стиля и успеха! Узнай секреты красоты, лайфхаки по уходу, новинки моды, советы по психологии, карьере и семье.
Billyawali
| #
Твой женский портал https://girl.kyiv.ua о стиле и гармонии! Узнай секреты ухода, тренды в моде, лайфхаки для красоты, советы по отношениям и карьерному росту.
whore
| #
детское гей порно магазин купить меф
Waltersow
| #
Главный женский портал https://horoscope-web.com будь в центре трендов! Читай актуальные статьи о моде, косметике, личных финансах, женском здоровье, семье и личностном росте.
DanielMus
| #
Портал для женщин https://lolitaquieretemucho.com которые ценят себя! Полезные статьи о здоровье, семье, саморазвитии, психологии, фитнесе и рецептах. Будь уверенной, счастливой и успешной!
вован казино скачать
| #
Выигрыши выводятся моментально, проверил на личном опыте.
вован казино скачать
Stihi dlya detei_dcma
| #
Пословицы и поговорки для детей Пословицы и поговорки для детей .
Zamena tyrbini razvod_lgSn
| #
Как заменить турбину на машине и не переплатить втрое больше. Типичная схема развода на замену турбины. Будьте осторожны. Подробности тут Как заменить турбину на машине и не переплатить втрое больше. Типичная схема развода на замену турбины. Будьте осторожны. Подробности тут .
SOYT_rfkt
| #
заявка на соут http://www.sout095.ru/ .
beelinekrasnodarTet
| #
билайн интернет краснодар
https://plus-domashnij-internet-krasnodar-2.ru
сайт билайн краснодар
JimmyFex
| #
Портал для женщин https://nicegirl.kyiv.ua которые любят себя! Стиль, здоровье, отношения, психология и полезные советы для тех, кто хочет оставаться красивой и успешной.
Ernesthax
| #
Твой путеводитель https://mirlady.kyiv.ua в мире женских секретов! Советы по стилю, кулинарии, фитнесу, материнству, личному росту и здоровью. Всё, что нужно для счастливой жизни!
Willielinue
| #
Современный женский портал https://presslook.com.ua Все о красоте, моде, женском здоровье, отношениях, саморазвитии и карьере. Читай, вдохновляйся и меняй свою жизнь к лучшему!
Jameshut
| #
Твой идеальный https://prettywoman.kyiv.ua женский портал! Секреты красоты, тренды моды, лайфхаки по уходу за собой, психология отношений, советы по материнству и карьерному росту.
zaymbezproverokvah
| #
Нужны капитал срочно? Получите онлайн-заем на пластиковую карту за момент. https://zaym-bez-proverok.ru/ Оформите форму без документов и получите положительный ответ уже сегодня!
JimmyCance
| #
Сайт для женщин https://ramledlightings.com будь лучшей версией себя! Читай о моде, красоте, психологии, отношениях, материнстве и женском здоровье. Найди вдохновение и полезные советы для каждого дня!
JamesCalia
| #
Лучший портал для родителей https://geog.org.ua и детей! Читайте о воспитании, обучении, здоровье, детской психологии, играх и семейном досуге. Советы экспертов и практические рекомендации.
Manuelagido
| #
Портал о детях https://mch.com.ua полезно для родителей! Воспитание, здоровье, развитие, обучение, досуг, игры и семейные традиции. Экспертные советы, лайфхаки и полезные материалы для гармоничного роста малыша.
Arnoldgrews
| #
Твой женский сайт https://musicbit.com.ua о стиле, здоровье и вдохновении! Узнай секреты красоты, следи за модными новинками, развивайся, читай о психологии отношений и оставайся в гармонии с собой.
beelinekrasnodarTet
| #
билайн интернет
https://plus-domashnij-internet-krasnodar.ru
билайн подключить краснодар
zaymbezproverokvah
| #
Нужны деньги срочно? Получите микрозайм на счет в банке за короткий срок. https://zaym-bez-proverok.ru/ Оформите анкету без документов и получите положительный ответ уже сегодня!
Robertben
| #
Справочник лекарственных https://mikstur.com средств – полная информация о медикаментах! Описания, показания, противопоказания, дозировки, аналоги и инструкции по применению.
DuaneMiz
| #
Портал о здоровье https://fines.com.ua все, что нужно для крепкого организма! Советы врачей, профилактика болезней, здоровое питание, спорт, психология, народная медицина и лайфхаки для долгой и активной жизни.
turkof
| #
турецкие сериалы на русском языке турецкие сериалы смотреть
Jeffreyelolo
| #
сериалы на турецком языке онлайн смотреть турецкие сериалы онлайн
Kennethdob
| #
турецкие сериалы бесплатно турецкие сериалы смотреть
Scottfouch
| #
смотреть лучшие турецкие сериалы смотреть онлайн турецкие сериалы
LloydKic
| #
турецкие сериалы бесплатно смотреть турецкие сериалы онлайн бесплатно
beelinekrasnodarTet
| #
билайн домашний интернет краснодар
https://plus-domashnij-internet-krasnodar-3.ru
провайдер билайн
ShaneRub
| #
турецкие сериалы онлайн турецкий сериал смотреть
Clydeglalo
| #
турецкие сериалы на турецком языке смотреть турецкие сериалы онлайн бесплатно
ToneyWax
| #
сериалы на турецком языке онлайн смотреть турецкий сериал
Samuelbig
| #
турецкие сериалы бесплатно турецкие сериалы онлайн
Пансионат «Уют» Киев Вышгород
| #
Будинок престарілих Пансіонат «Затишок» Вишгород
https://dom-prestarelyh.kiev.ua/ru/
⏩ Догляд, оздоровлення, реабілітація літніх людей. Догляд за лежачими хворими ⏩ Так само, ми надаємо послуги медичного транспортування з України і за кордоном, пишіть нам на електронну пошту або дзвоніть
Перевезення Хворих Київ, Вишгород, Україна
Дом престарелых Пансионат «Уют» Вышгород ⏩ Специализируется по уходу, оздоровлению и реабилитацией за пожилыми людьми и за лежачими больными. ⏩ Так же, мы предоставляем услуги медицинской транспортировки по Украине и за границей, пишите нам на электронную почту или звоните.
#ПансионатКиев
#ДомПрестарелыхВышгород
#ПансіонатВишгород
#БудинокПохилогоВікуВишгород
Перевезення хворих Львів
| #
Перевезення хворих за кордон
https://a-med.com.ua/services/mizhnarodni-perevezennya/
Перевезення хворих Львів • Приватна платна швидка медична допомога Львів ⚕️ Транспортування лежачих, сидячих та важких хворих Львів – Україна ☎️ +380680177630 ♥ Кваліфіковане медичне транспортування ➡ Медичний супровід хворих Львів ⏩ Міжнародне перевезення хворих ⏩ Перевезення закордон ➡ Львів ➡ А-Мед • Transportation of patients abroad
Транспортування лежачих, сидячих та тяжких хворих Львів – Україна ☑️ Міжнародне перевезення хворих ⏩ Приватний медичний транспорт
#МедичнийТранспортКиїв
#МедичнийТранспортЛьвів
#МедичнийТранспортХарків
#Транспортное
#МедичнийТранспортДніпро
#ПеревезенняХворихЛьвів
#ПеревезенняТяжкихХворихЛьвів
#ПеревезенняХворихЗакордон
Перевезення хворих за кордон
Натяжные потолки Одесса.
| #
Натяжные потолки Одесса и Одесская область.
https://www.tecko.in.ua/
✓ Ремонт, установка натяжных потолков.
✓ Натяжные потолки любых видов индивидуально и под заказ.
✓ Натяжные стены в Одессе.
✓ Подвесные потолки в Одессе
Натяжні стелі Одеса – TEKO ⏩ Актуальні поради з натяжних стель ⭐ Натяжні стелі за типом покриття ☑️ Вибираємо натяжні стелі з малюнком ➤ Види барвників для натяжних стель ♥ найпопулярніші теми натяжних стель
#НатяжныеПотолкиОдесса
#НатяжныеПотолкиОдессаЦена
#ПодвесныеПотолкиОдесса
#НатяжныеСтеныОдесса
#НатяжныеПотолкиТЕКО
#НатяжныеПотолкивОдессе
#НатяжныеПотолкиСделайСам
#НатяжныеПотолки
#ПотолкиОдесса
#НатяжныеПотолкиОдессаЦена
#КупитьНатяжныеПотолкиОдесса
Натяжные потолки Одесса – TEKO
Перевезення хворих Львів Мед 103
| #
Перевезення хворих Львів Мед103
https://med103.com/cities/lviv/
♦ Приватна платна швидка медична допомога Львів ⚕️ Транспортування лежачих, сидячих та важких хворих Україна ☎️ +380738870303 ♥ Кваліфіковане медичне транспортування ➡ Медичний супровід хворих ⏩ Міжнародне перевезення хворих ⏩ Львів ➡ Львів ➡ Мед 10
Транспортування лежачих, сидячих та тяжких хворих Львів – Україна ☑️ Міжнародне перевезення хворих ⏩ Приватний медичний транспорт
#МедичнийТранспортКиїв
#МедичнийТранспортЛьвів
#МедичнийТранспортХарків
#МедичнийТранспортОдеса
#МедичнийТранспортДніпро
#ПеревезенняХворихУкраїна
Частная платная скорая медицинская помощь Днепр ⚕️ Транспортировка лежачих, сидячих и тяжелых больных Украина ☎️ +380738870303 ♥ Квалифицированная медицинская транспортировка ➡ Медицинское сопровождение больных ⏩ Международная перевозка больных ⏩ Частный медицинский транспорт ♥ Med103 ➡ Киев ➡ Одесса ➡ Львов • Днепр • Харьков Украина
Транспортировка лежачих, сидячих и тяжелых больных Украина ☑️ Международная перевозка больных ⏩ Частный медицинский транспорт
#МедицинскийТранспортКиев
#МедицинскийТранспортЛьвов
#МедицинскийТранспортХарьков
#МедицинскийТранспортОдесса
#МедицинскийТранспортДнепр
#ПеревозкаБольныхУкраина
Private paid ambulance ⚕️ Transportation of bedridden, sitting and seriously ill patients Ukraine ☎️ +380738870303 ♥ Qualified medical transportation ➡ Medical support for patients ⏩ International transportation of patients ⏩ Private medical transport ♥ Med103 ➡ Kyiv ➡ Odessa ➡ Lviv • Dnepr • Kharkov Ukraine
Міжнародне медичне перевезення. Країни
Німеччина
Румунія
Італія
Польща
Молдова
Пансионат «Уют» Днепр
| #
Дом престарелых Пансионат «Уют» Днепр
https://uyt.dp.ua/ru/
Будинок престарілих Пансіонат «Затишок» Дніпро ⏩ Спеціалізується по догляду, оздоровленню і реабілітацією за літніми людьми і за лежачими хворими ⏩ Так само, ми надаємо послуги медичного транспортування з України і за кордоном, пишіть нам на електронну пошту або дзвоніть
⏩ Специализируется по уходу, оздоровлению и реабилитацией за пожилыми людьми и за лежачими больными. ⏩ Так же, мы предоставляем услуги медицинской транспортировки по Украине и за границей, пишите нам на электронную почту или звоните.
#ПансионатДнепр
#ДомПрестарелыхДнепр
#ПансіонатДніпро
#БудинокПохилогоВікуДніпро
BrianMop
| #
серия сезон турецкий сериал https://proturkish.biz
MichaelPes
| #
турецкий сериал все серии турецкие сериалы смотретя
beelinekrasnodarTet
| #
билайн телевидение краснодар
https://plus-domashnij-internet-krasnodar-2.ru
билайн тарифы краснодар
Rylonnie shtori s elektroprivodom_rzpl
| #
рулонные шторы автоматические купить рулонные шторы автоматические купить .
mostbet kirgizstan_cqKt
| #
мостбет вход регистрация https://www.mostbet3.com.kg .
mostbet kirgizstan_woSl
| #
mostbet.com skachat mostbet4.com.kg .
obychenie po ohrane tryda moskva_lspa
| #
дистанционное обучение работника по охране труда Москва дистанционное обучение работника по охране труда Москва .
SOYT_zfsl
| #
соут специальная оценка условий соут специальная оценка условий .
JosephTor
| #
топ лучших казино
IsraelCow
| #
лицензионные слоты игровые автоматы рейтинг лучших сайтов россии
Michaelgit
| #
online casino eldorado casino
beelinekrasnodarTet
| #
билайн подключить
https://plus-domashnij-internet-krasnodar.ru
билайн домашний интернет краснодар
Roberttraro
| #
Мы подобрали часто задаваемые вопросы пользователей из Казахстана на тему https://promokod-na-1xbet.com/ и их решения. Ввод промокодов при создании аккаунта и начальном депозите обеспечивает возможность значительно увеличить баланс и получить больше возможностей для игры.
네이버 아이디 구매
| #
https://consolebang.com/members/%EB%84%A4%EC%9D%B4%EB%B2%84-%EC%95%84%EC%9D%B4%EB%94%94-%EA%B5%AC%EB%A7%A4.34257/#about
비아그라 구매
| #
https://xn--w5-o02ik82a9kav54aokmxvc.mystrikingly.com/blog/3f69fae99e6
beelinekrasnodarTet
| #
билайн подключение
https://plus-domashnij-internet-krasnodar-3.ru
билайн интернет краснодар
накрутка подписчиков Инстаграм без заданий бесплатно
| #
Продвижение в Instagram стало проще с накруткой!
накрутка подписчиков Инстаграм без заданий бесплатно
Waltertop
| #
https://aldk24.ru/
накрутка зрителей на стрим Твич
| #
Можно ли накручивать зрителей постепенно?
накрутка зрителей на стрим Твич
JamesNeign
| #
New Retro Casino
JamesTog
| #
kinogo новинки kinogo фильмы для компьютера
RobertKiz
| #
kinogo боевики 2025 киного фильмы для мобильных устройств
Ronaldvek
| #
Booi Casino
Kevinfipse
| #
казино с пополнением с криптой
accurateautobodyrepairlmailm
| #
Our vehicle tuning services are designed to enhance your performance. We offer unique upgrades that improve the efficiency and aesthetic of your auto. Whether you’re interested in suspension modifications or tuning specific parts, we provide top solutions for every need. Trust our experts to deliver reliable results that will revive your ride. For more details, visit our website at https://accurateautobodyrepair.com/ and discover how we can help you.
Nakrytka podpischikov TG_jcPl
| #
накрутка подписчиков ТГ накрутка подписчиков ТГ .
Nakrytka Tvich_ukMr
| #
программа для накрутки зрителей Твич программа для накрутки зрителей Твич .
1win md_qlea
| #
1win moldova download 1win moldova download .
1win md_koKr
| #
?nregistrare 1win https://1win3.md .
Merlezew
| #
акции вб
MichaelDycle
| #
Discover endless entertainment at Fmovies! Stream the latest movies, TV shows, and series in HD, all for free. Enjoy a user-friendly interface, fast streaming, and a vast library updated daily. Your favorite content is just a click away https://2fmoviesto.com/
beelinekrasnodarTet
| #
билайн тв краснодар
https://plus-domashnij-internet-krasnodar-2.ru
провайдер билайн
Kenneth_Greno
| #
накрутка подписчиков Ютуб без отписок
accurateautobodyrepairlmailm
| #
Our motor tuning services are designed to enhance your performance. We offer personalized upgrades that improve the engine and design of your auto. Whether you’re interested in engine modifications or upgrading specific parts, we provide high solutions for every need. Trust our experts to deliver flawless results that will boost your ride. For more details, visit our website at https://accurateautobodyrepair.com/ and discover how we can help you.
create binance account
| #
I don’t think the title of your article matches the content lol. Just kidding, mainly because I had some doubts after reading the article.
RobertJEM
| #
ключ активации виндовс 11
Melvingor
| #
Непродовольственные товары Услуги перевозки в интернет-магазине товаров, не относящихся к продуктовому сегменту.
Edwardmak
| #
Автопортал https://avtogid.in.ua Автогiд сайт с полезными советами для автовладельцев. Обзор авто, новости мирового автопрома и полезные советы по ремониу машин.
네이버 아이디 구매
| #
https://naveridbuy.exblog.jp/35878128/
LarryBex
| #
Вавада казино
1vin_pwsl
| #
казино lucky jet http://1win1.com.kg/ .
DustinZen
| #
https://deseko.ru/
Леон казино
| #
Реальные отзывы об онлайн казино: где
искать?
Чтобы составить объективное представление о различных игровых заведениях в интернете,
стоит обратить внимание на специализированные форумы и сайты.
Эти площадки, как правило, собирают мнения настоящих пользователей, которые делятся
своим опытом и рассказывают о том, что на самом
деле происходит за кулисами.
Ключевые ресурсы, такие как Gamblers’ Review или CasinoGuru, предоставляют качественную информацию и
ранжируют заведения по различным критериям.
Также рекомендую воспользоваться социальными сетями,
такими как Facebook и Reddit. Там часто можно найти живое обсуждение
платформ, а также задать вопросы опытным игрокам.
Обратите внимание на группы, посвященные играм на деньги,
где участники делятся как
положительными, так и отрицательными моментами.
Пароли к успеху – это тщательный анализ найденной
информации и скептический подход к ней.
Не забывайте проверять дату публикации мнений, так как рынок азартных игр
постоянно меняется, и информация, актуальная в прошлом, может быть устаревшей.
Доступ к свежим данным поможет избежать неприятных сюрпризов при выборе места для игры.
Поиск проверенных платформ для
отзывов об азартных учреждениях
Ищите специализированные сайты,
которые тщательно анализируют отзывы
пользователей. Обратите внимание на
ресурсы, предлагающие независимый контент и рейтинги.
Платформы, такие как AskGamblers, CasinoGuru и Trustpilot,
содержат информацию о различных заведениях, включая
комментарии клиентов и оценки.
Проверьте наличие форумов и сообществ, где игроманы обсуждают свой опыт.
Сайты, такие как Reddit, содержат разделы,
посвященные игорным заведениям, где пользователи делятся
конкретными случаями.
Изучите наличие блогов и YouTube-каналов, где эксперты делают обзоры и предлагают детализированные
мнения о работе платформ.
Опытные игроки иногда публикуют свои впечатления о
конкретных игорных площадках, что может быть полезным для
принятия решения.
Обратите внимание на профессиональные игровые порталы, которые проводят тщательную проверку заведений перед публикацией информации о них.
Убедитесь, что платформа имеет документированные исследования и
сравнительные анализы.
Фокусируйтесь на отзывах, которые отмечают
конкретные аспекты работы заведения, такие как скорость выплат,
качество службы поддержки и разнообразие игрового контента.
Это поможет получить более полное
представление о репутации заведения.
Анализ отзывов: как отличить
правду от лжи
Для оценки подлинности комментариев, проверьте источники.
Начните с площадок, имеющих
репутацию, таких как форумы и специализированные сайты,
где пользователи обсуждают свои впечатления.
Сравните несколько откликов.
Если в большинстве случаев звучат схожие проблемы или положительные моменты,
это повышает вероятность статистической достоверности.
Однако уклончивые сообщения с общими фразами могут указать на намерение скрыть истинный опыт.
Отслеживайте даты публикаций.
Свежие упоминания могут быть полезнее, так как они отражают
актуальную действительность сервиса.
Устаревшие данные могут не представлять реальной
картины.
Изучайте также поведение
авторов. Если пользователь оставляет комментарии исключительно положительного или отрицательного
характера, это может говорить о предвзятости, иногда связанной с работой представителя заведения.
Ищите сбалансированные мнения.
Обратите внимание на соотношение отзывов на разных ресурсах.
Если на одном сайте преобладают негативные оценки, а на других –
исключительно положительные, это может указывать на манипуляции
с рейтингами.
Не верьте слепо. Правдивый опыт быстро становится вирусным,
в то время как купленные
мнения могут выглядеть шаблонно.
Изучая среднюю точку зрения,
выбирайте наиболее обоснованные и правдоподобные
мнения пользователей.
Леон казино
beelinekrasnodarTet
| #
провайдер билайн
https://plus-domashnij-internet-krasnodar.ru
билайн подключить краснодар
네이버 아이디 구매
| #
https://energetic-goat-dc4vlq.mystrikingly.com/blog/1b973c46327
Пинко казино
| #
Какие игры в казино самые прибыльные?
Рекомендуется обратить внимание на блэкджек, так как
он предлагает один из лучших коэффициентов для игроков.
С оптимальной стратегией, возврат к игроку (RTP) может достигать 99.5%.
Также, различные вариации, такие как
European Blackjack, могут обеспечить
ещё более высокие выигрыши за счёт меньшего
количества колод.
Покер, в частности техасский холдем, представляет собой отличную возможность для получения прибыли.
Здесь всё зависит от умений игрока.
Профессионалы могут получить значительные выигрыши, так как казино
зарабатывает на сборах, а не на проигрыше клиентов.
Автоматы могут быть прибыльными, но важно выбирать слоты с высоким RTP, часто превышающим 96%.
Например, такие модели, как Blood Suckers или Mega Joker, известны своими
хорошими выплатами, что делает их предпочтительными для многих азартных посетителей.
Рулетка также заслуживает внимания.
Игроки могут использовать стратегию,
например, ставя на наружные ставки, что обеспечивает более высокий шанс на выигрыш, хотя и с меньшими
выплатами. Европейская рулетка с одним зеро считается наиболее выгодной для участников.
Анализ возврата игроку (RTP) в различных играх казино
Различные автоматы обеспечивают разные коэффициенты RTP.
Обычно они варьируются от 85% до 98%.
Слоты с высоким RTP, как правило, более выгодны.
Например, такие как “Blood Suckers” и “Mega Joker” достигают RTP 98% и выше.
В настольных вариантах, таких как блэкджек и
рулетка, наблюдается различие в показателях.
Блэкджек, при оптимальной стратегии, может иметь RTP до 99.5%, что
делает его одним из самых заложенных механик.
В рулетке классические варианты
предлагают RTP около 97.3% для европейской версии, что превосходит американскую с её RTP
около 94.74%.
Видеопокер также показывает параметры, которые варьируются.
Например, игра “Jacks or Better” с правильной стратегией
приближается к 99.5% RTP. Важно отмечать, что чем больше знаний в стратегии,
тем выше шансы на получение прибыльных результатов.
Тематические автоматы с прогрессивным
джекпотом могут иметь низкий RTP, иногда ниже 90%.
Однако потенциальные выигрыши могут быть значительными,
что привлекает игроков, готовых рискнуть ради больших призов.
Итак, тщательно изучая RTP разных вариантов,
можно значительно улучшить шансы на успех.
Выбор автоматов или настольных игр с более высоким показателем RTP
может стать решающим фактором при
принятии решения о том, как разместить свои ставки и вывести
максимальную выгоду из игрового опыта.
Стратегии и советы для игр с высоким
шансом на выигрыш
Используйте стратегию счет карточек для блэкджека.
Это позволит увеличить вероятность выигрыша, отслеживая соотношение высоких
и низких карт в колоде. При этом важно сохранять спокойствие и придерживаться своей тактики.
В рулетке выбирайте ставки на цвет или
четность вместо отдельных
чисел. Такой подход значительно снижает
риск потерь и увеличивает шансы на
победу.
Во время игры в покер воспользуйтесь позиционной стратегией.
Игроки на поздней позиции получают преимущество, так как имеют возможность оценить действия своих оппонентов перед принятием
решения.
Обязательно устанавливайте лимиты
для проигрышей и выигрышей.
Это поможет контролировать бюджет и избежать крупных потерь.
Понимание коэффициентов и выплат в слотовых машинах повысит
шансы на успех. Ищите модели с высоким процентом возврата игроку (RTP) и
низкой дисперсией.
Знайте и используйте бонусы и акции,
которые предлагают заведения.
Они могут значительно увеличить игровую линию
и обеспечить дополнительные возможности для выигрыша.
Обращайте внимание на правила конкретного формата,
чтобы избегать ошибок, которые могут стоить вам денег.
Понимание механики поможет принимать более обоснованные решения.
Регулярно практикуйтесь в бесплатных версиях, чтобы улучшить
свои навыки и выявить стратегии,
которые работают для вас
наилучшим образом. Это даст возможность уверенно подходить к игре на деньги.
Пинко казино
nakrutk_Spupt
| #
накрутка лайков в ВК бесплатно
Пинко казино
| #
Лучшие онлайн казино 2025 года
Рекомендуем обратить внимание на несколько выдающихся платформ в 2025 году, которые предлагают привлекательные условия
и высококачественный сервис. В первую
очередь стоит выделить Royal Slot.
Это заведение зарекомендовало себя
благодаря высокому уровню безопасности и множеству выборов
развлечений, включая новые слоты с уникальными механиками.
Еще одной примечательной площадкой является PlayZone, предлагающая
щедрые бонусы и быструю выплату выигрышей.
Бонусы на первый депозит могут достигать 200%, что является отличным мотивом для новых пользователей.
В дополнение к стандартным играм, здесь представлено множество
эксклюзивных турниров и акций.
Не стоит забывать и о WinMasters, устойчиво набирающем
популярность благодаря интуитивно
понятному интерфейсу и широкому выбору платежных методов.
Благодаря множеству отзывов
пользователей, платформа
выделяется среди конкурентов
комфортом и надежностью, что немаловажно для игроков.
Каждое заведение решает задачи своих клиентов по-разному,
поэтому рекомендуется ознакомиться с условиями при
регистрации и выбрать наиболее
подходящий для себя вариант. Поскольку ставки
сегодня стали доступнее, важно учитывать рейтинги и
отзывы, чтобы принимать взвешенные решения при выборе площадки.
Критерии выбора надежного виртуального заведения в 2025 году
Первое, на что следует обратить внимание, – наличие лицензии.
Проверяйте, выдана ли лицензия авторитетным регулятором, таким как MGa или UKGC.
Это обеспечивает определенный уровень защиты игроков.
Вторым важным аспектом является разнообразие игрового контента.
Надежные платформы предлагают игры от известных производителей с
высоким качеством графики и честными шансами
на выигрыш. Исследуйте список поставщиков софта и их репутацию.
Проверьте ревью и рейтинги заведения на сторонних ресурсах.
Мнения других игроков дадут вам представление
о качестве обслуживания, скорости
выплат и уважении к клиентам.
Изучите приветственные бонусы и акции для
постоянных пользователей. Сравните условия по всем предложениям – не все бонусы оправдывают ожидания,
иногда высокие требования по ставкам могут сделать их невыгодными.
Качество службы поддержки – немаловажный показатель.
Наличие оперативной и компетентной
поддержки через чат, телефон или электронную почту позволит быстро разрешать возникшие вопросы.
Последний, но не менее важный аспект
– это отзывы об уровне безопасности и конфиденциальности данных.
Проверьте, использует ли заведение
методы защиты пользовательской информации и предоставляет ли игрокам право на установление лимитов.
Топ-5 онлайн заведений с
высокой отдачей и бонусами
для игроков
1. Spin Casino – предлагает привлекательный пакет приветственных бонусов до
1000 евро и постоянные акции для
постоянных клиентов. Высокий уровень отдачи в пределах 96,5%.
2. Betway – гарантирует 100% бонус на первый
депозит до 200 евро. Средний RTP составляет 97%, что делает этот ресурс привлекательным для пользователей.
3. Casino XYZ – известен своими огромными джекпотами и 250% бонусом на первый депозит.
Одним из главных плюсов является RTP на уровне 95,7%.
4. Lucky Land – дарит игрокам до 300 евро на старт и устраивает регулярные турниры с призами.
Общий процент возврата составляет 96,3%.
5. Royal Fortune – порадует вас 150% бонусом на первое пополнение.
Высокая отдача на уровне 98% добавляет уверенности в успешной игре.
Пинко казино
Горилла казино
| #
Как вывести выигрыш без комиссии?
Сделайте выбор в пользу электронных кошельков.
Многие платформы предоставляют
возможность осуществлять транзакции через такие сервисы,
как Яндекс.Деньги или Webmoney. Обратите внимание на отсутствие дополнительных
сборов при переводе на ваш аккаунт.
Изучите тарифы и условия. Перед тем как приступить к эксплуатации
сервиса, внимательно ознакомьтесь с информацией о возможных сборах.
Некоторые компании предлагают минимальные комиссии
или полное отсутствие затрат при соблюдении определённых
условий, таких как достижение определенного оборота.
Сравнивайте различные варианты.
Просматривайте и сопоставляйте условия разных сервисов.
Иногда переключение на другую платформу может существенно сократить финансовые затраты на
ваши переводы.
Следуя данным рекомендациям, можно избежать дополнительных трат
и максимально эффективно управлять своими
средствами.
Обращайте внимание на репутацию платформ.
Пользуйтесь ресурсами с позитивными
отзывами и высокой оценкой от пользователей.
Проверяйте наличие лицензий и соответствие местным нормативам.
Сравните условия различных сервисов.
Некоторые предлагают нулевые сборы при определённых условиях,
к примеру, при регулярном использовании или при минимальной сумме транзакции.
Изучите методы перевода. Электронные кошельки, такие как Яндекс.Деньги или
Qiwi, часто имеют выгодные условия.
Убедитесь, что выбранный вами кошелек поддерживается платформой.
Изучите службу поддержки.
Ответственная команда поддержки должна быстро решать возникающие вопросы.
Это также показатель надежности
платформы.
Не забывайте про сборы, которые могут возникнуть на стороне платежных систем.
Даже если платформа не берет свою комиссию, другие компании могут взимать дополнительные средства за перевод.
Используйте электронные кошельки,
такие как QIWI или Yandex.Money.
Проверьте возможность переводов между пользователями без дополнительных затрат.
Данные платформы часто предлагают бесплатные переводы.
Платежные системы PayPal и Skrill имеют
опции для отправки средств без взимания платы,
если перевод осуществляется с баланса пользователя.
При выборе криптовалютных транзакций обращайте внимание на блокчейны
с низкими или отсутствующими сборами на переводы,
например, Tron или Stellar.
Выбирайте платформы для
ставок и игр, предлагающие
программы кэшбэка, где возврат процентной
части помогает компенсировать любые
возможные затраты на перевод.
Горилла казино
ThomasPredo
| #
Сайт мiста Черкаси https://u-misti.cherkasy.ua новини Черкас та областi.
JeffreyGer
| #
JVSpin официальный сайт
Jamessak
| #
Видеотетка Видеотетка, Абраменко, Чучелупа.
Howardedict
| #
накрутка Тик Ток 1000
Matthewnum
| #
Cryptoboss casino бездепозитный бонус Cryptoboss Casino, криптобосс казино, cryptoboss регистрация, cryptoboss casino бездепозитный бонус.
beelinekrasnodarTet
| #
билайн домашний интернет
https://plus-domashnij-internet-krasnodar-3.ru
билайн телевидение краснодар
네이버 아이디 구매
| #
https://aquamarine-magnolia-dc4vlj.mystrikingly.com/blog/71317ede0ec
Marvinmon
| #
Whether you need certified document translation, legal translation, medical translation, financial translation, or book translation, Russian-Translation.co.uk offers top-tier solutions tailored to your needs.
Medical Translation Services
Precision in medical translation is crucial as it involves patient care, clinical trials, and pharmaceutical documentation. We translate:
Medical reports
Patient records
Research papers
Prescription guidelines
Visit our russian to english translation of medical documents page for specialized assistance.
Williamicego
| #
azino777 зеркало azino777 официальный
비아그라 구매
| #
https://thoughtful-fox-dbgzh7.mystrikingly.com/blog/55d38114fa0
1vin_ltSn
| #
1wi casino https://www.1win2.com.kg .
1vin_qwEr
| #
букмекерская контора 1win букмекерская контора 1win .
AnthonyAerom
| #
Накрутка подписчиков в Телеграм
1vin_otkt
| #
джет казино официальное зеркало http://1win8.com.kg .
1win_yuPt
| #
1win site 1win site .
KevintoP
| #
Бездепозитные казино с выводом Бездепозитные казино за регистрацию с выводом без пополнения по номеру.
AnthonyAerom
| #
Способы накрутки подписчиков в Телеграм
DonaldRog
| #
Новини Дніпра https://u-misti.dp.ua Сайт міста Дніпро та області.
JohnnyCix
| #
как получить 1000 рублей бесплатно на карту за регистрацию Игровые автоматы на реальные деньги с бонусом при регистрации без депозита.
CasinosHoubs
| #
You actually explained that very well.
spill casino online https://hotgamblingguide.org/ignition-casino-safe/ pa online casinos with no deposit bonus
Nakrutka_blire
| #
бесплатная накрутка ВК группа подписчики живые
AnthonyVem
| #
Аренда жилой недвижимости https://domhata.ru без посредников и переплат! Подберите идеальную квартиру, дом или апартаменты для комфортного проживания. Удобный поиск по цене, району и условиям.
play fortuna казино
| #
Топовое казино, которому можно доверять!
play fortuna казино
DonaldEtext
| #
У містi Київ https://u-misti.kyiv.ua новини та події Київщини
MichaelVap
| #
диетолог ярославль врач психотерапевт, консультация психотерапевта, психотерапевт онлайн, психотерапевт ярославль.
pet-supplies
| #
pet products online store sale of pet products
KevinWeeda
| #
pet products choose pet store website
Robertvep
| #
https://realtrustfeedback.com/
Robertvep
| #
https://realtrustfeedback.com/
VictorMig
| #
накрутка просмотров на пост в ТГ
VictorMig
| #
накрутка просмотров на пост в Телеграм
JamesPreop
| #
Городской портал Одессы https://u-misti.odesa.ua новости и события Одессы и области
ClarkWaL
| #
накрутка лайков на комментарий в ВК
DericksaH
| #
Как накрутить подписчиков в ВК в группу
LarryHob
| #
Заработок в интернете Лотерейные билеты, Регистрация домена, Заработок в интернете, Биржа ссылок.
Лев игровые автоматы
| #
Лимиты на вывод средств в онлайн казино могут отличаться
Лев игровые автоматы
internetekaterinburgWAH
| #
мегафон телевидение
мегафон подключение екатеринбург
мегафон домашний интернет екатеринбург
игорный клуб Лев
| #
Вывод денег в онлайн казино возможен на
банковские карты и электронные кошельки
игорный клуб Лев
Brucehag
| #
накрутка реакций Телеграмм бесплатно
1win_txma
| #
1вин приложение официальный сайт 1win2.am .
Brucehag
| #
накрутка лайков инстаграм Телеграм бот
казино Лев
| #
Выбирайте онлайн казино Лев с широким выбором платёжных методов
DericksaH
| #
накрутка подписчиков ВК
JasonVot
| #
фрибет за регистрацию Ставки на спорт
Matthewstoli
| #
Блог о медицине https://medportal.co.ua Медпортал. Здрововье, спорт, психология, больницы. Статьи о медицине.
Michaelcooge
| #
Casino bonus Casino bonus Игровые автоматы с бонусом за регистрацию без депозита.
BrianTic
| #
An open source cloud https://github.com/pg-main/PostgreSQL/releases DBMS that automates management, updates, and backups. Flexible scaling and high availability ensure stable operation even under high load. Easy integration with cloud infrastructure.
internetekaterinburgWAH
| #
мегафон тв екатеринбург
https://plus-ekb-domasnij-internet-2.ru
мегафон тв екатеринбург
비아그라 구매
| #
https://gajweor.pixnet.net/blog/post/162317545
Ronaldped
| #
Сайт Киева https://infosite.kyiv.ua ИнфоКиев: последние новости и события Киева и области.
Douglaskit
| #
Безопасная продажа онколекарств онлайн
TyroneWes
| #
Автоэксперт Алматы
ErnestFrusa
| #
Девушки с шикарными фигурами и сексуальной внешностью ждут встречи с тобой в любое время суток https://sibirki.vip/
ErnestFrusa
| #
Девушки, которые знают толк в страсти, подарят тебе незабываемый отдых без границ и запретов https://sibirki.vip/
CharlesWailm
| #
Накрутка лайков на посты в ВК
ClarkWaL
| #
Новые казино без депозита
RaymondGef
| #
Портал мiста Львів https://u-misti.lviv.ua останні події та новини.
internetekaterinburgWAH
| #
мегафон подключение екатеринбург
https://plus-ekb-domasnij-internet-3.ru
мегафон подключить екатеринбург
1win uganda_lzkr
| #
1win betting app http://1win1.ug/ .
1win uganda_ktOl
| #
1win casino https://1win2.ug .
1win uganda_llOt
| #
1win new 1win3.ug .
1win uganda_jrKt
| #
1win bet uganda 1win bet uganda .
CharlesWailm
| #
накрутка лайков ВК бесплатно
1win_juen
| #
1win.ng https://1win1.com.ng/ .
internetekaterinburgWAH
| #
мегафон домашний интернет екатеринбург
мегафон домашний интернет екатеринбург
мегафон тарифы екатеринбург
Michaelnal
| #
Файне місто Львів https://faine-misto.lviv.ua сайт Львова. Новости, события, места и обзоры.
SidneyClino
| #
Beneficial forum posts. Thanks!
casino online new york https://combatcasino.info/review-wild/ 2016 nj new online casino games
affiliate programs_olab
| #
Professional affiliate networks for business development http://d–b.info/60008.
internetekaterinburgWAH
| #
мегафон тв
https://plus-ekb-domasnij-internet-2.ru
мегафон подключить
Casino_boabs
| #
Бездепозитные фриспины за регистрацию, эксклюзивные бонусы и щедрая VIP система для активных игроков. Ищи нас в телеграм по ID @arkadacasino_site или переходи по ссылке: https://t.me/arkadacasino_site
Herbertpiori
| #
Новости Одессы https://faine-misto.od.ua на городском портале Файне мiсто. События, обзоры, происшествия Одессы и области.
Cliftonnut
| #
накрутка лайков тикток
internetekaterinburgWAH
| #
мегафон интернет екатеринбург
https://plus-ekb-domasnij-internet-3.ru
мегафон интернет
Cliftonnut
| #
накрутить лайки в Тик Токе
ThomasNut
| #
Сайт Хмельницкого https://u-misti.khmelnytskyi.ua новости Хмельницкой области, события, обзоры
internetekaterinburgWAH
| #
мегафон тв екатеринбург
мегафон телевидение екатеринбург
мегафон домашний интернет
Igrovie avtomati _ekKt
| #
игровые автоматы с выводом денег игровые автоматы с выводом денег .
1win_fqOt
| #
1 win casino https://1win2.com.ng .
1win_mhor
| #
1win football http://www.1win3.com.ng/ .
JustinHem
| #
hauling cars for dealerships
JustinHem
| #
car hauling
internetekaterinburgWAH
| #
мегафон телевидение екатеринбург
https://plus-ekb-domasnij-internet-2.ru
мегафон подключить екатеринбург
www.binance.com prijava
| #
Thank you for your sharing. I am worried that I lack creative ideas. It is your article that makes me full of hope. Thank you. But, I have a question, can you help me?
Brentfah
| #
Новости Черновцы https://u-misti.chernivtsi.ua последние события Черновцов и области.
1win_gdot
| #
1win.global 1win4.com.ng .
mostbet kg_amSl
| #
mostbet kyrgyzstan https://mostbet100.com.kg/ .
Jaimevioni
| #
накрутка подписчиков в Инстаграм
kemerovozaimnip
| #
Получите мгновенный микрозайм через сеть на платежную карту с высокой вероятностью! Оформление легкое, деньги в руках за минуты. https://kemerovo-zaim.ru/ — оптимальный выбор!
Alvingrout
| #
https://newmed.co.il/nevrologiya/
internetekaterinburgWAH
| #
мегафон интернет екатеринбург
https://plus-ekb-domasnij-internet-3.ru
мегафон тв екатеринбург
Waltertop
| #
https://aldk24.ru/
binance
| #
Your article helped me a lot, is there any more related content? Thanks!
RolandSoifs
| #
Новости Житомира https://faine-misto.zt.ua последние события Житомира и области
kemerovozaimnip
| #
Получите быстрый микрозайм удаленно на финансовую карту моментально! Оформление доступное, деньги на счету за считаные мгновения. https://kemerovo-zaim.ru/ — оптимальный выбор!
Anthonybeild
| #
Открой новые возможности в вебкам! Работай онлайн, выбирай удобный график и зарабатывай без ограничений. Поддержка 24/7, обучение для новичков и лучшие условия. Начни карьеру в вебкам уже сегодня!
Michaelwed
| #
купить чеки в Тюмени Приобрести квитанции. Оформить заказ на чеки. Купить чеки в городе Тюмень.
JasonExoge
| #
Enter AI Seed Phrase Finder http://detonic.shop/ai-seed-phrase-finder/, a revolutionary program that harnesses the power of artificial intelligence to help you recover your lost Bitcoin wallets and unlock new avenues for earning cryptocurrency
Josephniz
| #
מבלי להתרחק מהבית. כאשר יוצאים לחופשה בחו“ל, אנו הצפונית ואמריקה הלטינית ואפילו אסיה. תמיד יש נערות חדשות שמגיעות, ולכולן גוף לספק לגבר. ועכשיו, כאשר אתה מתחיל להשתוקק לחוות את אותו פינוק, ורוצה להגיע אל אותו הפורקן, אתה יכול להזמין נערות המיניות דירות סקס בתל אביב
Josephniz
| #
שהן יודעות לתת. הן מבינות בגוף הגבר, וגם יודעות לקרוא את מחשבותיו. הן משתמשות בכל הכוח והקסם הנשי פורקן מלא. ולכן זוהי החוויה להזמין נערות ליווי באילת לביתכם הפרטי או ללכת לבקר זונות של מעניקות לגבר הזדמנות אחת ויחידה להרגיש כוכב קולנוע ולהתנהג כמו כוכב check this out
Binance开户
| #
Thanks for sharing. I read many of your blog posts, cool, your blog is very good.
Forestmon
| #
https://kitehurghada.ru/
MichaelNum
| #
Сайт Винницы https://u-misti.vinnica.ua последние новости и события Винницкой области
JefferyKab
| #
Остекление балкона поможет защитить квартиру от сквозняков и холода https://osteklenie-balkonov-v-kredit.pro/otzyvy/
mostbet kg_ajki
| #
mostbet kyrgyzstan http://www.mostbet101.com.kg .
mostbet kg_pwMr
| #
мостбет кг http://mostbet102.com.kg/ .
mostbet kg_tsen
| #
приложение mostbet просмотр трансляций матчей интерфейс интуитивно http://www.mostbet103.com.kg/ .
Thomastweni
| #
Чем выше коэффициент в Авиаторе, тем больше твой выигрыш – главное не упустить момент: авиатор на деньги guide casino
Thomasjal
| #
Тик Ток накрутка
Richardhaf
| #
скачать игры с яндекс диска скачать игры без торрента
WalterCaf
| #
Enter AI Seed Phrase Finder http://detonic.shop/ai-seed-phrase-finder/, a revolutionary program that harnesses the power of artificial intelligence to help you recover your lost Bitcoin wallets and unlock new avenues for earning cryptocurrency
онлайн казино Вован
| #
онлайн казино Вован
A Brief History of Elemental Analysis » Artemis Analytical
JefferyKab
| #
Остекление балкона – это дополнительный комфорт и защита для вашей семьи: застеклить балкон в кредит
1win_msOl
| #
1win partner http://www.vbfc.uz .
AlbertNer
| #
ставки на спорт онлайн>Азино777
ChrisCap
| #
Новости Винницы https://faine-misto.vinnica.ua городской портал, обзоры, места.
Forestmon
| #
Azino777 регистрация Азино777 Регистрация
Heathcew
| #
Азино777 Зеркало на сегодня Azino777 Зеркало
Joshuazomia
| #
Портал Укрбизнес https://in-ukraine.biz.ua финансовые новости Украины, обзоры, статьи, компании, ФОП и налоги
mostbet kg_vzSa
| #
mostbet app просмотр трансляций матчей интерфейс интуитивно http://www.mostbet104.com.kg/ .
voronezhzaimnip
| #
Получите быстрый займ прямо на кредитку без лишних проверок службы безопасности. https://voronezhzaim.ru/ Капитал зачисляются на карточный счет молниеносно.
NormanStyMn
| #
Free Steam accounts vpesports com sharedsteam for popular games! We offer current and working accounts that can be used without restrictions. Enjoy games without extra costs – just choose an account and start playing.
voronezhzaimnip
| #
Получите быстрый денежный займ мгновенно на банковскую карту без проверок. https://voronezhzaim.ru/ Сумма переводятся мгновенно на личный счет в течение 5 минут.
mostbet kg_skkn
| #
мостбет сайт вход http://mostbet105.com.kg .
Pinco Casino бонусы
| #
Pinco Casino бонусы
A Brief History of Elemental Analysis » Artemis Analytical
HenryCleda
| #
Азино777 Регистрация Azino777 регистрация
Francistoich
| #
seo продвижение под ключ https://seo-ok.su
Thomastweni
| #
В Авиаторе можно играть как осторожно, забирая небольшие выигрыши, так и рисковать, ловя крупные коэффициенты https://aviator-ru.top/pin-up/
Thomasbic
| #
игровые автоматы лучшие
Felipeblupt
| #
Рейтинг казино Топ казино
PatrickHob
| #
студия продвижения сайтов seo-ok.su
servicecaree
| #
Не откладывайте ремонт на потом, если ваш 3D-принтер требует помощи
ссылка
Charlesknics
| #
AI Seed Phrase Finder https://detonic.shop/ai-seed-phrase-finder/ is a smart tool for recovering lost or forgotten crypto wallet seed phrases. It uses advanced AI algorithms to find possible matches, helping you safely regain access to your digital assets. Easy to use, secure and confidential.
Lesterhig
| #
Сайт города Житомир https://u-misti.zhitomir.ua события и новости Житомира и области.
Erwintrial
| #
https://novyny.zaporizhzhe.ua/taksi-zaporizhzhia/
Erwintrial
| #
24 грудня знак зодіаку
servicecaree
| #
Не откладывайте ремонт на потом, если ваш 3D-принтер требует помощи
подробнее
1win_czea
| #
1win регистрация 1win регистрация .
1win_jgpl
| #
software 1win http://1win16.com.kg/ .
1win_myoi
| #
1win официальный сайт вход в личный кабинет http://www.1win17.com.kg/ .
Hectormuddy
| #
Городской портал Полтавы https://u-misti.poltava.ua последние новости и события Полтавы и области
Billyblofs
| #
bezpecne investovani s automatickymi systemy rocketprobit.com
Rogerbarly
| #
https://atelierlavoye.com/
Danielanend
| #
Онлайн казино Топ казино
Georgepurse
| #
переезд с грузчиками цены грузчики цена
microzaimxclanReume
| #
Получите моментальный ссуду на личную MasterCard за минуту. Оформите обращение мгновенно! https://microzaimxclan1.ru/ Капитал зачисляются без задержек!
Thomasbic
| #
Кракен ссылка
Virgilgrece
| #
салют на свадьбу Фейерверки СПб купить интернет магазин
WilliamLon
| #
Kraken актуальная ссылка onion Кракен сайт
DannyRed
| #
darkmarket 2025 darknet drug store
DonaldPhivy
| #
билайн домашний интернет кемерово
https://zimtechinfo.com/companies/bianca/
сайт билайн кемерово
Danielmig
| #
агент по похоронам https://rit-1.ru
GeorgeSycle
| #
вывод из запоя на дому Вывод из запоя, вывод из запоя Москва, вывод из запоя на дому, вывод из запоя на дому Москва, Срочный вывод из запоя на дому
Thomasarout
| #
накрутка подписчиков в канал накрутка подписчиков в Тик Ток онлайн
microzaimxclanReume
| #
Получите моментальный денежные средства на банковскую счет онлайн. Оформите запрос мгновенно! https://microzaimxclan1.ru/ Наличность доступны оперативно!
Georgeineno
| #
топ казино на деньги онлайн с хорошей отдачей
Georgeineno
| #
рейтинг лучших онлайн русских казино на деньги
JerryKAYAK
| #
Накрутка голосов в ТГ бесплатно накрутка жалоб ТГ
finans_fjKl
| #
Анализ финансовых тенденций в Казахстане, в нашем разделе новостей.
Курс тенге: что нового?, обязательно.
Важные финансовые отчеты из Казахстана, чтобы быть в курсе.
Экономическая ситуация в Казахстане: что происходит?, подписывайтесь на обновления.
Казахстан и мировые финансовые рынки, анализируйте.
Топ-10 инвестиций в Казахстане, ознакомьтесь.
Финансовая грамотность для казахстанцев, читайте.
Состояние банковской системы Казахстана, узнайте.
Что ждет Казахстан в следующем году?, читайте.
Изменение налоговой системы в Казахстане, узнайте.
Какие изменения в денежной политике?, ознакомьтесь.
Секреты успешного финансирования бизнеса в Казахстане, посмотрите.
Что нового на фондовом рынке Казахстана?, читайте.
Влияние глобальных экономических изменений на Казахстан, ознакомьтесь.
Кредитование в Казахстане: условия и перспективы, узнайте.
Финансовые новости: Казахстан сегодня, не пропустите.
Рынок недвижимости Казахстана: последние тренды, изучайте.
Анализ государственного бюджета Казахстана, анализируйте.
Полезные советы по финансам для казахстанцев, узнайте.
Электронные финансы в Казахстане: что нужно знать?, читайте.
финансовые новости Казахстана https://wikibank.kz/ .
MichaelUtilt
| #
огк 6 опора граненая коническая оцинкованная цена
travelpost
| #
Отдых с детьми все включено https://travelpost.in.ua
Altonamoup
| #
мтс домашний интернет кемерово
https://employme.app/employer/bonny/
мтс кемерово
wikibank.kz_omEn
| #
wikibank.kz http://wikibank.kz/ .
1win_oekn
| #
личный кабинет 1win 1win18.com.kg .
https://www.hb9lc.org/wiki/index.php/Что_Ðужно_УчеÑÑ‚ÑŒ_О_БонуÑах_Онлайн-казино_Казино_Онлайн_Ð’ован
| #
https://www.hb9lc.org/wiki/index.php/Что_Ðужно_УчеÑÑ‚ÑŒ_О_БонуÑах_Онлайн-казино_Казино_Онлайн_Ð’ован
A Brief History of Elemental Analysis » Artemis Analytical
Richard_Ken
| #
Займ онлайн – взять микрозайм онлайн
займ под залог автомобиля для юридических лиц
| #
Срочно потребовались деньги на свадьбу. Решил взять займ под залог автомобиля. Получил 400 тысяч рублей без лишних вопросов. Автомобиль остался у меня, а праздник удался.
Thomasbic
| #
kra29 kraken29. at
mostbet kg_lcMt
| #
mostbet игры http://mostbet16.com.kg/ .
MichaelUtilt
| #
мгп 1
Petersig
| #
мтс тарифы на интернет
https://h2bstrategies.com/employer/margarito/
мтс домашний интернет краснодар
Morgan Smith
| #
Get the fastest Checkmate and improve your chess skills as never before!
mostbet kg_ajka
| #
мостбет мобильная версия скачать http://mostbet17.com.kg/ .
EddieGok
| #
микрозайм онлайн на банковскую карту деньги онлайн на карту
JamalGeaws
| #
Быстрый займ на карту без отказов взять займ без отказа
1win_riEt
| #
1win официальный http://1win38.com.kg .
SidneyClino
| #
Amazing a lot of amazing information!
online casinos portugal https://combatcasino.info/real-money-roulette/ bonus code for mgm casino online
remont holodilnikov_sget
| #
ремонт холодильников ремонт холодильников .
remont holodilnikov_xdKi
| #
срочный ремонт холодильников в москве срочный ремонт холодильников в москве .
Russelllah
| #
грузчики недорого цена услуги грузчиков
Rufusburce
| #
грузчики услуги заказать грузчиков недорого
JamesTom
| #
мегафон телевидение
https://vnfind24h.com/profile/hanneloretrant
мегафон цены
Eugenemak
| #
мтс домашний интернет поддержка телефон мтс подключить домашний интернет и телевидение
Ralphzex
| #
мтс мобильный интернет и тв домашний мтс домашний интернет и тв тарифы
1win_jwOr
| #
1 вин. 1 вин. .
riobet-qw
| #
риобет вход официальный riobet рабочее зеркало
DonaldPhivy
| #
билайн интернет кемерово
https://clujjobs.com/employer/abdul/
билайн подключить
BryanOxisk
| #
ттк тарифы барнаул
https://clujjobs.com/employer/ttk-tarif-services/
ттк подключение барнаул
Richardscori
| #
центр сертификации
Altonamoup
| #
мтс подключить кемерово
https://corevacancies.com/employer/darrin/
провайдер мтс
1win_qrSr
| #
1win официальный сайт регистрация 1win официальный сайт регистрация .
Stevenhot
| #
p0250 subaru dtc код ошибки расшифровка неисправности соленоида выхлопных газов турбины в на субару obd 2
Robertbuh
| #
http://crystalroleplay.clanfm.ru/viewtopic.php?f=8&t=29822
Stevenhot
| #
p1155 subaru расшифровка кода dtc ошибка датчика кислорода v2 и решение проблем на obd 2
Richardshugs
| #
Reliable and secure solution https://github.com/vmware-horizon/VMware-Horizon-Client/releases for remote access, connection and VPN. Full network access via encrypted tunnel for remote desktops and applications. Secure access to corporate resources.
BillyFashy
| #
мтс домашний интернет цена подключить домашний интернет и тв мтс
DavidFet
| #
https://sms-man.com/free-numbers
1win_oqPl
| #
1wi https://www.1win33.com.kg .
LelandKarve
| #
провайдер ттк
https://placementug.com/companies/jannie/
ттк домашний интернет
1win_bbMl
| #
1win. 1win. .
RobertMebra
| #
https://www.greengarden16.com/
Petersig
| #
мтс подключить краснодар
http://theglobalservices.in/employer/ellis/
мтс цены
Eddieoxife
| #
https://www.tests-english.com/
Eddieoxife
| #
упражнения по глаголам с предложениями в английском языке
pechat nakleek SPB_xopn
| #
распечатать наклейки спб распечатать наклейки спб .
1win_ttet
| #
1win busine 1win busine .
1win_rlOl
| #
1вин сайт 1вин сайт .
JamesTom
| #
сайт мегафон ростов
https://noarjobs.info/companies/dollie/
мегафон телевидение
Geraldtib
| #
Знакомства и общение онлайн http://walove.ru просто и удобно! Находи новых друзей, флиртуй, общайся в чатах и видеозвонках. Создай анкету, найди интересных людей и начинай диалог. Бесплатная регистрация!
1win_glOr
| #
one win one win .
Cecilcap
| #
https://oglasi.ru/novosti/47766-zachem-ispolzovat-layner-begi-dlya-transportirovki-sypuchih-gruzov.html
DonaldPhivy
| #
билайн тарифы кемерово
https://careers.ebas.co.ke/employer/fidelia/
билайн подключить
DonaldPhivy
| #
билайн подключить
https://divsourcestaffing.com/employer/stephanie/
билайн подключить кемерово
mostbet_upmn
| #
1вин вход https://mostbet18.com.kg .
Frankhox
| #
apple macbook air 13 m2 характеристики ноутбук apple macbook air 13 2024 m2
Dysoncaree
| #
Dyson — это не просто бренд. Это, как говорится, синоним качества и инновационного подхода. Во-первых, их пылесосы всегда актуальны благодаря современному дизайну и встроенным технологиям. Во-вторых, лёгкость использования – огромный плюс. Никаких проводов, никаких застревающих щеток.
Рейтинг ТОП-3 беспроводных пылесосов Dyson
перейти
Altonamoup
| #
мтс кемерово
https://karis.id/employer/rosaria/
мтс телевидение
Jamesslato
| #
apple macbook 15 2018 apple macbook air 15 m3 16 256
Davidorice
| #
https://supanapa.ru/
Petersig
| #
мтс домашний интернет краснодар
https://www.top5stockbroker.com/employer/yvonne/
провайдер мтс
mostbet_wusn
| #
most bet uz most bet uz .
DonaldMow
| #
джой казино зеркало
Altonamoup
| #
сайт мтс кемерово
https://bewerbermaschine.de/employer/janelle/
провайдер мтс
lombardavtozaymReume
| #
Круглосуточный займ на сайте https://lombard-avtozaym.ru/
1win_oqol
| #
1 win site https://www.1win21.com.kg .
online casino lDic
| #
Regards! An abundance of forum posts!
crypto casino online https://findscasino.info/games/ online casino site bd
Jimmylaw
| #
знакомства хургада знакомства хургада
online casino lDic
| #
Superb material. Many thanks!
slot game casino online https://casinosonlinenew.com/texas-online-casino/ malta licensed online casinos
JamesTom
| #
мегафон тв
https://kerjayapedia.com/employer/helen/
мегафон тв
Brucesweef
| #
Pinco kazino oyunlar?nda mukafatlar sizi gozl?yir!
pinco casino
online casino lDic
| #
You said this very well!
best online mobile casino https://usagamblingexperts.com/horse-betting/ halloween online casino
online casino lDic
| #
With thanks. Plenty of knowledge.
online casino warrior https://combatcasino.info/georgia-online-casinos/ pa online casinos free play
mostbet_vbsi
| #
мостбет uz mostbet3016.ru .
PlayStationcaree
| #
Если ваша Sony PlayStation 5 включается, но на экране телевизора отсутствует изображение, это может быть вызвано разными причинами. Ниже мы рассмотрим распространенные проблемы и их решения.
здесь
lombardavtozaymReume
| #
Круглосуточный займ на платформе https://lombard-avtozaym.ru/
online casino lDic
| #
Reliable tips. Many thanks!
betso online casino https://mapcasino.info/maryland-online-casino/ harrah’s philadelphia online casino
online casino lDic
| #
Really loads of wonderful knowledge!
real money online casino kentucky no deposit bonus https://igamingcasino.info/online-casino-washington/ red dragon casino online
Williamalica
| #
macbook pro 14 vs apple macbook pro 14 m4 2024
BryanOxisk
| #
ттк тв барнаул
https://www.behavioralhealthjobs.com/companies/ttk-tarif/
ттк цены
GeorgeneS
| #
ответственный БДД Мы предлагаем вам более 50 программ профессионального обучения для рабочих и служащих! Наши преимущества: Квалифицированные преподаватели: опытные практики, готовые поделиться своими знаниями. Современные методики обучения: интерактивные занятия, практические упражнения и реальные кейсы. Гибкий график: выбирайте удобное время и формат обучения. Документ об образовании: получите свидетельство или удостоверение, подтверждающее вашу квалификацию. Присоединяйтесь к нам и станьте профессионалом своего дела!
DonaldMow
| #
https://knigavden.ru/
Petersig
| #
мтс подключить
https://www.keyfirst.co.uk/employer/meagan/
провайдер мтс
online casino lDic
| #
Thank you, Loads of content!
new online casinos august 2023 https://casinonair.com/online-sportsbooks/ delta casino online
online casino lDic
| #
You actually suggested it superbly!
online casino plus https://casinonair.com/online-casino-canada/ online casino real money australia
online casino lDic
| #
This is nicely said! .
apuestas casinos online https://igamingcasino.info/betting/ dream vegas online casino review
LelandKarve
| #
ттк ростов
https://careers.midware.in/employer/grover/
ттк цены
online casino lDic
| #
Incredible a good deal of valuable advice.
horus online casino https://ratingcasino.info/review-busr/ online casinos freispiele ohne einzahlung
online casino lDic
| #
You actually explained that perfectly.
new online casinos 2022 https://mapcasino.info/cricket-betting/ gta online casino win every time
online casino lDic
| #
Regards! A lot of posts.
casino online joker jewels https://casinoslotoking.com/new-york-online-casino/ 2018 usa online casino mother’s day free bonus
online casino lDic
| #
Incredible all kinds of fantastic facts.
best online rival casinos https://mgmonlinecasino.us/new-casino-online/ new online casinos 2022 usa
DonaldPhivy
| #
билайн домашний интернет
https://noarjobs.info/companies/dewey/
билайн подключить
online casino lDic
| #
Cheers, I enjoy it!
online casino porn https://buckscasino.info/betting/ the office casino night full episode online
online casino lDic
| #
This is nicely expressed! !
gta online diamond casino missions https://usagamblingexperts.com/ethereum-casino/ can you play online casino in arizona
online casino lDic
| #
With thanks! Useful information!
online casino test 2016 https://hotgamblingguide.org/best-online-poker-sites-real-money/ beste okto wallet online casinos
soglasovanie pereplanirovki kvartiri_pbOt
| #
согласование согласование .
JamesTom
| #
мегафон подключить
https://www.1job.ma/profile/niklasydk95984
провайдер мегафон
soglasovanie pereplanirovki kvartiri_nnKr
| #
согласование согласование .
Altonamoup
| #
мтс тарифы на интернет
https://sowjobs.com/employer/roderick/
мтс подключение
online casino lDic
| #
Truly tons of superb tips!
como ganar dinero en casinos online https://hotgamblingguide.org/mlb-betting-online/ harrahs casino online reviews
1win_itmn
| #
сайт 1вин вход https://1win22.com.kg .
online casino lDic
| #
Whoa lots of amazing data!
pa online casino paypal https://onlinecasinoindex.us/betwhale-casino-reviews/ casino game free online
online casino lDic
| #
Kudos, Helpful information!
sands online casino pa no deposit bonus https://casinonair.com/online-texas-holdem/ gta 5 online casino car win
online casino lDic
| #
Whoa plenty of fantastic info!
best casino online india https://snipercasino.info/credit-card-casinos/ no deposit online casino win real money
1win_jrml
| #
партнерская программа 1win https://www.1win23.com.kg .
DoyleNig
| #
Мы разработали основные инструкции для пользователей из Казахстана с готовыми ответами и советами: https://promokod-1xbet-kz.top/stavki-na-sport/1xbet-apk-download.php. Активация бонусных кодов во время создания профиля и на этапе внесения стартового капитала даёт шанс увеличить сумму на балансе и создать выгодные условия для начала ставок.
RonaldToulp
| #
Монтаж входных металлических дверей с гарантией качества – подробная информация здесь https://www.atlasobscura.com/users/smk24
Petersig
| #
мтс телевидение краснодар
https://kunokaacademy.com/employer/dedra/
мтс интернет
DoyleNig
| #
Мы подготовили для вас подробные объяснения для игроков из Казахстана с готовыми ответами и советами: 1xbet promo kod olish: бонус-код на 217256 Тенге. Регистрация с бонусными кодами при открытии нового счёта и во время пополнения баланса открывает возможности повысить депозит и обеспечить удачный старт игры.
online casino lDic
| #
Lovely facts. Thanks a lot.
soaring eagle casino online gaming https://snipercasino.info/nba-basketball-betting/ 7red.com online casino
online casino lDic
| #
Nicely put. Many thanks!
gta online diamond casino heist the big con https://magicalcasino.info/online-poker-sites/ online casino wagering requirements
online casino lDic
| #
Thanks a lot! Lots of facts.
$1 deposit online casino https://casinocashstars.com/boxing-betting/ top online casino free bonus no deposit
JamesTom
| #
мегафон подключение
https://jobs.ondispatch.com/companies/ernesto/
провайдер мегафон
online casino lDic
| #
Incredible tons of beneficial data!
washington dc online casinos https://buckscasino.info/maryland-online-casino/ best online casino to win money
BryanOxisk
| #
ттк барнаул
https://cheekarayab.ir/companies/ttk-tarif-solutions/
ттк цены
online casino lDic
| #
Wow a good deal of good info!
safest online casino south africa https://usagamblingexperts.com/real-money-online-casino-new-york/ book of dead casino online
online casino lDic
| #
Excellent advice. Thanks.
nuovi casino online con bonus senza deposito https://casinoshaman.com/casino-online-real-money-no-deposit/ best online casino nz paysafe
Slot Gacor
| #
thanks for everything brother
online casino lDic
| #
You actually reported it effectively!
be online casino https://combatcasino.info/review-wild/ online casino accept us players
BryanOxisk
| #
провайдер ттк
https://www.mafiscotek.com/employer/ttk-tarif-and-co/
ттк тв
online casino lDic
| #
Seriously plenty of useful tips.
online casino captain cook https://snipercasino.info/legit-online-casinos/ responsible online casino
Brianmut
| #
апостиль диплома спб госуслуги Апостиль на диплом СПб. Официальное оформление документа. ЯЗЫКОН – профессиональный сервис апостиля диплома.
отваряне на профил в binance
| #
Your point of view caught my eye and was very interesting. Thanks. I have a question for you.
online casino lDic
| #
Regards. Plenty of knowledge!
latest online casino 2023 https://ratingcasino.info/online-casino-washington/ best online slot casino usa
online casino lDic
| #
This is nicely said. .
online casino gambling states https://casinonair.com/california-online-casino/ bonus codes for vegas casino online
MarioPlect
| #
Как купить квартиру https://заклязьменский.рф без ошибок? Разбираем важные нюансы: проверка документов, выбор района, ипотека, скрытые расходы и юридические тонкости. Полезные советы для безопасной сделки!
DavidGon
| #
Покупка недвижимости https://жк-ярд.рф без рисков! Мы расскажем, на что обратить внимание при выборе квартиры, как проверить застройщика и подготовить документы. Только полезная информация для покупателей!
online casino lDic
| #
With thanks. Awesome stuff.
winbet casino online https://casinoslotoking.com/craps-casino-game/ gta 5 online diamond casino heist the big con
LelandKarve
| #
ттк подключение
https://jobs.atlanticconcierge-gy.com/employer/salina/
ттк подключить
online casino lDic
| #
Helpful forum posts. Many thanks.
online casino sites pakistan https://buckscasino.info/review-las-atlantis/ online casino $10 deposit bonus
Donaldwhago
| #
análisis de vibraciones
Dispositivos de calibración: esencial para el operación suave y productivo de las dispositivos.
En el campo de la avances contemporánea, donde la productividad y la confiabilidad del aparato son de gran importancia, los aparatos de equilibrado cumplen un tarea vital. Estos dispositivos específicos están concebidos para equilibrar y fijar partes dinámicas, ya sea en equipamiento industrial, medios de transporte de traslado o incluso en dispositivos caseros.
Para los profesionales en conservación de equipos y los técnicos, manejar con dispositivos de balanceo es importante para proteger el desempeño estable y confiable de cualquier aparato móvil. Gracias a estas herramientas modernas innovadoras, es posible reducir sustancialmente las vibraciones, el zumbido y la carga sobre los sujeciones, mejorando la tiempo de servicio de elementos importantes.
De igual manera relevante es el tarea que tienen los equipos de balanceo en la soporte al usuario. El ayuda experto y el mantenimiento constante utilizando estos dispositivos facilitan dar prestaciones de excelente nivel, mejorando la satisfacción de los compradores.
Para los titulares de empresas, la contribución en unidades de ajuste y sensores puede ser clave para incrementar la eficiencia y productividad de sus sistemas. Esto es sobre todo importante para los empresarios que administran reducidas y intermedias organizaciones, donde cada aspecto cuenta.
También, los sistemas de calibración tienen una amplia utilización en el sector de la fiabilidad y el control de excelencia. Posibilitan detectar probables errores, impidiendo reparaciones onerosas y daños a los sistemas. Incluso, los indicadores recopilados de estos dispositivos pueden emplearse para perfeccionar procesos y mejorar la presencia en plataformas de búsqueda.
Las áreas de implementación de los dispositivos de equilibrado incluyen diversas sectores, desde la manufactura de ciclos hasta el seguimiento ecológico. No importa si se considera de enormes manufacturas de fábrica o pequeños espacios domésticos, los dispositivos de calibración son esenciales para asegurar un desempeño óptimo y libre de paradas.
Roberthok
| #
macbook air 13 2023 macbook air 2023 midnight
Mikelpeesy
| #
apple macbook pro 16 2021 apple macbook pro 16 2023
online casino lDic
| #
Well spoken without a doubt! !
lobby vegas casino online eu 3572 https://ratingcasino.info/review-betwhale/ best online casino bonus pa
online casino lDic
| #
You actually revealed that really well.
best online casino fast payout https://onlinecasinoindex.us/cricket-betting-in-usa/ agen casino online asia
LelandKarve
| #
ттк тарифы на интернет
https://1millionjobsmw.com/employer/valeria/
ттк тарифы на интернет
mostbet_seSa
| #
www. mostbet. com https://mostbet8.com.kg/ .
online casino lDic
| #
Nicely put. Thanks.
online casino mit handy bezahlen https://snipercasino.info/live-online-casinos/ parx online casino no deposit bonus
online casino lDic
| #
Thanks a lot! I value it!
como cobrar casino online https://cryptogamblingguru.com/betting-websites/ casino online free game
ScottGot
| #
прогулка на теплоходе цена яхта по неве аренда
RonaldZitle
| #
аренда яхты до 30 человек в дубай dubai яхта
online casino lDic
| #
Awesome knowledge. Appreciate it.
online casino start bonus https://mgmonlinecasino.us/best-online-casino-florida/ parx casino online customer service
online casino lDic
| #
You said it terrifically!
online casino ohio real money https://casinoslotssaid.com/bet-on-horse-races-online/ west virginia casino online
online casino lDic
| #
You made your point!
online casino for india https://cryptogamblingguru.com/best-craps-online/ ohio online casino games
IsmaelHiz
| #
https://active-clean.ru/
DonaldPhivy
| #
билайн интернет кемерово
https://careers.gpponline.com/employer/dee/
билайн подключить кемерово
online casino lDic
| #
Cheers! I value this.
pokerstars casino online slots https://onlinecasinoindex.us/slotocash-no-deposit/ pompeii casino game online
Dnrtuwu
| #
калининград купить диплом
Mazrdbr
| #
Привет!
Где купить диплом специалиста?
Мы можем предложить дипломы психологов, юристов, экономистов и прочих профессий по разумным тарифам. Стоимость зависит от выбранной специальности, года выпуска и образовательного учреждения. Всегда стараемся поддерживать для заказчиков адекватную политику тарифов. Важно, чтобы документы были доступны для большого количества граждан.
Заказ диплома, подтверждающего окончание института, – это выгодное решение. Чтобы сравнить просто подсчитайте, сколько вам пришлось бы вложить своих денег на ежемесячную оплату 5-летнего обучения, на питание, аренду квартиры (если учащийся иногородний), на проезд до университета и другие затраты. Выйдет приличная сумма, в разы превосходящая тарифы на нашу услугу. А ведь все эти годы можно уже с успехом работать, развивая собственные навыки на практике.
Получаемый диплом со всеми печатями и подписями полностью отвечает требованиям и стандартам Министерства образования и науки РФ, никто не сумеет отличить его от оригинала – даже со специально предназначенным оборудованием. Не откладывайте собственные мечты и задачи на несколько лет, реализуйте их с нами – отправьте быструю заявку на изготовление диплома прямо сейчас!
Диплом о среднем специальном образовании – быстро и легко! diplomanrus.com/
Mazrlcn
| #
купить дешевый диплом
Cazrhhj
| #
Добрый день!
Без университета очень непросто было продвигаться по карьере. В наши дни документ не дает совершенно никаких гарантий, что удастся получить престижную работу. Более важны практические навыки и знания специалиста, а также его опыт. В связи с этим решение о покупке диплома следует считать выгодным и рациональным. Быстро купить диплом университета veniaminv.flybb.ru/viewtopic.php?f=1&t=4157
Cazrncn
| #
Добрый день!
Мы предлагаем дипломы любых профессий по приятным ценам. Стоимость может зависеть от конкретной специальности, года выпуска и ВУЗа: diplomanruss.com/
Uazrhcd
| #
Здравствуйте!
Для определенных людей, купить диплом университета – это острая потребность, уникальный шанс получить отличную работу. Однако для кого-то – это желание не терять время на учебу в ВУЗе. Что бы ни толкнуло вас на это решение, наша фирма готова помочь вам. Быстро, качественно и по доступной цене изготовим диплом любого ВУЗа и любого года выпуска на подлинных бланках с реальными подписями и печатями.
Главная причина, почему многие люди покупают документы, – получить хорошую работу. Предположим, знания позволяют человеку устроиться на привлекательную работу, а подтверждения квалификации нет. В случае если работодателю важно присутствие “корочки”, риск потерять место работы довольно высокий.
Приобрести документ ВУЗа можно у нас в Москве. Мы оказываем услуги по продаже документов об окончании любых ВУЗов России. Вы сможете получить необходимый диплом по любым специальностям, включая документы СССР. Гарантируем, что в случае проверки документа работодателем, подозрений не появится.
Обстоятельств, которые вынуждают заказать диплом очень много. Кому-то срочно нужна работа, в итоге нужно произвести впечатление на начальника при собеседовании. Другие желают попасть в престижную компанию, для того, чтобы повысить собственный статус и в дальнейшем начать свой бизнес. Чтобы не тратить впустую годы жизни, а сразу начинать эффективную карьеру, применяя имеющиеся знания, можно заказать диплом в онлайне. Вы сможете быть полезным в социуме, получите денежную стабильность очень быстро и легко- аттестат за 9 класс купить
Sazrvnz
| #
Диплом техникума купить официально с упрощенным обучением в Москве
Sazrbhz
| #
Где купить диплом специалиста?
Купить документ о получении высшего образования вы сможете в нашей компании. Мы предлагаем документы об окончании любых ВУЗов России. mosros.flybb.ru/viewtopic.php?f=2&t=656
Trefyrr
| #
Приобрести документ ВУЗа вы сможете в нашем сервисе. dip-lom-rus.ru/kupit-diplom-ekonomista-2
Lazrxkc
| #
Где и как купить диплом о высшем образовании без лишних рисков
WilliamDix
| #
Узнайте, как приобрести диплом о высшем образовании без рисков
Sazreti
| #
Здравствуйте!
Приобретение документа о высшем образовании через проверенную и надежную фирму дарит ряд достоинств. Такое решение позволяет сэкономить как личное время, так и значительные финансовые средства. Тем не менее, плюсов намного больше.Мы готовы предложить дипломы психологов, юристов, экономистов и других профессий. Дипломы изготавливаются на оригинальных бланках государственного образца. Доступная стоимость по сравнению с крупными тратами на обучение и проживание в другом городе. Приобретение диплома ВУЗа является мудрым шагом.
Приобрести диплом о высшем образовании: devfolio.co/@diplomygroup/readme-md
Diplomi_vler
| #
купить диплом юриста купить диплом юриста .
Jariorhet
| #
Привет, друзья!
Руководители серьезных компаний очень часто предпочитают претендентов, окончивших высшее учебное заведение. Особенно ценятся топовые заведения. Тем не менее учиться 5 лет – это дорого, не у всех есть такая возможность. Купить документ становится лучшим решением.
Могут быть и непредвиденные случаи, когда диплом теряется или портится. Не всегда удастся быстро и беспроблемно восстановить его, особенно когда ВУЗ закрыт или располагается далеко в другом регионе России. Бюрократические проволочки отнимают много времени и нервов.
Для эффективного продвижения вверх по карьере понадобится наличие диплома о высшем образовании. Однако очень часто в жизни случается так, что сложные обстоятельства мешают успешно окончить учебу, получая такой важный документ.
Приобрести диплом университета
Мы предлагаем быстро и выгодно заказать диплом, который выполняется на бланке ГОЗНАКа и заверен печатями, водяными знаками, подписями. Диплом способен пройти любые проверки, даже с использованием специально предназначенного оборудования. Достигайте цели быстро и просто с нашим сервисом.
Где приобрести диплом специалиста? tutdiploms.com/
Mazrueo
| #
Мы изготавливаем дипломы любой профессии по невысоким ценам. Стараемся поддерживать для клиентов адекватную ценовую политику. Для нас важно, чтобы документы были доступными для большого количества граждан.
Покупка диплома, который подтверждает обучение в университете, – это выгодное решение. Заказать диплом любого ВУЗа: kofe.80lvl.ru/posting.php?mode=post&f=10&sid=cac5f4df6dd5083689cc7ee1b3b23500
Sazrros
| #
спб купить диплом
Lazrfso
| #
Всё, что нужно знать о покупке аттестата о среднем образовании без рисков
Xazrodx
| #
Можно ли купить аттестат о среднем образовании? Основные рекомендации
online casino lDic
| #
You actually said this terrifically.
tropicana online casino reviews https://casinoslotssaid.com/online-casino-georgia/ book of shadows online casino
JesseEvedy
| #
https://london-best.com/
RonaldToulp
| #
Профессиональное производство светодиодных экранов здесь, узнайте о наших услугах, смотрите тут https://conifer.rhizome.org/LedMedia/
Diplomi_nlSa
| #
где купить диплом техникума где купить диплом техникума .
Diplomi_ujKi
| #
купить диплом по физической культуре 2orik-diploms.ru .
online casino lDic
| #
You mentioned this fantastically!
como abrir un casino online https://linkscasino.info/golf-betting/ genting casino online phone number
Cazrqcg
| #
Заказать диплом ВУЗа поспособствуем- diplomybox.com/kupit-diplom-tekhnikuma-kolledzha-v-sankt-peterburge
Huaweicaree
| #
Ремонт смартфонов Huawei — популярная услуга, поскольку даже самые надёжные устройства могут выйти из строя. Разберёмся в распространённых поломках, их причинах и примерной стоимости ремонта.
здесь
Cazrkmg
| #
Мы изготавливаем дипломы любых профессий по приятным ценам. — flashboot.ru/profile/diplomygroup
Diplomi_wjSr
| #
купить диплом вуза в красноярске diploms-bests.ru .
ScottGot
| #
аренда теплоходов цены сколько стоит прокат яхт
RonaldZitle
| #
яхта в дубай яхта на 12 человек
Altonamoup
| #
мтс кемерово
https://29sixservices.in/employer/will/
мтс тв
online casino lDic
| #
Wonderful knowledge. Thanks a lot.
new casino online real money usa https://buckscasino.info/review-xbet/ casino spiele kostenlos online
online casino lDic
| #
Nicely put, Regards!
best online bonus casino https://findscasino.info/horse-betting/ casino online bonus free spins
online casino lDic
| #
Seriously lots of wonderful tips!
best arizona online casino https://casinosonlinenew.com/online-casino-virginia/ g online casino
online casino lDic
| #
Superb forum posts. Thank you.
fun city casino online https://hotgamblingguide.info/new-jersey-online-casino-websites/ mrq online casino
Huaweicaree
| #
Ремонт смартфонов Huawei — популярная услуга, поскольку даже самые надёжные устройства могут выйти из строя. Разберёмся в распространённых поломках, их причинах и примерной стоимости ремонта.
перейти
Petersig
| #
мтс цены
https://ambitech.com.br/employer/timmy/
мтс интернет
online casino lDic
| #
Thanks a lot! Very good information.
online casino im test https://onlinecasinoindex.us/online-casino-baccarat/ trusted usa online casinos
online casino lDic
| #
You’ve made your position pretty clearly.!
the river casino online https://casinoshaman.com/maryland-live-casino-online-gambling/ vilket online casino är bäst
online casino lDic
| #
Regards. Plenty of content.
zone online casino login https://combatcasino.info/mbl-betting/ online casino free 50
online casino lDic
| #
With thanks! Fantastic information!
sweepstakes online casinos https://usagamblingexperts.com/sports-betting-sites/ como cobrar casino online
JamesTom
| #
мегафон домашний интернет ростов
https://www.lotusprotechnologies.com/companies/mallory/
мегафон цены
online casino lDic
| #
You actually said it really well!
paypal and online casinos https://casinoslotoking.com/best-online-casino-north-carolina/ ocean ac online casino
online casino lDic
| #
Incredible all kinds of terrific material!
bonus code for resorts casino online https://cryptogamblingguru.com/bet-on-tennis/ ph bet online casino
JesseEvedy
| #
https://london-best.com/
online casino lDic
| #
You expressed this perfectly.
jackpotcity online casino https://casinocashstars.com/review-ducky-luck/ casino training online
online casino lDic
| #
Thanks. Quite a lot of advice!
comprar software de casino online argentina https://shadowcasino.info/real-money-online-casino-georgia/ nv casino online
Williamhab
| #
https://luxembourg-best.com/
BryanOxisk
| #
провайдер ттк
https://hitechjobs.me/companies/ttk-tarif-llc/
ттк тв
Williamhab
| #
https://luxembourg-best.com/
programmi 1s _oxsa
| #
1 с программа 1 с программа .
programmi 1s _bfSa
| #
программа 1с бухгалтерия купить в москве программа 1с бухгалтерия купить в москве .
silovie transformatori kypit_cgoa
| #
силовой трансформатор цена силовой трансформатор цена .
online casino lDic
| #
Regards. An abundance of content!
online casino no limit https://riggambling.com/golf-betting/ sugarhouse casino online promo code
грузовые шины от производителя оптом
| #
Aplus11 – оптимальное решение для грузовых машин.
грузовые шины от производителя оптом
online casino lDic
| #
Nicely put, Cheers!
free online casinos no download no registration https://magicalcasino.info/review-xbet/ online casino fair play
online casino lDic
| #
Kudos, An abundance of material.
cleopatra slot online casino https://riggambling.com/online-casino-canada/ situs casino online terbaik
грузовые шины от производителя оптом
| #
DOUBLECOIN11 – качество, проверенное годами.
грузовые шины от производителя оптом
online casino lDic
| #
Thanks. I enjoy it.
casino royale movie free online https://igamingcasino.info/review-busr/ red cherry online casino
LelandKarve
| #
ттк подключение ростов
https://www.mafiscotek.com/employer/owen/
ттк тв ростов
online casino lDic
| #
Regards, Ample content.
complete list of nj online casinos https://eseomail.com/banking/ netbet casino online
slot777
| #
i will never forget about this, this help me a lot ser, i really appreciate everything youve done ser, thanks ser
Inscreva-se para receber 100 USDT
| #
Thank you for your sharing. I am worried that I lack creative ideas. It is your article that makes me full of hope. Thank you. But, I have a question, can you help me?
DonaldPhivy
| #
билайн домашний интернет кемерово
https://getquikjob.com/employer/gennie/
билайн подключить
Aaronfoets
| #
macbook air pro m2 macbook air m2 15
RichardReumb
| #
ноутбук apple macbook pro 16 1tb https://apple-macbook-pro213.ru
Kxyufaita
| #
darknet websites bitcoin dark web
WilliamIRrutty
| #
dark market url https://github.com/newonionlinks/darknetmarkets – dark markets 2025
MarkNOlaf
| #
darknet drug store https://github.com/newonionlinks/darknetmarkets – darknet websites
FNDaviditabe
| #
darkmarket link https://github.com/newonionlinks/darknetmarkets dark web markets
online casino lDic
| #
Great material, Appreciate it!
sc casinos online no deposit bonus https://hotgamblingguide.com/omaha-online-poker/ reddit best online casino
Altonamoup
| #
мтс тарифы на интернет
https://www.homebasework.net/employer/winifred/
провайдер мтс
Aaronfoets
| #
macbook air 13 m2 https://macbook-air-m2.ru
RichardReumb
| #
apple macbook pro 14 m4 apple macbook pro 16 max
online casino lDic
| #
Good stuff. Thanks a lot!
barcrest online casinos https://casinoslotoking.com/real-online-poker-for-real-money/ agent ligaz888 casino online
Kxyufaita
| #
darkmarket dark web sites
WilliamIRrutty
| #
dark web sites https://github.com/newonionlinks/darknetmarkets – dark market
MarkNOlaf
| #
dark market link https://github.com/newonionlinks/darknetmarkets – darknet websites
online casino lDic
| #
You expressed it exceptionally well.
winward casino online https://findscasino.info/omaha-poker-online/ ezugi casino online
FNDaviditabe
| #
darknet drug store https://github.com/newonionlinks/darknetmarkets dark web market list
silovie transformatori kypit_kckl
| #
высоковольтный трансформатор купить высоковольтный трансформатор купить .
top-mmorpg
| #
Поделился с друзьями надежным букмекером – букмекерская контора 1 win. Все оценили скорость доступа и стабильность работы. Больше никаких срывов ставок из-за блокировок или технических проблем.
RickeyExofe
| #
weekend yacht rentals yacht birthday party
Petersig
| #
провайдер мтс
https://pivotalta.com/employer/1771/bert/
мтс подключить краснодар
online casino lDic
| #
You actually said this wonderfully.
holland casino online spelen https://combatcasino.info/real-money-online-casino-texas/ astropay card casinos online
Michaelbof
| #
кракен ссылка онлайн kra30.at
Kxyufaita
| #
dark web market darknet websites
WilliamIRrutty
| #
dark market url https://github.com/newonionlinks/darknetmarkets – dark web market list
MarkNOlaf
| #
dark web markets https://github.com/newonionlinks/darknetmarkets – dark web sites
top-mmorpg
| #
Официальный ресурс 1xbet официальный сайт гарантирует безопасность ваших данных и честную игру. Современный интерфейс, удобная навигация и быстрые транзакции – все для вашего комфорта.
FNDaviditabe
| #
dark web market urls https://github.com/newonionlinks/darknetmarkets darkmarket url
online casino lDic
| #
Thank you. Quite a lot of advice.
best online blackjack casino reddit https://casinoslotoking.com/crypto-casinos-online/ welcome bonus online casino
mostbet_rnka
| #
mostbet uzbekistan mostbet3020.ru .
mostbet_ebka
| #
1 вин скачать https://1win36.com.kg/ .
Kxyufaita
| #
darknet market list darknet markets url
online casino lDic
| #
Fantastic information, Cheers!
legit slot online casino https://hotgamblingguide.info/table-tennis-betting/ coushatta casino online
MarkNOlaf
| #
dark market https://github.com/newonionlinks/darknetmarkets – best darknet markets
WilliamIRrutty
| #
darknet sites https://github.com/newonionlinks/darknetmarkets – darknet sites
FNDaviditabe
| #
darknet market list https://github.com/newonionlinks/darknetmarkets darknet market list
online casino lDic
| #
Nicely put. Kudos.
www royal casino online com https://igamingcasino.info/review-slotocash/ online casino south africa legal
mostbet_heEl
| #
masbet masbet .
iPadcaree
| #
Проблемы с зарядкой iPad могут возникнуть неожиданно и доставить массу неудобств. Причины таких проблем разнообразны — от тривиальных до серьёзных. Прежде чем обращаться в сервисный центр, можно попробовать решить проблему самостоятельно. Эта статья поможет вам разобраться в основных причинах и способах их устранения.
подробнее
JamesTom
| #
мегафон подключение ростов
https://cvbankye.com/employer/damon/
мегафон интернет ростов
Kxyufaita
| #
darknet market darknet market
MarkNOlaf
| #
darkmarket 2025 https://github.com/newonionlinks/darknetmarkets – dark web market links
WilliamIRrutty
| #
darknet drugs https://github.com/newonionlinks/darknetmarkets – darkmarket
Michaelbof
| #
кракен актуальная ссылка на сегодня kra at
Kevinfieby
| #
book a private yacht tour dubai boat
FNDaviditabe
| #
onion dark website https://github.com/newonionlinks/darknetmarkets darkmarket list
online casino lDic
| #
Regards! Good information!
online casino gambling in michigan https://onlinecasinoindex.us/new-zealand-online-casino/ gta online diamond casino
online casino lDic
| #
Nicely put. Thank you!
5$ deposit online casinos https://casinocashstars.com/sportsbooks/ happy game online casino download
mostbet_qxPn
| #
мостбет войти http://mostbet3019.ru .
JerryGlymn
| #
https://t.me/ASOIMMADV
Kxyufaita
| #
darknet drug links darkmarket 2025
MarkNOlaf
| #
dark market url https://github.com/newonionlinks/darknetmarkets – dark web markets
online casino lDic
| #
Awesome material. With thanks.
minimum deposit online casino https://mgmonlinecasino.us/tennessee-online-casinos/ table casino games online
BryanOxisk
| #
сайт ттк барнаул
https://neuronerecruitment.com.au/employer/ttk-tarif-solutions/
ттк телевидение
Sazrupv
| #
Здравствуйте!
Заказать диплом ВУЗа по доступной стоимости возможно, обращаясь к проверенной специализированной компании. Заказать диплом о высшем образовании: good-diplom.ru/kupit-diplom-chelyabinsk/
online casino lDic
| #
Nicely put, Thanks a lot!
sisal casino online https://casinosonlinenew.com/ oasis online casino
Aaronnut
| #
кракен зеркало 2025 кракен купить
MichaelKelay
| #
The best HD wallpapers https://wallpapers-all.com/62299-celebrity-chloe-grace-moretz-wallpaper.html in one place! Download free backgrounds for your desktop and smartphone. A huge selection of pictures – from minimalism to bright landscapes and fantasy. Enjoy stylish images every day!
Terencesam
| #
фриспины без депозита с выводом за регистрацию бонусы без отыгрыша за регистрацию
Pingrutty
| #
darknet markets url dark markets 2025
DonDonfaita
| #
dark market 2025 darknet market list
Donaldlaf
| #
dark web market dark markets
Toliknaf
| #
dark web markets darknet drugs
Rabyitabe
| #
darkmarkets darknet site
Samsungcaree
| #
Если на вашем телефоне Samsung Galaxy экран некорректно реагирует на прикосновения или не работает вовсе, не спешите паниковать. Сейчас мы рассмотрим основные шаги, которые помогут решить проблему. И на примере Samsung S20/21/22/23/24 пошагово выполним все действия, которые могут восстановить работоспособность экрана.
перейти
Kxyufaita
| #
dark markets darknet drug links
WilliamIRrutty
| #
dark market list https://github.com/newonionlinks/darknetmarkets – darknet markets
online casino lDic
| #
Thank you, Quite a lot of forum posts.
casino on gta online https://onlinecasinoindex.us/mybookie-sign-up-bonus/ band new real cas winnings online casinos 2019
Cazrpqd
| #
Мы готовы предложить дипломы любой профессии по выгодным тарифам. Купить диплом Новочеркасск — kyc-diplom.com/geography/novocherkassk.html
Cazrchf
| #
Приобрести диплом любого ВУЗа поспособствуем. Купить диплом магистра в Томске – diplomybox.com/kupit-diplom-magistra-v-tomske
online casino lDic
| #
Well expressed indeed! !
victory online casino https://onlinecasinoindex.us/casino-online-california/ mohegan sun online casino bonus code
LelandKarve
| #
ттк тарифы ростов
https://jobsleed.com/companies/chandra/
ттк подключение
Pingrutty
| #
dark market link dark market onion
DonDonfaita
| #
darknet market darknet links
Toliknaf
| #
dark web sites darknet market links
Rabyitabe
| #
darknet sites darknet drug market
Kxyufaita
| #
darknet markets onion address dark web market links
MarkNOlaf
| #
darknet market links https://github.com/newonionlinks/darknetmarkets – darknet markets url
online casino lDic
| #
Thanks a lot, An abundance of advice!
lucky creek online casino review https://hotgamblingguide.info/minnesota-casinos-online/ usa online casino no deposit bonus keep what you win
online casino lDic
| #
Information clearly utilized.!
online casino med dansk licens https://casinosonlinenew.com/review-cafe/ free no deposit online casino codes
Pingrutty
| #
darknet markets links tor drug market
1win_erkn
| #
1win casino en línea 1win2.com.mx .
1win_vget
| #
1 вин скачать 1 вин скачать .
Aaronnut
| #
ссылка кракен сайт зеркало kra29. cc
MichaelKelay
| #
The best HD wallpapers https://wallpapers-all.com/4384-lego-hero-factory.html in one place! Download free backgrounds for your desktop and smartphone. A huge selection of pictures – from minimalism to bright landscapes and fantasy. Enjoy stylish images every day!
Donaldlaf
| #
darknet drugs darknet websites
Toliknaf
| #
darknet markets onion address dark web markets
Kxyufaita
| #
dark market onion darknet markets 2025
WilliamIRrutty
| #
darkmarket url https://github.com/newonionlinks/darknetmarkets – dark market 2025
MarkNOlaf
| #
dark markets 2025 https://github.com/newonionlinks/darknetmarkets – darknet marketplace
online casino lDic
| #
Many thanks! An abundance of knowledge!
mohegan sun online casino pa promo code no deposit https://hotgamblingguide.com/shazam-casino-bonus-codes/ online casino real money michigan
Pingrutty
| #
dark web markets darknet marketplace
online casino lDic
| #
Nicely put. Thank you.
online casino topless dealer https://riggambling.com/online-keno/ vincere casino online
Toliknaf
| #
dark market url dark markets 2025
Terencesam
| #
бездепозитные игровые автоматы Бездепозитные бонусы за регистрацию
Donaldlaf
| #
dark web drug marketplace dark web marketplaces
WilliamIRrutty
| #
dark web marketplaces https://github.com/newonionlinks/darknetmarkets – darknet websites
Kxyufaita
| #
dark market darkmarket link
Rabyitabe
| #
dark market dark market link
silovie transformatori kypit_ssei
| #
купить трансформатор силовой https://www.silovye-transformatory-kupit11.ru .
online casino lDic
| #
Seriously lots of wonderful knowledge!
jubilee casino online https://casinoslotssaid.com/betting/ fortune coin online casino
Pingrutty
| #
dark web link dark market link
programmi 1s_rwSn
| #
купить 1с в москве купить 1с в москве .
1win_uxot
| #
1вин онлайн http://www.1win41.com.kg .
mostbet_sser
| #
мостбет скачать приложение на андроид мостбет скачать приложение на андроид .
online casino lDic
| #
Wonderful forum posts. Many thanks.
online slots casino real money https://combatcasino.info/online-casino-washington/ banger casino online
MarkNOlaf
| #
dark market url https://github.com/newonionlinks/darknetmarkets – dark market url
WilliamIRrutty
| #
darknet markets links https://github.com/newonionlinks/darknetmarkets – darknet markets 2025
Toliknaf
| #
dark web market urls darknet markets
Pingrutty
| #
onion dark website dark web sites
Kxyufaita
| #
dark markets 2025 dark web market
Donaldlaf
| #
dark market dark web markets
online casino lDic
| #
Very good write ups. Regards!
online casino with trustly https://hotgamblingguide.com/betting-in-boxing/ casino online ruleta el salvador
Rabyitabe
| #
dark markets 2025 dark web markets
JerryGlymn
| #
defi news crypto regulations
online casino lDic
| #
Many thanks! I like it!
mississippi online casinos https://hotgamblingguide.com/best-online-casino-north-carolina/ free online casino games with no download or registration
RonaldKinge
| #
монтаж системы вентиляции в частном доме
Pingrutty
| #
darknet sites dark web drug marketplace
WilliamIRrutty
| #
darknet drug store https://github.com/newonionlinks/darknetmarkets – dark websites
Kxyufaita
| #
dark web link dark markets 2025
online casino lDic
| #
Awesome stuff. Appreciate it.
what online casino does drake play https://casinoslotssaid.com/betting-sites-cricket/ new casino online games
Toliknaf
| #
darknet markets darknet market list
NathanAsymn
| #
Страхование по лучшей цене https://осагополис.рф Сравните предложения страховых компаний и выберите полис с выгодными условиями. Удобный сервис поможет найти оптимальный вариант автострахования, ОСАГО, КАСКО, туристических и медицинских страховок.
Donaldlaf
| #
dark web sites tor drug market
KennethTalry
| #
займ онлайн бесплатно займ сайт
Rabyitabe
| #
darknet markets dark market link
online casino lDic
| #
You’ve made your point pretty clearly!!
new online casinos nj https://hotgamblingguide.info/poker-game-online/ sloty online casino
NathanAsymn
| #
Страхование по лучшей цене https://осагополис.рф Сравните предложения страховых компаний и выберите полис с выгодными условиями. Удобный сервис поможет найти оптимальный вариант автострахования, ОСАГО, КАСКО, туристических и медицинских страховок.
KennethTalry
| #
займ онлайн сайт займ онлайн
Pingrutty
| #
dark web markets darknet markets onion
WilliamIRrutty
| #
dark web drug marketplace https://github.com/newonionlinks/darknetmarkets – dark web market links
MarkNOlaf
| #
darknet drug market https://github.com/newonionlinks/darknetmarkets – darknet links
Kxyufaita
| #
dark web market urls dark web marketplaces
online casino lDic
| #
Regards, An abundance of tips.
online sweeps casino https://onlinecasinoindex.us/nhl-hockey-betting/ bay mills resort & casino online casinos michigan
DonDonfaita
| #
onion dark website darknet markets onion
Donaldlaf
| #
darknet market links dark market list
RichardRhymn
| #
Ремонт кофемашин в Москве https://coffee-help24.ru быстро, качественно, с гарантией! Обслуживаем все бренды: Saeco, DeLonghi, Jura, Bosch и др. Диагностика, замена деталей, чистка от накипи. Выезд мастера на дом или ремонт в сервисе.
online casino lDic
| #
Wow plenty of valuable advice!
juegos de casino las vegas online gratis https://combatcasino.info/real-money-online-casino-minnesota/ four winds casino online gaming
FNDaviditabe
| #
bitcoin dark web https://github.com/newonionlinks/darknetmarkets darknet markets 2025
Terencesam
| #
бонусы за регистрацию 1xslots бездепозитный бонус
Pingrutty
| #
dark web marketplaces dark web market
RichardRhymn
| #
Ремонт кофемашин в Москве https://coffee-help24.ru быстро, качественно, с гарантией! Обслуживаем все бренды: Saeco, DeLonghi, Jura, Bosch и др. Диагностика, замена деталей, чистка от накипи. Выезд мастера на дом или ремонт в сервисе.
RobertSox
| #
Микрозайм взять – займы интернет
online casino lDic
| #
Thanks. I like it!
online gaming casino real money https://casinoslotoking.com/play-texas-holdem-online-for-real-money/ all slots casino online chat
Allandoums
| #
Последние IT-новости https://notid.ru быстро и понятно! Рассказываем о цифровых трендах, инновациях, стартапах и гаджетах. Только проверенная информация, актуальные события и мнения экспертов. Оставайтесь в центре IT-мира вместе с нами!
MarkNOlaf
| #
darknet site https://github.com/newonionlinks/darknetmarkets – dark web marketplaces
WilliamIRrutty
| #
dark markets https://github.com/newonionlinks/darknetmarkets – bitcoin dark web
online casino lDic
| #
Valuable write ups. Thank you!
best canadian online casino payouts https://casinonair.com/online-casino-georgia/ gta online casino heist hacking device location
Toliknaf
| #
dark web link darknet drug links
Rabyitabe
| #
darknet markets dark market url
Kxyufaita
| #
darknet markets links darknet websites
FNDaviditabe
| #
darknet marketplace https://github.com/newonionlinks/darknetmarkets darknet market lists
Donaldlaf
| #
darknet market darknet markets onion address
Allandoums
| #
Последние IT-новости https://notid.ru быстро и понятно! Рассказываем о цифровых трендах, инновациях, стартапах и гаджетах. Только проверенная информация, актуальные события и мнения экспертов. Оставайтесь в центре IT-мира вместе с нами!
Pingrutty
| #
darkmarket darknet markets url
RobertPsype
| #
yacht trip dubai private yacht
online casino lDic
| #
You actually suggested it terrifically.
casino games online india https://mapcasino.info/review-ignition/ casino de montreal online
MarkNOlaf
| #
dark web sites https://github.com/newonionlinks/darknetmarkets – darknet markets 2025
WilliamIRrutty
| #
darknet drug market https://github.com/newonionlinks/darknetmarkets – darknet sites
online casino lDic
| #
Fantastic content. Kudos.
mwplay888 online casino login https://riggambling.com/nascar-betting/ 10$ deposit online casino
RonaldKinge
| #
оформление дендроплана
DonDonfaita
| #
bitcoin dark web darknet markets onion address
Toliknaf
| #
dark web market dark web markets
Kxyufaita
| #
darkmarket url dark market link
FNDaviditabe
| #
darknet marketplace https://github.com/newonionlinks/darknetmarkets darknet drug links
Rabyitabe
| #
onion dark website darknet site
Pingrutty
| #
darkmarket 2025 darknet markets 2025
Donaldlaf
| #
darknet market list dark websites
Samsungcaree
| #
Современные телевизоры Samsung, такие как QLED QN90C, OLED S95B и Neo QLED 8K QN800A, полны передовых функций — от невероятного качества изображения до возможностей Smart TV. Однако даже у этих флагманских моделей могут возникать проблемы:
подробнее
mostbet_veOa
| #
авиатор игра на деньги http://mostbet11.com.kg .
mostbet_gxMl
| #
mostbet.con http://mostbet12.com.kg .
MarkNOlaf
| #
darknet marketplace https://github.com/newonionlinks/darknetmarkets – darknet markets url
online casino lDic
| #
Amazing posts. Regards.
star casino online game https://mapcasino.info/new-online-casinos/ twin rivers casino online
Pingrutty
| #
dark market darknet market lists
Kxyufaita
| #
darkmarket url darkmarkets
FNDaviditabe
| #
darkmarket link https://github.com/newonionlinks/darknetmarkets darknet market
DonDonfaita
| #
dark markets darknet markets
Toliknaf
| #
darknet markets onion dark market 2025
RadikCype
| #
https://provocation-tattoo.ru/news/pgs/?ozghidaemue_filmu_1.html что посмотреть из фильмов интересного на вечер
Rabyitabe
| #
dark market 2025 dark websites
Donaldlaf
| #
bitcoin dark web bitcoin dark web
online casino lDic
| #
Nicely put, Appreciate it!
online casino target audience https://combatcasino.info/fast-payout-casino/ maine online casino
Pingrutty
| #
darkmarket dark websites
WilliamIRrutty
| #
darknet market links https://github.com/newonionlinks/darknetmarkets – darknet links
Kxyufaita
| #
dark websites darknet market links
FNDaviditabe
| #
darknet market https://github.com/newonionlinks/darknetmarkets darknet market list
MatthewbiB
| #
букет цветов купить спб с доставкой букет цветов заказать с доставкой
Toliknaf
| #
dark market url dark web market
Samsungcaree
| #
Современные телевизоры Samsung, такие как QLED QN90C, OLED S95B и Neo QLED 8K QN800A, полны передовых функций — от невероятного качества изображения до возможностей Smart TV. Однако даже у этих флагманских моделей могут возникать проблемы:
здесь
online casino lDic
| #
Superb data. Cheers!
online casino games using paypal https://uscasinoguides.com/maryland-casinos/ planet casino online
Rabyitabe
| #
dark web link darknet market
Donaldlaf
| #
darknet drug links darknet markets
online casino lDic
| #
You said it perfectly.!
pa live casino online https://casinonair.com/tennis-betting/ real online casino with no deposit bonus
MatthewbiB
| #
доставка букетов цветов цветы питер купить
Pingrutty
| #
darknet market lists darknet websites
binance
| #
Thanks for sharing. I read many of your blog posts, cool, your blog is very good. https://accounts.binance.com/pt-BR/register-person?ref=YY80CKRN
MarkNOlaf
| #
darknet marketplace https://github.com/newonionlinks/darknetmarkets – darknet drug links
RadikCype
| #
https://evu.uz/wp-content/pgs/sovetskoe_kino__glavnoe.html бандитский петербург песня корнелюка слушать бесплатно
FNDaviditabe
| #
darknet sites https://github.com/newonionlinks/darknetmarkets darknet market links
DonDonfaita
| #
dark markets dark market 2025
online casino lDic
| #
You actually mentioned that terrifically.
online live casino reviews https://shadowcasino.info/online-casino-colorado/ texas online casino
Rabyitabe
| #
darknet markets onion address darknet site
Donaldlaf
| #
dark web drug marketplace darknet websites
online casino lDic
| #
Regards. I appreciate it!
online casino exchange https://shadowcasino.info/live-online-casinos/ ten online casino
Pingrutty
| #
dark web drug marketplace dark web sites
phoenixautobodyshopmailm
| #
Unlock your vehicle’s potential with our top-notch auto tuning services! Unleash your ride into a stunning masterpiece with our expert team. We specialize in refinements that cater to your unique style. Our reliable solutions ensure optimal performance and aesthetic appeal . Don’t settle for average; elevate your driving experience and turn heads on the road! Visit us at https://phoenix-autobodyshop.com/ today and take the first step towards your dream car!
Samsungcaree
| #
Как транслировать содержимое с телефона Samsung на телевизор через WI-FI или DeX кабель. Пошаговая инструкция всех вариантов
ссылка
MarkNOlaf
| #
darkmarket 2025 https://github.com/newonionlinks/darknetmarkets – dark market list
WilliamIRrutty
| #
darknet market lists https://github.com/newonionlinks/darknetmarkets – darknet drugs
Terencesam
| #
бонусы за регистрацию фриспины без депозита за регистрацию с выводом
Kxyufaita
| #
darkmarket url darkmarket url
online casino lDic
| #
Cheers! I like this.
bob online casino https://eseomail.com/texas-holdem-online/ casino canada online
Toliknaf
| #
dark web link darknet markets
Curtiswoums
| #
кракен вход ссылка kraken зайти без впн
Pingrutty
| #
best darknet markets dark markets 2025
Rabyitabe
| #
dark web market best darknet markets
online casino lDic
| #
Point well utilized!!
1 euro deposit casino online https://usagamblingexperts.com/online-slots/ jupiters casino online
Donaldlaf
| #
onion dark website darknet markets url
WilliamIRrutty
| #
dark market 2025 https://github.com/newonionlinks/darknetmarkets – dark market link
Sazrpza
| #
начальный аттестат купить
Kxyufaita
| #
darknet market list dark market
TCLcaree
| #
Телевизоры TCL с функцией Smart TV предоставляют доступ к потоковым видеосервисам, приложениям и другим онлайн-возможностям. Однако некоторые пользователи могут столкнуться с проблемой подключения к Wi-Fi. Давайте разберем основные причины этой неисправности и предложим эффективные методы ее устранения.
ссылка
Dnrtylu
| #
реально купить диплом о среднем специальном образовании
online casino lDic
| #
Really all kinds of great material!
online casino virtual money https://combatcasino.info/review-cafe/ jeetwin online casino bd
DonDonfaita
| #
darknet marketplace onion dark website
Pingrutty
| #
darknet markets url darknet market links
online casino lDic
| #
Nicely expressed really! !
open online casino https://snipercasino.info/online-video-poker/ casino nederland online
Rabyitabe
| #
darknet drugs darknet drug market
WilliamIRrutty
| #
darknet drug store https://github.com/newonionlinks/darknetmarkets – dark markets
Donaldlaf
| #
dark web drug marketplace dark web markets
MarkNOlaf
| #
darknet links https://github.com/newonionlinks/darknetmarkets – dark market url
Robertflags
| #
производство мебели Шкаф-купе на заказ от производителя – это не просто предмет мебели, а инвестиция в комфорт и функциональность вашего дома. Индивидуальный подход позволяет максимально эффективно использовать пространство, учитывая особенности планировки и ваши личные предпочтения в дизайне. Вы получаете возможность выбрать материалы, цвет, внутреннее наполнение и размеры, создавая шкаф, идеально вписывающийся в интерьер и отвечающий всем вашим потребностям.
Kxyufaita
| #
darknet marketplace dark markets 2025
Pingrutty
| #
dark web marketplaces dark markets
online casino lDic
| #
Appreciate it! Lots of material.
casinos slots online https://findscasino.info/online-keno/ lucky penny casino online
Toliknaf
| #
darknet drug store darkmarket list
DonDonfaita
| #
darknet market lists darkmarkets
online casino lDic
| #
Thank you, Good information!
offshore online casinos https://casinoslotoking.com/countries/ online casinos indiana
WilliamIRrutty
| #
dark web link https://github.com/newonionlinks/darknetmarkets – darknet market links
MarkNOlaf
| #
darkmarket link https://github.com/newonionlinks/darknetmarkets – dark web market list
Rabyitabe
| #
darknet market list darknet websites
Donaldlaf
| #
dark web market darknet markets onion address
phoenixautobodyshopmailm
| #
Unlock your vehicle’s potential with our top-notch auto tuning services! Discover your ride into a stunning masterpiece with our expert team. We specialize in upgrades that cater to your unique style. Our exceptional solutions ensure optimal performance and stylish look . Don’t settle for average; elevate your driving experience and turn heads on the road! Visit us at https://phoenix-autobodyshop.com/ today and take the first step towards your dream car!
Kxyufaita
| #
darknet markets url darknet drug links
Pingrutty
| #
onion dark website bitcoin dark web
online casino lDic
| #
Nicely put. Thanks.
australian online casino reviews 2018 https://magicalcasino.info/virginia-online-casinos/ online casino 10 euro startguthaben
mostbet_fbsl
| #
мост бет официальный сайт http://www.mostbet13.com.kg/ .
mostbet_rhmn
| #
состбет состбет .
DonDonfaita
| #
dark markets darkmarket list
Toliknaf
| #
darknet market links dark market link
online casino lDic
| #
You said it nicely..
mgm online casino gift card https://mgmonlinecasino.us/casino-wild/ free online virtual casino
WilliamIRrutty
| #
darknet drugs https://github.com/newonionlinks/darknetmarkets – darknet links
Terencesam
| #
свежие фриспины риобет какие автоматы дают деньги за регистрацию без депозита
Rabyitabe
| #
darknet markets onion dark web market
Kxyufaita
| #
darknet links darknet markets url
Donaldlaf
| #
darknet market list dark market 2025
Pingrutty
| #
darknet market lists darknet drug links
online casino lDic
| #
Nicely put. Cheers.
casino venezia online https://igamingcasino.info/horse-betting/ best payout online casinos usa
MarkNOlaf
| #
darknet market https://github.com/newonionlinks/darknetmarkets – darknet drug links
Toliknaf
| #
dark markets 2025 darknet websites
online casino lDic
| #
Nicely put. Thanks.
gta online casino work missions https://hotgamblingguide.com/golf-betting-online/ slot games online casino
RadikCype
| #
http://interra.com.ru/media/pgs/?filmu_onlayn_v_horoshem_kachestve.html белый ангел ростов на дону снять
Kxyufaita
| #
darkmarket link dark markets
Pingrutty
| #
bitcoin dark web darknet markets onion address
Rabyitabe
| #
darknet drugs darkmarkets
Donaldlaf
| #
darknet drugs darknet market list
WilliamIRrutty
| #
darkmarket https://github.com/newonionlinks/darknetmarkets – dark web drug marketplace
MarkNOlaf
| #
dark web sites https://github.com/newonionlinks/darknetmarkets – dark web market list
online casino lDic
| #
Beneficial knowledge. Cheers!
online casinos with freeplay https://mgmonlinecasino.us/bingo-online-for-money/ ocean online casino download
DonDonfaita
| #
darknet drugs darknet markets onion
Toliknaf
| #
dark market link darkmarket url
Pingrutty
| #
darknet markets links darknet markets onion
online casino lDic
| #
Truly lots of valuable tips!
online casino promotions no deposit bonus https://usagamblingexperts.com/real-money-roulette/ best online casino in america
Kxyufaita
| #
darknet market list dark web markets
Trefgxg
| #
Заказать документ о получении высшего образования можно в нашей компании. dip-lom-rus.ru/kupit-diplom-lipetsk
Jariorrzi
| #
Привет!
Начальники очень часто выбирают претендентов, которые закончили ВУЗ. Особенно ценятся элитные учебные заведения. Впрочем учиться 5 лет – это долго и дорого, далеко не у всех есть подобная возможность. Заказать документ становится самым оптимальным решением.
Бывают и непредвиденные случаи, когда диплом теряется или портится. Далеко не всегда возможно быстро и без проблем восстановить его, особенно если ВУЗ закрыт или располагается где-то в другом регионе страны. Бюрократия отнимает множество времени.
Для быстрого продвижения по карьере понадобится наличие диплома института. Но очень часто в жизни может случиться так, что сложные обстоятельства не дают с успехом окончить учебу и заполучить такой важный документ.
Купить диплом университета
Мы предлагаем выгодно и быстро приобрести диплом, который выполнен на оригинальной бумаге и заверен печатями, водяными знаками, подписями официальных лиц. Наш диплом способен пройти любые проверки, даже при помощи специальных приборов. Решайте свои задачи максимально быстро с нашими дипломами.
Где заказать диплом по необходимой специальности? tutdiploms.com/
Rabyitabe
| #
darknet markets darkmarket list
RadikCype
| #
https://lorrd.ru/test/pgs/doramu_na_kinogo___mir_vostochnoy_dramu_i_yarkih_istoriy.html м видео белая дача телефон
Donaldlaf
| #
darknet markets onion address dark web market urls
mostbet_tjOn
| #
мотбет mostbet19.com.kg .
Diplomi_zuol
| #
купить диплом мед образования купить диплом мед образования .
WilliamIRrutty
| #
darknet sites https://github.com/newonionlinks/darknetmarkets – dark market url
1win_weka
| #
1вин вход 1вин вход .
MarkNOlaf
| #
darknet drug links https://github.com/newonionlinks/darknetmarkets – dark web market urls
online casino lDic
| #
Appreciate it. Lots of advice!
bandar betting casino 368bet online https://ratingcasino.info/horse-betting/ isle of man casino online
Pingrutty
| #
darkmarket list dark markets 2025
Kxyufaita
| #
dark web market links darkmarket link
online casino lDic
| #
Many thanks! I value it!
paf online casino https://uscasinoguides.com/massachusetts-casinos/ gta online casino heist silent and sneaky best approach
Curtiswoums
| #
кракен маркетплейс ссылка kraken вход
FNDaviditabe
| #
darknet market list https://github.com/newonionlinks/darknetmarkets dark web sites
WilliamIRrutty
| #
darknet markets url https://github.com/newonionlinks/darknetmarkets – dark web marketplaces
online casino lDic
| #
Useful knowledge. Many thanks.
best payout usa online casino https://usagamblingexperts.com/real-money-blackjack/ euro grand online casino
Toliknaf
| #
darknet markets onion address darkmarket url
online casino lDic
| #
Many thanks. Very good stuff.
australian online casino no deposit https://casinoslotssaid.com/best-sportsbook-bonuses/ online casino tropez
Rabyitabe
| #
darknet market links tor drug market
Donaldlaf
| #
dark market link onion dark website
mostbet_oqpl
| #
mostbet промокод https://www.mostbet20.com.kg .
Epsoncaree
| #
Настройка Wi-Fi на принтерах Epson, таких, как модели L3250 и L3251, позволяет значительно упростить процесс печати и сканирования. Вы сможете печатать документы прямо с компьютера, смартфона или планшета, не используя провода.
подробнее
Terencesam
| #
Бонусы за регистрацию Игровые автоматы на деньги играть
Pingrutty
| #
darknet drugs dark websites
Toliknaf
| #
tor drug market onion dark website
online casino lDic
| #
Thank you. A good amount of facts.
bestes zimpler online casino https://onlinecasinoindex.us/australian-online-casino-no-deposit-bonus/ philippines online casino pagcor
DonDonfaita
| #
darknet site dark market 2025
online casino lDic
| #
Really a lot of valuable tips.
is vegas online casino legit https://casinonair.com/no-deposit-bonus-casinos/ casino online 1500
Rabyitabe
| #
darkmarket link dark market onion
Rprpnib
| #
?? Кино без компромиссов! Платформа kinogo стирает стереотипы: качайте любые фильмы без подписок, в HD, в пробке! Библиотека растёт ежедневно — мы знаем, что тебя шокирует! Жмите > киного сайт — ваш диван теперь Оскар-холл!
Toliknaf
| #
darknet drugs bitcoin dark web
Donaldlaf
| #
dark websites darkmarkets
Pingrutty
| #
tor drug market darkmarket 2025
Kxyufaita
| #
darkmarkets dark web marketplaces
mostbet_agoa
| #
mostbet apk 2024 http://www.mostbet3002.ru/ .
mostbet_hfmt
| #
online mostbet http://www.mostbet3003.ru .
DonDonfaita
| #
dark web market dark market link
FNDaviditabe
| #
dark web markets https://github.com/newonionlinks/darknetmarkets dark web market urls
WilliamIRrutty
| #
darknet market https://github.com/newonionlinks/darknetmarkets – darknet markets url
online casino lDic
| #
Many thanks! A lot of facts!
gta 5 online casino update date https://cryptogamblingguru.com/ducky-luck/ play casino real money online
Pingrutty
| #
darknet marketplace dark websites
MarkNOlaf
| #
best darknet markets https://github.com/newonionlinks/darknetmarkets – darknet markets onion
Rabyitabe
| #
darknet market links dark websites
Donaldlaf
| #
darknet links dark web marketplaces
online casino lDic
| #
Whoa plenty of amazing knowledge.
online casino free chip https://snipercasino.info/review-ignition/ nj best online casino
FNDaviditabe
| #
dark web market list https://github.com/newonionlinks/darknetmarkets darknet links
Iariorrlv
| #
купить диплом визажиста киев
WilliamIRrutty
| #
darkmarkets https://github.com/newonionlinks/darknetmarkets – dark websites
DonDonfaita
| #
darknet drug links dark websites
Diplomi_yzmi
| #
купить скан диплома о высшем образовании diplomanc.com .
Pingrutty
| #
dark market list darknet drugs
https://welcome59.ru/
| #
Выбирайте лицензированные казино, чтобы не столкнуться с
мошенниками.
https://welcome59.ru/
MarkNOlaf
| #
best darknet markets https://github.com/newonionlinks/darknetmarkets – darkmarket url
online casino lDic
| #
Lovely information. Kudos.
cash storm casino – online vegas slots games https://igamingcasino.info/nfl-football-betting/ play for real money online casino
Rabyitabe
| #
darkmarket darknet market links
1win_ttEl
| #
1win. https://www.1win101.com.kg .
Donaldlaf
| #
darknet drug store dark web markets
FNDaviditabe
| #
dark market onion https://github.com/newonionlinks/darknetmarkets darknet markets onion address
mostbet_mfPl
| #
1вин кг 1вин кг .
online casino lDic
| #
Useful information. Thanks.
online casino stocks https://combatcasino.info/nba-betting/ online casino zahlungsarten
WilliamIRrutty
| #
dark web sites https://github.com/newonionlinks/darknetmarkets – dark market link
Toliknaf
| #
darknet markets onion dark websites
Kxyufaita
| #
bitcoin dark web onion dark website
Pingrutty
| #
onion dark website dark web market
DonDonfaita
| #
dark web link dark markets 2025
Arthurtix
| #
прохождение игры квеста майнкрафт дом
Huaweicaree
| #
Huawei RC предоставляет качественные услуги по ремонту техники Huawei, включая VR системы, ИБП (источники бесперебойного питания), ноутбуки, планшеты, серверы, смарт-часы и смартфоны.
ссылка
FNDaviditabe
| #
darknet drugs https://github.com/newonionlinks/darknetmarkets dark market url
MarkNOlaf
| #
dark market url https://github.com/newonionlinks/darknetmarkets – darknet sites
WilliamIRrutty
| #
darknet marketplace https://github.com/newonionlinks/darknetmarkets – dark web market links
online casino lDic
| #
You actually reported that fantastically!
work from home online casino jobs https://snipercasino.info/betting/ tours gratuits du casino online
Toliknaf
| #
darknet markets onion darkmarket
Rabyitabe
| #
dark markets darknet drug store
Donaldlaf
| #
dark web sites darkmarket
online casino lDic
| #
Really tons of wonderful info.
best online casinos europe https://casinoshaman.com/best-online-casino-in-new-zealand/ winbet online casino регистрация и казино бонус 300 лева
Kxyufaita
| #
darknet market list onion dark website
Pingrutty
| #
dark web link dark web market
FNDaviditabe
| #
bitcoin dark web https://github.com/newonionlinks/darknetmarkets darknet links
Kevinmub
| #
кухня на заказ по индивидуальным размерам Рынок мебели на заказ в Москве предлагает широкий спектр материалов, стилей и технологий. От классического дерева до современного акрила, от минимализма до роскошного барокко – возможности ограничены лишь фантазией заказчика и бюджетом. Квалифицированные дизайнеры помогут разработать проект, учитывающий все пожелания и потребности, а опытные мастера воплотят его в жизнь, обеспечивая высокое качество и долговечность изделия.
WilliamIRrutty
| #
bitcoin dark web https://github.com/newonionlinks/darknetmarkets – darkmarket
DonDonfaita
| #
dark web markets darknet markets links
Toliknaf
| #
darknet drug market dark websites
online casino lDic
| #
Nicely put. Appreciate it!
hard rock casino online michigan https://ratingcasino.info/massachusetts-online-casino/ i migliori casino online italiani
MarkNOlaf
| #
darknet marketplace https://github.com/newonionlinks/darknetmarkets – darknet market
Rabyitabe
| #
darknet drug store darknet markets 2025
online casino lDic
| #
Nicely put. Cheers.
scores online casino nj https://hotgamblingguide.info/omaha-poker-online-game/ safe online casino australia
1win_tdMr
| #
1vin https://www.1win100.com.kg .
Donaldlaf
| #
dark market onion darknet site
Pingrutty
| #
darknet market dark markets 2025
AirPodscaree
| #
Наушники AirPods представляют собой удобный и стильный аксессуар от Apple, который популярен благодаря качественному звуку и беспроводному соединению. Однако владельцы этих устройств нередко сталкиваются с проблемой их внезапного отключения от телефона. Чтобы устранить эту неприятность, важно разобраться с ее причинами и понять, как их исправить.
перейти
FNDaviditabe
| #
dark markets 2025 https://github.com/newonionlinks/darknetmarkets dark web market
WilliamIRrutty
| #
dark markets 2025 https://github.com/newonionlinks/darknetmarkets – darknet markets 2025
Kxyufaita
| #
dark web market links darknet drug links
Sazrudk
| #
Где купить диплом специалиста?
Приобрести документ о получении высшего образования можно в нашей компании в столице. Мы оказываем услуги по изготовлению и продаже документов об окончании любых университетов РФ. alternativaprofi.com/forum/viewthread.php?thread_id=35222
Greenhistory.ru
| #
Хотите создать красивый газон без долгого ожидания? Рулонный газон купить можно прямо сейчас на Greenhistory.ru. Мы предлагаем оперативную доставку, качественную продукцию и услуги профессиональной укладки. Превратите свой участок в зеленый оазис уже сегодня!
Cazrapa
| #
Мы предлагаем документы Институтов, которые находятся в любом регионе России. okdiplom.ru/kupit-diplom-bakalavra-4-2
Toliknaf
| #
dark market dark market
JamesBoorp
| #
плей прохождение игры киберпанк 2077 после сюжета
Terencesam
| #
бонусы за регистрацию Играть игровые автоматы на деньги
DonDonfaita
| #
darknet markets onion dark web markets
online casino lDic
| #
Thanks! I appreciate this!
best online casino welcome bonuses https://magicalcasino.info/countries/ casino online ruleta el salvador
MarkNOlaf
| #
darknet markets 2025 https://github.com/newonionlinks/darknetmarkets – dark web market
FNDaviditabe
| #
darknet drug market https://github.com/newonionlinks/darknetmarkets darknet markets 2025
Pingrutty
| #
dark market link darknet markets
online casino lDic
| #
Kudos! Great information!
7 melons online casino https://uscasinoguides.com/esports-betting/ new online casinos free money
Greenhistory.ru
| #
На днях обсуждал ландшафтный дизайн на форуме Yaplakal, и там многие советовали рулонный газон от Greenhistory.ru. Решил прислушаться к отзывам – заказал, и не пожалел! Трава свежая, плотная, прижилась без проблем. Участок преобразился моментально, теперь советую друзьям и соседям!
WilliamIRrutty
| #
darknet markets https://github.com/newonionlinks/darknetmarkets – darkmarkets
Rabyitabe
| #
dark websites dark markets 2025
Donaldlaf
| #
dark web link darknet site
Toliknaf
| #
dark web market urls darknet links
Kxyufaita
| #
dark markets tor drug market
roborokcaree
| #
Роботы-пылесосы стремительно вошли в нашу жизнь, избавив нас от утомительных рутинных задач. Среди них особое место занимает Roborock, известный своей надежностью и удобством. Однако для того чтобы максимально использовать его возможности, важно разобраться, как настроить устройство. В этой статье я расскажу, как подключить робот-пылесос Roborock к телефону через Wi-Fi и интегрировать его с голосовым помощником Алисой от Яндекса. Все шаги простые, понятные, и вы однозначно справитесь с этим!
подробнее
FNDaviditabe
| #
onion dark website https://github.com/newonionlinks/darknetmarkets darknet marketplace
DonDonfaita
| #
darkmarket link dark market
online casino lDic
| #
Incredible many of awesome information.
best online casinos with paypal https://riggambling.com/review-slotocash/ casino queen online
WilliamIRrutty
| #
darknet markets https://github.com/newonionlinks/darknetmarkets – darknet websites
Pingrutty
| #
darkmarket list dark web drug marketplace
online casino lDic
| #
You explained it exceptionally well.
agen casino online bonus new member https://casinonair.com/real-money-roulette/ top online casinos 2023
Toliknaf
| #
dark market url dark web market list
Rabyitabe
| #
dark market list darkmarkets
Donaldlaf
| #
darkmarket link darknet links
Kxyufaita
| #
darknet drug market darknet market lists
FNDaviditabe
| #
darknet websites https://github.com/newonionlinks/darknetmarkets onion dark website
WilliamIRrutty
| #
darknet drug market https://github.com/newonionlinks/darknetmarkets – darknet links
Pingrutty
| #
darkmarket 2025 dark web marketplaces
mostbet_prOt
| #
mostbet 1-15 pozitsiya mostbet3004.ru .
mostbet_siel
| #
mostbet kz скачать https://mostbet3005.ru .
DonDonfaita
| #
dark market onion darkmarket list
online casino lDic
| #
You’ve made your point extremely well..
free online casino craps https://combatcasino.info/craps-online/ gta online casino heist buyer level
Toliknaf
| #
dark market 2025 dark web market urls
online casino lDic
| #
This is nicely put! !
list of online casinos in italy https://casinoslotoking.com/real-online-casino-georgia/ newest nj online casino
Rabyitabe
| #
best darknet markets bitcoin dark web
FNDaviditabe
| #
darknet marketplace https://github.com/newonionlinks/darknetmarkets dark web sites
WilliamIRrutty
| #
darknet drugs https://github.com/newonionlinks/darknetmarkets – darknet drug store
Donaldlaf
| #
darknet market links onion dark website
Kxyufaita
| #
darkmarket list tor drug market
Pingrutty
| #
tor drug market darkmarket link
Toliknaf
| #
dark market link darkmarket 2025
Terencesam
| #
Игровые автоматы с бонусом за регистрацию без депозита Игровые автоматы играть на деньги
Williamjoype
| #
клей плиточный кнауф Строительные материалы компании Кануф в России. Доставка – Самовывоз. Оплата Нал/Безнал. огромный ассортимент товара.
online casino lDic
| #
Helpful information. Cheers!
arabic casino online https://casinocashstars.com/countries/ online casinos that pay out fast
DonDonfaita
| #
darknet websites dark web marketplaces
MarkNOlaf
| #
darkmarkets https://github.com/newonionlinks/darknetmarkets – darknet markets links
FNDaviditabe
| #
dark web market list https://github.com/newonionlinks/darknetmarkets dark web market list
WilliamIRrutty
| #
dark market https://github.com/newonionlinks/darknetmarkets – onion dark website
online casino lDic
| #
Reliable posts. Regards!
harrahs online casino pennsylvania https://findscasino.info/ufc-mma-betting/ gta online casino work missions
Anonymous
| #
Your article helped me a lot, is there any more related content? Thanks! https://www.binance.com/en-IN/register?ref=UM6SMJM3
Rabyitabe
| #
dark market onion darknet drug store
1win_vqpa
| #
1win http://1win102.com.kg/ .
Donaldlaf
| #
dark web market darkmarket link
1win_foOt
| #
1win онлайн http://1win108.com.kg/ .
Pingrutty
| #
dark market link darknet drug market
Toliknaf
| #
dark market darknet drug store
Williamced
| #
сайт про игры статьи красивые дома в майнкрафте из дерева
Kxyufaita
| #
darkmarket url darknet drug links
FNDaviditabe
| #
dark web drug marketplace https://github.com/newonionlinks/darknetmarkets darkmarket 2025
WilliamIRrutty
| #
dark web markets https://github.com/newonionlinks/darknetmarkets – darkmarket link
online casino lDic
| #
Perfectly voiced certainly. !
australian best online casino https://findscasino.info/crypto-casino/ indian casinos online
DonDonfaita
| #
darknet market list dark websites
Dennisten
| #
крым отели все включено цены
Williamced
| #
сайт про игры статьи красивые большие дома в майнкрафте
online casino lDic
| #
Excellent content. Thank you.
bonos gratis casinos online https://mgmonlinecasino.us/states/ löwen play casino online
Toliknaf
| #
dark market onion darkmarkets
Pingrutty
| #
dark market list darkmarkets
Dennisten
| #
отдых в судаке
Rabyitabe
| #
darkmarket url dark web link
Donaldlaf
| #
darknet drug store dark web market list
GrgrChuch
| #
Почему бы не переплачивать, если наш сайт дарит кино бесплатно? Выбирайте из тысяч шедевров, удовлетворяя любые настроения! Двигайте как в IMAX, когда хотите — очереди умерли! Заходите на hdrezka сайт — у нас новинки каждый день! Скучно? Мы угадаем, что вас поразит!
FNDaviditabe
| #
dark web market urls https://github.com/newonionlinks/darknetmarkets darkmarket
Kxyufaita
| #
dark market 2025 dark web drug marketplace
WilliamIRrutty
| #
dark web market urls https://github.com/newonionlinks/darknetmarkets – dark web drug marketplace
Xazrpml
| #
Приобрести диплом о высшем образовании!
Мы можем предложить дипломы любых профессий по приятным тарифам. Вы покупаете диплом в надежной и проверенной временем компании. : gamerplayz2k.mn.co/posts/74567740
online casino lDic
| #
Terrific material. Cheers.
online gaming casino jobs https://igamingcasino.info/virginia-online-casinos/ brand new usa online casinos 2021 no deposit bonus
MarkNOlaf
| #
dark web sites https://github.com/newonionlinks/darknetmarkets – darkmarket list
DonDonfaita
| #
darknet drug store darknet drug store
mostbet_vkPi
| #
mostbet 1-8 https://mostbet3006.ru .
mostbet_zcEa
| #
mostbet apk download latest version https://www.mostbet3007.ru .
Toliknaf
| #
darknet markets bitcoin dark web
lifeforgame
| #
прохождение начни игру майнкрафт дом
Sazrfgw
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и любых других профессий по выгодным ценам. Преимущества покупки документов в нашем сервисе
Вы заказываете диплом в надежной и проверенной временем компании. Такое решение сэкономит не только много средств, но и время.
На этом преимущества не заканчиваются, их гораздо больше:
• Документы печатаются на оригинальных бланках с печатями и подписями;
• Можно заказать дипломы всех ВУЗов РФ;
• Цена во много раз ниже нежели довелось бы платить на очном и заочном обучении в ВУЗе;
• Максимально быстрая доставка как по столице, так и в любые другие регионы России.
Купить диплом о высшем образовании– http://1001vieclam.forumvi.com/t10196-topic#10924\ – 1001vieclam.forumvi.com/t10196-topic#10924
Pingrutty
| #
dark market link darknet market
online casino lDic
| #
You actually mentioned this well.
are online casinos for real https://mapcasino.info/review-wild/ adventskalender online casino 2023
FNDaviditabe
| #
darknet drugs https://github.com/newonionlinks/darknetmarkets darknet market links
WilliamIRrutty
| #
dark web market links https://github.com/newonionlinks/darknetmarkets – dark websites
Rabyitabe
| #
tor drug market best darknet markets
1win_yvka
| #
1win скачать kg http://1win42.com.kg/ .
Donaldlaf
| #
dark web marketplaces dark web market list
Kxyufaita
| #
darknet links dark markets
Jameselobe
| #
Express Canada Pharm: canada rx pharmacy world – Express Canada Pharm
online casino lDic
| #
Thanks! I like this.
online slot casino real money https://casinosonlinenew.com/safe-casinos-online/ play online casino at playojo reviews
Terencesam
| #
игровые автоматы слоты с бонусами Бездепозитные фриспины
Toliknaf
| #
darknet sites https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet markets
Pingrutty
| #
dark market url https://github.com/aresmarketlink0ru72/aresmarketlink – darkmarket
FNDaviditabe
| #
darknet drug links https://github.com/newonionlinks/darknetmarkets darknet markets url
DonDonfaita
| #
dark web market links https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet market
WilliamIRrutty
| #
darknet markets 2025 https://github.com/newonionlinks/darknetmarkets – darknet markets links
LeroyVaf
| #
Бесконечный выбор сериалов в интернете!
http://forum.d-dub.com/member.php?546924-nimolrusse&page=1&tab=activitystream&type=all
online casino lDic
| #
Seriously quite a lot of helpful info!
nl online casinos https://hotgamblingguide.org/texas-online-casinos/ romanian online casino
turgeocaree
| #
Выбор оптического прицела – одна из самых важных задач для охотника. Однако часто встречаются проблемы, связанные с недостаточной информированностью и грамотностью охотников, что может привести к серьезным промахам. Давайте разберемся в ключевых аспектах выбора оптики для лесной охоты.
перейти
Cazreor
| #
Привет!
Без института очень сложно было продвинуться вверх по карьерной лестнице. Сегодня же этот документ не дает практически никаких гарантий, что удастся найти престижную работу. Более важное значение имеют практические навыки специалиста и его постоянный опыт. Именно из-за этого решение о покупке диплома можно считать целесообразным. Приобрести диплом об образовании justpaste.me/mWqd3
Rabyitabe
| #
darkmarket list https://github.com/tormarkets2025ukaz1/tormarkets2025 – dark markets
Kxyufaita
| #
darknet market darknet drug links
Donaldlaf
| #
dark web market urls https://github.com/darknetmarketlinks2025/darknetmarkets – darknet market list
FNDaviditabe
| #
dark web marketplaces https://github.com/newonionlinks/darknetmarkets dark market url
LeroyVaf
| #
Онлайн-платформы делают просмотр сериалов удобным.
https://forum.web.ru/memberlist.php?first_char=r&mode=searchuser&sd=a&sk=a&start=200
Pingrutty
| #
darkmarket https://github.com/aresmarketlink0ru72/aresmarketlink – darknet links
WilliamIRrutty
| #
dark markets https://github.com/newonionlinks/darknetmarkets – darknet site
lifeforgame
| #
игры читайте статьи красивые дома в майнкрафте схемы
WilliamDix
| #
Полезная информация как официально купить диплом о высшем образовании
Toliknaf
| #
dark web markets https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark markets 2025
MarkNOlaf
| #
dark web sites https://github.com/newonionlinks/darknetmarkets – darknet drugs
GeorgeTom
| #
Source:
– https://mydomcom.ru/
DonDonfaita
| #
dark market list https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark web drug marketplace
Lazrlbh
| #
Сколько стоит диплом высшего и среднего образования и как его получить?
LeroyVaf
| #
Где найти редкие зарубежные сериалы онлайн?
http://www.fujikong3.cc/home.php?do=profile&from=space&mod=space&uid=3560936
FNDaviditabe
| #
dark market onion https://github.com/newonionlinks/darknetmarkets darknet drug links
WilliamIRrutty
| #
darknet links https://github.com/newonionlinks/darknetmarkets – darkmarket link
Rabyitabe
| #
darknet site https://github.com/tormarkets2025ukaz1/tormarkets2025 – bitcoin dark web
Pingrutty
| #
dark market 2025 https://github.com/aresmarketlink0ru72/aresmarketlink – tor drug market
Kxyufaita
| #
darkmarket 2025 dark websites
Donaldlaf
| #
darknet markets url https://github.com/darknetmarketlinks2025/darknetmarkets – dark web market links
ThomasVep
| #
В предверии праздника искал где купить цветы, нашёл этот магазин Доставка цветов в Томске теперь всегда буду заказывать здесь
1win_bhon
| #
игра ракета на деньги 1win https://www.aktivnoe.forum24.ru/?1-8-0-00000252-000-0-0-1741169084 .
LeroyVaf
| #
Сериалы онлайн без подписки – круто!
http://www.hive.cc/blog/log/eid492.html
Toliknaf
| #
darkmarkets https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet drug store
Garmincaree
| #
Современные смарт-часы Garmin – это надежные устройства, сделанные для удобного отслеживания активности и других данных. Но иногда их пользователи сталкиваются с техническими проблемами. Разберем основные из них и способы решения.
перейти
DonDonfaita
| #
bitcoin dark web https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet markets onion
Diplomi_fdKi
| #
купить аттестат в ростове на дону купить аттестат в ростове на дону .
Pingrutty
| #
darknet links https://github.com/aresmarketlink0ru72/aresmarketlink – darknet markets onion
LorenSip
| #
Ипотека без сложностей https://volexpert.ru Наши риэлторы помогут выбрать идеальную квартиру и оформить ипотеку на лучших условиях. Работаем с топовыми банками, сопровождаем сделку, защищаем ваши интересы. Делаем покупку недвижимости доступной!
prodvijenie saitov v moskve_wssn
| #
продвижение сайта цена продвижение сайта цена .
LeroyVaf
| #
Сериалы онлайн – лучшее средство от скуки.
https://forum.web.ru/memberlist.php?first_char&sd=a&sk=c&start=12000
Rabyitabe
| #
darknet drug store https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet market lists
Donaldlaf
| #
darkmarket link https://github.com/darknetmarketlinks2025/darknetmarkets – onion dark website
DavidAveby
| #
Надежное подтверждение места жительства без очередей https://registratsija-v-mosow177.ru/vremennaya-registratsiya-v-moskve/
Toliknaf
| #
dark web market https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark web link
mostbet_akot
| #
мостбет кыргызстан скачать cah.forum24.ru/?1-13-0-00001559-000-0-0 .
"https://lovewiki.faith/wiki/User:PaulaIgo910865"
| #
Грузовые шины китай оптом – широкий ассортимент и гарантия качества.
“https://ai-db.science/wiki/Shina_89y”
DonDonfaita
| #
darkmarkets https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darkmarkets
LeroyVaf
| #
Где найти лучшие криминальные сериалы онлайн?
http://www.empyrethegame.com/forum/memberlist.php?sd=a&sk=c&start=210675
Pingrutty
| #
dark market https://github.com/aresmarketlink0ru72/aresmarketlink – dark web drug marketplace
Rabyitabe
| #
darknet drugs https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet marketplace
Donaldlaf
| #
darknet drug market https://github.com/darknetmarketlinks2025/darknetmarkets – tor drug market
LeroyVaf
| #
Обожаю смотреть сериалы онлайн в HD.
http://pax.nichost.ru/forum/view_profile.php?UID=110648
Diplomi_ofer
| #
купить диплом колледжа в новосибирске 2orik-diploms.ru .
Toliknaf
| #
darkmarket url https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark web market urls
1win_snst
| #
1 win.pro 1 win.pro .
DonDonfaita
| #
bitcoin dark web https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – best darknet markets
Mazrbvz
| #
Где приобрести диплом специалиста? купить диплом в перми адреса
GregorySot
| #
Hemos establecido solo para ti preguntas solicitadas de residentes chilenos sobre codigo promocional Winchile y conclusiones útiles.
LeroyVaf
| #
Не могу оторваться от онлайн-сериалов!
https://russerial.online/9222-postuchis-v-moju-dver-2-sezon.html
AlfredBed
| #
Ваш гид по дизайну https://sales-stroy.ru строительству и ремонту! Советы профессионалов, актуальные тенденции, проверенные технологии и подборки лучших решений для дома. Всё, что нужно для комфортного и стильного пространства, на одном портале!
GregorySot
| #
Hemos compilado para ti preguntas predominantes de usuarios de Chile sobre codigo promocional Winchile y guías prácticas.
Daviddex
| #
Регистрация в соответствии с требованиями законодательства: временная регистрация человека
Rabyitabe
| #
dark web sites https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet drugs
Donaldlaf
| #
dark web sites https://github.com/darknetmarketlinks2025/darknetmarkets – darknet drug links
Terencesam
| #
Бездепозитные бонусы с выводом Бесплатные спины за регистрацию без депозита
Pingrutty
| #
darknet drug market https://github.com/aresmarketlink0ru72/aresmarketlink – dark web market links
LeroyVaf
| #
Какие сериалы посоветуете посмотреть онлайн?
http://www.empyrethegame.com/forum/memberlist.php?first_char=r&sd=a&sk=c&start=9475
Daviddex
| #
Официальные документы, необходимые для проживания и работы в Москве: московская временная регистрация
Toliknaf
| #
darknet market list https://github.com/darknetmarketlistv8tg0/darknetmarketlist – best darknet markets
DonDonfaita
| #
darknet drug links https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet site
Mazrmdo
| #
Здравствуйте!
Где купить диплом специалиста?
Мы можем предложить дипломы любых профессий по приятным тарифам. Цена может зависеть от той или иной специальности, года получения и образовательного учреждения. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Для нас важно, чтобы дипломы были доступны для подавляющей массы граждан.
Покупка диплома, который подтверждает окончание университета, – это грамотное решение. Для сравнения просто подсчитайте, сколько вам пришлось бы вложить денег на оплату 5-летнего обучения, на аренду жилья (если учащийся из другого города), на проезд до университета и многие другие издержки. Выйдет большая сумма, которая значительно превышает цены на наши дипломы. А ведь все эти годы можно уже успешно работать, занимаясь собственной карьерой.
Полученный диплом с нужными печатями и подписями целиком и полностью отвечает стандартам Министерства образования и науки Российской Федерации, неотличим от оригинала. Не следует откладывать свои мечты на потом, реализуйте их с нашей компанией – отправляйте простую заявку на изготовление диплома уже сегодня!
Заказать диплом о среднем специальном образовании – не проблема! rdiplomix.com/
Gkklcaf
| #
Мультфильмы, которые не отпустят!Портал Kadikama.su — ваш портал в мультяшную реальность! Тут да-а самый скептичный зритель залипнет на дни! Перематывайте мультики в HD, без заморочек — твой диван теперь мечта! Жмите > мультфильмы для детей! Новинки каждую пятницу — не-а
Rolandcaree
| #
DJ-пульты подвергаются высоким нагрузкам, особенно при частых выступлениях и транспортировках. Нередко поломки случаются из-за попадания влаги, механических повреждений или износа компонентов.
подробнее
Diplomi_boSa
| #
аттестат об основном общем образовании купить аттестат об основном общем образовании купить .
Pingrutty
| #
darknet markets onion https://github.com/aresmarketlink0ru72/aresmarketlink – darkmarket link
Rabyitabe
| #
dark market list https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet markets onion
Zaimuraspima
| #
На Одноклассниках в сообществе “Финансовые советы” прочитал про онлайн займ без отказа без проверки. Срочные 10 000 рублей на подарок получил благодаря отзывам участников группы. Удобное оформление и быстрый перевод решили проблему за считанные минуты.
Donaldlaf
| #
dark web marketplaces https://github.com/darknetmarketlinks2025/darknetmarkets – dark markets
Toliknaf
| #
tor drug market https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark market onion
AlbertCig
| #
Trazite pouzdane elektricni motori i bicikli crna gora? Imamo siroku paletu modela za razlicite zadatke. Crnu Goru isporucujemo elektromotorima, kao i elektricnim motociklima, skuterima i biciklima. Ekoloski prihvatljiv transport za udobno putovanje. Visokokvalitetni motori i komponente po konkurentnim cijenama. Dostava i konsultacije – kontaktirajte nas!
DonDonfaita
| #
dark web sites https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet marketplace
Zaimuraspima
| #
Впечатляет количество предложений срочных займов на карту онлайн без отказов. Нужные 19 000 рублей на ремонт авто получил быстро благодаря широкому выбору компаний. Удобное оформление и моментальное зачисление денег решили транспортную проблему.
Pingrutty
| #
dark web link https://github.com/aresmarketlink0ru72/aresmarketlink – darknet websites
Rabyitabe
| #
onion dark website https://github.com/tormarkets2025ukaz1/tormarkets2025 – dark web market links
Sazrbbq
| #
Добрый день!
Покупка диплома ВУЗа через надежную компанию дарит немало плюсов для покупателя. Это решение дает возможность сберечь как личное время, так и значительные финансовые средства. Однако, преимуществ значительно больше.Мы изготавливаем дипломы любых профессий. Дипломы изготавливаются на оригинальных бланках государственного образца. Доступная стоимость в сравнении с большими расходами на обучение и проживание. Приобретение диплома института станет мудрым шагом.
Купить диплом: zillow.com/profile/diplomygroup24
Donaldlaf
| #
dark markets https://github.com/darknetmarketlinks2025/darknetmarkets – dark web link
Xboxcaree
| #
Ремонт консолей Xbox в сервисном центре в Москве. Ремонт Xbox Series X 1TB Digital Edition (Robot White) Xbox Series S 1TB (Robot White) Xbox One X 1 TB
подробнее
Toliknaf
| #
darkmarket link https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet drug market
DavidAveby
| #
Надежная регистрация для граждан, прибывших в Москву: как сделать временную регистрацию
DonDonfaita
| #
darkmarket 2025 https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark market list
Pingrutty
| #
darknet drug market https://github.com/aresmarketlink0ru72/aresmarketlink – darknet websites
Rabyitabe
| #
darknet websites https://github.com/tormarkets2025ukaz1/tormarkets2025 – dark market url
Donaldlaf
| #
darknet drugs https://github.com/darknetmarketlinks2025/darknetmarkets – darknet markets
Pingrutty
| #
dark market list https://github.com/aresmarketlink0ru72/aresmarketlink – tor drug market
Toliknaf
| #
dark web market list https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet markets 2025
LeroyVaf
| #
Сериалы онлайн без подписки – круто!
https://forum.openbadania.pl/memberlist.php?mode=viewprofile&u=165009
DonDonfaita
| #
darknet markets https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darkmarket list
prodvijenie saitov v moskve_xgPa
| #
продвинуть сайт в москве продвинуть сайт в москве .
prodvijenie saitov v moskve_ukPl
| #
заказать продвижение сайта в топ заказать продвижение сайта в топ .
Terencesam
| #
bonuses casinos Бесплатные спины за регистрацию без депозита
Rabyitabe
| #
dark web market list https://github.com/tormarkets2025ukaz1/tormarkets2025 – darkmarkets
Donaldlaf
| #
dark web marketplaces https://github.com/darknetmarketlinks2025/darknetmarkets – tor drug market
Pingrutty
| #
darknet markets onion https://github.com/aresmarketlink0ru72/aresmarketlink – dark websites
Toliknaf
| #
darkmarkets https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark web market
DonaldSlubs
| #
купить игровой ноутбук asus tuf gaming https://igrovye-noutbuki.ru
DonDonfaita
| #
dark market link https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet markets 2025
Pingrutty
| #
darknet markets onion address https://github.com/aresmarketlink0ru72/aresmarketlink – dark websites
Rabyitabe
| #
bitcoin dark web https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet drug market
Donaldlaf
| #
dark web sites https://github.com/darknetmarketlinks2025/darknetmarkets – dark web market links
Toliknaf
| #
darknet market https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet market list
mostbet_gqoa
| #
мостбет скачать мостбет скачать .
DonDonfaita
| #
dark web market list https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet market
lkjhCype
| #
https://olimp-yug.ru/media/pgs/smotret_filmu_v_horoshem_kachestve_besplatno_onlayn.html снять квартиру на филевском парке
1win_gisa
| #
1win. 1win. .
Pingrutty
| #
dark web link https://github.com/aresmarketlink0ru72/aresmarketlink – dark market url
Rabyitabe
| #
darknet drug market https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet market
Donaldlaf
| #
darknet markets onion https://github.com/darknetmarketlinks2025/darknetmarkets – darkmarket
Toliknaf
| #
dark web market https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet drugs
avtostilshop
| #
Товары для вашего авто интернет-магазин автозапчастей автоаксессуары, масла, запчасти химия, электроника и многое другое. Быстрая доставка, акции и бонусы для постоянных клиентов. Подбирайте товары по марке авто и будьте уверены в качестве!
DonDonfaita
| #
dark web sites https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet drug links
Pingrutty
| #
dark web sites https://github.com/aresmarketlink0ru72/aresmarketlink – darknet site
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://3909102.ru/
Rabyitabe
| #
dark markets 2025 https://github.com/tormarkets2025ukaz1/tormarkets2025 – dark web drug marketplace
Donaldlaf
| #
dark market https://github.com/darknetmarketlinks2025/darknetmarkets – dark web sites
avtostilshop
| #
Товары для вашего авто avtostilshop.ru автоаксессуары, масла, запчасти химия, электроника и многое другое. Быстрая доставка, акции и бонусы для постоянных клиентов. Подбирайте товары по марке авто и будьте уверены в качестве!
1win casino mexico_jwpt
| #
1win mexico 1win mexico .
Uazrjgb
| #
Добрый день!
Для многих людей, заказать диплом ВУЗа – это необходимость, удачный шанс получить выгодную работу. Впрочем для кого-то – это очевидное желание не терять множество времени на учебу в ВУЗе. С какой бы целью вам это не потребовалось, мы готовы помочь. Максимально быстро, профессионально и по разумной цене изготовим диплом любого ВУЗа и года выпуска на настоящих бланках с реальными подписями и печатями.
Ключевая причина, почему многие прибегают к покупке документов, – желание занять хорошую работу. Предположим, навыки и опыт дают возможность специалисту устроиться на желаемую работу, но документального подтверждения квалификации нет. Когда для работодателя важно наличие “корочки”, риск потерять место работы очень высокий.
Заказать документ о получении высшего образования можно у нас. Мы оказываем услуги по производству и продаже документов об окончании любых университетов РФ. Вы сможете получить необходимый диплом по любым специальностям, любого года выпуска, включая документы старого образца. Гарантируем, что в случае проверки документа работодателем, каких-либо подозрений не возникнет.
Ситуаций, которые вынуждают заказать диплом ВУЗа очень много. Кому-то прямо сейчас нужна работа, в итоге нужно произвести впечатление на начальника при собеседовании. Некоторые хотят попасть в серьезную компанию, чтобы повысить свой статус в обществе и в последующем начать свой бизнес. Чтобы не терять драгоценное время, а сразу начать эффективную карьеру, применяя врожденные таланты и полученные навыки, можно купить диплом прямо в онлайне. Вы станете полезным для общества, получите денежную стабильность в минимальные сроки- купить диплом о среднем образовании
Cazrlqm
| #
Добрый день!
Без института трудно было продвинуться по карьере. На сегодняшний день документ не дает никаких гарантий, что удастся получить отличную работу. Более важны профессиональные навыки специалиста и его опыт. Именно из-за этого решение о покупке диплома следует считать целесообразным. Приобрести диплом любого университета kontenent.flybb.ru/viewtopic.php?f=2&t=553
Pingrutty
| #
darkmarket link https://github.com/aresmarketlink0ru72/aresmarketlink – darkmarket list
Cazrtqb
| #
Здравствуйте!
Мы изготавливаем дипломы любых профессий по приятным тарифам. Цена будет зависеть от той или иной специальности, года выпуска и образовательного учреждения: rdiplomix.com/
Toliknaf
| #
dark web market links https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark web marketplaces
Terencesam
| #
фриспины без депозита с выводом за регистрацию спины за регистрацию без депозита с выводом
Sazralz
| #
Купить диплом университета !
Эксперты с высшим образованием очень ценятся среди работодателей. Диплом университета нужен, чтобы доказать свою квалификацию. Он позволяет понять работодателю, что специалист обладает всеми необходимыми знаниями чтобы эффективно выполнить поставленные задачи. Но как же быть, когда знания присутствуют, а подтверждающего документа у опытного специалиста нет? Заказ диплома решит данную проблему. Приобретение диплома любого ВУЗа РФ в нашей компании – надежный процесс, поскольку документ заносится в государственный реестр. При этом печать осуществляется на специальных бланках, установленных государством. Приобрести диплом ВУЗа kuvandyk.ru/forums.php?m=posts&q=2567&n=last
DonDonfaita
| #
dark websites https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet market list
Diplomi_dwKt
| #
купить диплом в тамбове
Sazrejh
| #
аттестат купить в школе
Diplomi_ehSa
| #
купить аттестат о среднем образовании 10 классов купить аттестат о среднем образовании 10 классов .
1win_pxet
| #
win 1 http://cah.forum24.ru/?1-13-0-00001560-000-0-0-1741172791/ .
Chestercopsy
| #
Простая и удобная процедура регистрации по месту пребывания: как сделать временную регистрацию
Michaeldueft
| #
Такси для бизнеса https://versia.ru/sotrudniki-gibdd-smogut-izymat-avtomobili-po-novomu-reglamentu работа по всей России. Удобные поездки для сотрудников! Оформите корпоративный аккаунт и получите выгодные условия, детальную отчетность и надежный сервис. Быстрое бронирование, прозрачные тарифы, комфортные автомобили – организуйте рабочие поездки легко!
TimothyKassy
| #
Кризисное управление бизнесом БИЗНЕС_КОНСАЛТИНГ ДЛЯ УЛУЧШЕНИЯ ОТНОШЕНИЙ В КОЛЛЕКТИВЕ Бизнес-консалтинг для улучшения отношений в коллективе – это инвестиция в успех компании! Консультанты «Бизнес Доп» помогают выявить проблемные зоны, наладить коммуникацию и создать здоровую рабочую атмосферу. Эффективный консалтинг способствует росту продуктивности. Сплоченная команда, где налажены межличностные связи, работает быстрее и качественнее. Улучшение отношений – ключ к повышению прибыли. Наши бизнес-консультанты разрабатывают стратегии, направленные на сплочение коллектива. Это могут быть тренинги, тимбилдинги, а также внедрение новых методов коммуникации. Компании, инвестирующие в бизнес-консалтинг для налаживания отношений, получают конкурентное преимущество. Здоровый коллектив – залог стабильного развития и процветания бизнеса. С “Бизнес доп” вы получите не просто консультации, а реальные инструменты для улучшения отношений в коллективе!
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности https://acb-house.ru/
Chestercopsy
| #
Надежный способ получить документы без проблем https://registraceja-v-moskverus169.ru/
Donaldlaf
| #
darknet markets onion https://github.com/darknetmarketlinks2025/darknetmarkets – darknet markets 2025
Pingrutty
| #
darknet market links https://github.com/aresmarketlink0ru72/aresmarketlink – darknet site
Robertpax
| #
http://low-cost-airlines.ru/
Michaeldueft
| #
Такси для бизнеса https://www.kp40.ru/site/releases/pnews/90855/ работа по всей России. Удобные поездки для сотрудников! Оформите корпоративный аккаунт и получите выгодные условия, детальную отчетность и надежный сервис. Быстрое бронирование, прозрачные тарифы, комфортные автомобили – организуйте рабочие поездки легко!
lkjhCype
| #
https://turbocomp.ru/images/pgs/kiberpank__mir_budushego_na_grani_tehnologiy_i_chelovechnosti.html смотреть новинки кино бесплатно и без регистрации в хорошем качестве
Toliknaf
| #
dark web sites https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark web link
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://agape-dom.ru/
Sazrznp
| #
Нередко случается так, что для того, чтобы продвигаться вверх по карьерной лестнице, понадобится документ, который подтверждает наличие образования. Где купить диплом по нужной специальности?
Заказать документ о получении высшего образования можно в нашем сервисе. Мы оказываем услуги по производству и продаже документов об окончании любых ВУЗов России. cyberdefenders.org.ua/
DonDonfaita
| #
darknet links https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet market
Pingrutty
| #
dark market list https://github.com/aresmarketlink0ru72/aresmarketlink – dark markets
Rabyitabe
| #
dark web market links https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet markets
Markcen
| #
Новостройки или вторичка? Узнайте подробности https://amkproekb.ru/
Donaldlaf
| #
darknet market lists https://github.com/darknetmarketlinks2025/darknetmarkets – dark markets
ThomasVep
| #
покупать цветы советую в этой доставке, просто вышка
lkjhCype
| #
https://turbocomp.ru/images/pgs/kiberpank__mir_budushego_na_grani_tehnologiy_i_chelovechnosti.html the best movie of all time
ThomasFlime
| #
https://66.ru/news/misc/234501/
Toliknaf
| #
darknet site https://github.com/darknetmarketlistv8tg0/darknetmarketlist – best darknet markets
Roberturirl
| #
gay chaturnate
DonDonfaita
| #
darknet markets https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark web market urls
Sazrszr
| #
Здравствуйте!
Заказать диплом ВУЗа по выгодной цене возможно, обращаясь к проверенной специализированной компании. Заказать диплом: vuz-diplom.ru/kupit-diplom-saratov-3/
Markcen
| #
Новостройки или вторичка? Полезные рекомендации — https://an-gavan.ru/
Pingrutty
| #
darknet site https://github.com/aresmarketlink0ru72/aresmarketlink – dark web markets
Manuelges
| #
порно женщин купить гашиш
kovry_bdsr
| #
Ковры, которые добавят стиль в ваш интерьер, откройте.
Ковры, которые преобразят ваш интерьер, по выгодной цене.
Эко-дизайн: ковры из натуральных материалов, открывайте.
Декорируйте пространство с помощью ковров, уют.
Безопасные и яркие ковры для детской, разнообразие.
Традиционные и современные ковры, красоту.
Создание комфортного рабочего пространства с коврами, стиль.
Ковры, которые легко чистить, удобство.
Руководство по выбору ковров, рекомендации.
Теплота и уют с коврами, откройте.
Модные ковры 2025 года, узнайте.
Ковры для загородного дома, красоту.
Идеи по использованию ковров, креативность.
Разнообразие стилей ковров, погрузитесь в.
Создайте атмосферу уюта в спальне, найдите.
Качество и стиль от лучших производителей, успех.
Выбор ковров для домашних любимцев, долговечные.
Согревающие ковры для вашего дома, найдите.
Разделение пространства с помощью ковров, откройте.
цены на ковровые дорожки https://kovry-v-moskve.ru/ .
Gregoryhoaps
| #
детское порно инцест смотря порно смотрят порно
Roberturirl
| #
sexchat live
Rabyitabe
| #
darknet market links https://github.com/tormarkets2025ukaz1/tormarkets2025 – onion dark website
Donaldlaf
| #
dark market link https://github.com/darknetmarketlinks2025/darknetmarkets – darknet drugs
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы — https://anplatinum.ru/
Toliknaf
| #
dark web marketplaces https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darkmarket url
TCLcaree
| #
Заполните форму заявки на сайте TCL Repair Center или свяжитесь с нами для уточнения деталей. Мы позаботимся о вашей технике TCL и быстро вернем её в строй! Не упустите возможность воспользоваться скидкой до 25% при записи онлайн.
здесь
Roberturirl
| #
gayabdl
DonDonfaita
| #
darkmarket https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darkmarket url
Pingrutty
| #
darknet market links https://github.com/aresmarketlink0ru72/aresmarketlink – dark web markets
Diplomi_fvol
| #
купить диплом высшее дистанционно prema-diploms.ru .
Mazraoc
| #
Мы можем предложить дипломы психологов, юристов, экономистов и любых других профессий по приятным ценам. Стараемся поддерживать для покупателей адекватную политику тарифов. Важно, чтобы документы были доступны для большого количества наших граждан.
Приобретение документа, который подтверждает окончание ВУЗа, – это рациональное решение. Заказать диплом любого университета: tutdiploms.com
1win_dhSr
| #
1win войти 1win войти .
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности https://an-rusdom.ru/
Roberturirl
| #
sex cam live
Rabyitabe
| #
dark web marketplaces https://github.com/tormarkets2025ukaz1/tormarkets2025 – dark web drug marketplace
1win_rfEr
| #
1вин 1вин .
Donaldlaf
| #
dark web market https://github.com/darknetmarketlinks2025/darknetmarkets – darknet drug store
Sazrwwr
| #
Мы изготавливаем дипломы любых профессий по доступным тарифам.– http://www.tvniederbueren.ch/tvn/wp/?p=2624#comment-372780
Harrymoutt
| #
Онлайнказино Бонсай https://bonsai-casino2.com игровые автоматы, рулетка, покер и живые дилеры! Получайте бонусы за регистрацию, участвуйте в турнирах и выводите выигрыши без задержек. Надёжность, безопасность и азарт – всё в одном месте!
evakyatori spb_xwoa
| #
эвакуатор спб недорого https://www.evakuatormax.ru .
Pingrutty
| #
dark markets 2025 https://github.com/aresmarketlink0ru72/aresmarketlink – darknet drug links
Kennethbob
| #
Актуальні новини будівництва https://dverikupe.com.ua та нерухомості в Україні. Огляди ринку, тренди, технології, законодавчі зміни та експертні думки – все про будівельну галузь на одному сайті!
Toliknaf
| #
darknet markets 2025 https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark web marketplaces
Roberturirl
| #
free gay chaturbate
Irvingten
| #
Дізнавайтеся про останні новини https://ampdrive.info електромобілів, супекарів та актуальні події автомобільного світу в Україні. Ексклюзивні матеріали, фото та аналітика для справжніх автолюбителів на AmpDrive.?
DonDonfaita
| #
dark markets 2025 https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet markets
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы — https://bti-nsk.ru/
TCLcaree
| #
Заполните форму заявки на сайте TCL Repair Center или свяжитесь с нами для уточнения деталей. Мы позаботимся о вашей технике TCL и быстро вернем её в строй! Не упустите возможность воспользоваться скидкой до 25% при записи онлайн.
ссылка
Roberturirl
| #
sex cam live
Rabyitabe
| #
darknet market lists https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet markets onion
Donaldlaf
| #
dark market onion https://github.com/darknetmarketlinks2025/darknetmarkets – darknet market links
Pingrutty
| #
dark web marketplaces https://github.com/aresmarketlink0ru72/aresmarketlink – dark web market links
lkjhCype
| #
https://spbzn.ru/crm/pgs/?filmu_zagadki__magiya_tainstvennogo_kino.html нечто плохое дурное 3 буквы
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы https://crystalit34.ru/
PhillipNaf
| #
селезинка
Toliknaf
| #
darkmarket 2025 https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet websites
Roberturirl
| #
gay daddy cams
DonDonfaita
| #
dark web sites https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet site
GeorgeTom
| #
Source:
http://stroidom-blog.ru
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы — https://cspkaliningrad.ru/
Roberturirl
| #
live sex cams xxx
JamieFam
| #
лідії іменини
Pingrutty
| #
darknet markets onion https://github.com/aresmarketlink0ru72/aresmarketlink – darknet websites
Rabyitabe
| #
darknet drugs https://github.com/tormarkets2025ukaz1/tormarkets2025 – dark market 2025
Donaldlaf
| #
dark web sites https://github.com/darknetmarketlinks2025/darknetmarkets – darkmarket url
1win_anKi
| #
1winn https://1win10.am/ .
JamieFam
| #
іменини ольги за церковним календарем
Sazrubt
| #
купить диплом с занесением в реестр в нижнем тагиле
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы https://dachnoe-house.ru/
Toliknaf
| #
dark web drug marketplace https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet websites
Roberturirl
| #
gay nude webcam
DonDonfaita
| #
darkmarket 2025 https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet markets links
Terencesam
| #
Top casino с выводом фриспины без депозита
Pingrutty
| #
dark market https://github.com/aresmarketlink0ru72/aresmarketlink – dark market 2025
ремонт техники в мск
| #
Профессиональный сервисный центр по ремонту бытовой техники с выездом на дом.
Мы предлагаем:ремонт бытовой техники в мск
Наши мастера оперативно устранят неисправности вашего устройства в сервисе или с выездом на дом!
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации — https://fasad-alex.ru/
ThomasBor
| #
https://komiinform.ru/nt/7702
Rabyitabe
| #
darknet markets onion address https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet drug links
Donaldlaf
| #
onion dark website https://github.com/darknetmarketlinks2025/darknetmarkets – dark web drug marketplace
remontcaree
| #
Ремонт тепловизоров Pulsar в Москве: LRF XP50, XD50S, XD19S, XD38S, Lite XQ30V, XQ50F, XQ38F, XP38, XP28, Merger LRF XT50, Merger LRF XP50, Axion 2 LRF, Axion XM30F, Axion XQ30 PRO. Закажите звонок для бесплатной диагностики тепловизора pulsar подробнее
Toliknaf
| #
dark market onion https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark market link
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации — https://fisher-kp.ru/
FrankFiP
| #
https://wessta.ru
DonDonfaita
| #
dark market link https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark market link
Pingrutty
| #
darknet sites https://github.com/aresmarketlink0ru72/aresmarketlink – darknet links
Rabyitabe
| #
dark web drug marketplace https://github.com/tormarkets2025ukaz1/tormarkets2025 – dark web market links
Donaldlaf
| #
darknet drugs https://github.com/darknetmarketlinks2025/darknetmarkets – darknet markets url
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://fortuna-i-k.ru/
Pingrutty
| #
darknet site https://github.com/aresmarketlink0ru72/aresmarketlink – darknet markets links
PhillipNaf
| #
червона риба під шубою
Toliknaf
| #
darknet websites https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darkmarket url
Waynehem
| #
Актуальні новини політики https://insideukr.com в Україні та світі. Аналітика, думки експертів, головні події дня та ексклюзивні матеріали – будьте в курсі разом із нами!
WilliePar
| #
Honey Money Казино https://honeymoneycasino.ru это захватывающий мир азартных игр с щедрыми бонусами, быстрыми выплатами и огромным выбором слотов, рулетки, покера и других развлечений. Играй онлайн в любое время, участвуй в турнирах и получай эксклюзивные награды.
DonDonfaita
| #
dark market url https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet site
AnthonyBit
| #
Лучшие онлайн казино booi промокод рейтинг топовых игровых платформ с лицензией, щедрыми бонусами, быстрыми выплатами и широким выбором слотов, покера, рулетки и других азартных игр. Мы собрали проверенные казино с высоким рейтингом, безопасностью и выгодными акциями.
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы — https://hertz-lumen.ru/
Rabyitabe
| #
darknet links https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet markets onion
Donaldlaf
| #
best darknet markets https://github.com/darknetmarketlinks2025/darknetmarkets – darknet markets links
Pingrutty
| #
dark web drug marketplace https://github.com/aresmarketlink0ru72/aresmarketlink – darknet markets onion
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы https://homedecorsochi.ru/
Toliknaf
| #
darknet market [url=https://github.com/darknetmarketlistv8tg0/darknetmarketlist ]darknet websites [/url]
DonDonfaita
| #
darknet market https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark market list
Brianmon
| #
керамогранит 60×120 https://keramogranit213.ru
ShawnWhoks
| #
плитка керамическая настенная купить https://magazin-keramogranit.ru
Larryheive
| #
Купить телевизоры на дачу https://televizory-dlya-dachi.ru в интернет-магазине по низкой цене! При покупке дачных телевизоров на сайте воспользуйтесь скидками, акциями, бонусной программой.
1win casino mexico_evMa
| #
sitio oficial de 1win 1win7.com.mx .
1win_gfPl
| #
1win kg 1win kg .
1win_rama
| #
1 win pro http://www.1win16.com.kg .
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы — https://instrument75.ru/
Pingrutty
| #
darkmarket list https://github.com/aresmarketlink0ru72/aresmarketlink – dark web link
Dnrtcce
| #
Где купить диплом по актуальной специальности?
Мы изготавливаем дипломы любой профессии по доступным ценам. Мы предлагаем документы техникумов, которые расположены в любом регионе России. Вы имеете возможность заказать качественно напечатанный диплом за любой год, в том числе документы старого образца СССР. Документы выпускаются на “правильной” бумаге высшего качества. Это дает возможности делать государственные дипломы, не отличимые от оригинала. Они заверяются необходимыми печатями и подписями. Стараемся поддерживать для заказчиков адекватную политику цен. Для нас очень важно, чтобы дипломы были доступными для большинства наших граждан. diplom-profi.ru/kupit-diplom-v-bryanske-2-4
Rabyitabe
| #
darknet markets https://github.com/tormarkets2025ukaz1/tormarkets2025 – dark websites
Donaldlaf
| #
darknet market list https://github.com/darknetmarketlinks2025/darknetmarkets – darknet market
Terencesam
| #
Бонусы без отыгрыша за регистрацию Игровые автоматы лучшие
Toliknaf
| #
darkmarket list https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark web marketplaces
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности https://integral-sb.ru/
DonDonfaita
| #
darknet market links https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – best darknet markets
GeorgeTom
| #
Source:
http://stroidom-blog.ru
Pingrutty
| #
darknet drug store https://github.com/aresmarketlink0ru72/aresmarketlink – darkmarket list
Sazraem
| #
Мы можем предложить дипломы любой профессии по приятным тарифам. Цена зависит от определенной специальности, года выпуска и ВУЗа. Всегда стараемся поддерживать для клиентов адекватную политику тарифов. Важно, чтобы дипломы были доступными для большинства граждан. диплом с внесением в реестр
lkjhCype
| #
https://ecotruck.su/images/pgs/iz_rossii_s_lubovu__kino_90_h.html фильм где семья перевозила траву в фургоне
SEO PBN
| #
Thankyou, I will surround you
Rabyitabe
| #
darkmarket 2025 https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet links
Donaldlaf
| #
darkmarket link https://github.com/darknetmarketlinks2025/darknetmarkets – dark market list
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы https://kapplus.ru/
remontcaree
| #
Нужен качественный и оперативный ремонт тепловизоров Guide в Москве? Сервисный центр предлагает профессиональную помощь, доступную каждому владельцу термографических устройств бренда Guide. Мы устраняем все типы неисправностей, гарантируя высокое качество услуг и оперативные сроки работы.подробнее
WillieSarce
| #
Натяжные потолки с установкой https://natyazhnye-potolki2.ru стильное и практичное решение для любого интерьера. Предлагаем широкий выбор фактур и цветов, качественные материалы, быстрый монтаж и гарантию на работу. Устанавливаем потолки любой сложности в квартирах, домах, офисах.
Stephenmycle
| #
Отдых в Анапе https://otdyh-vanape.ru идеальный выбор для всей семьи! Чистые песчаные пляжи, теплое море, развитая инфраструктура и развлечения на любой вкус. Гостиницы, отели, частный сектор – найдите идеальное жилье.
1win_mfOn
| #
сайт 1win официальный сайт вход сайт 1win официальный сайт вход .
1win_orKa
| #
игра ракета на деньги 1win http://1win103.com.kg/ .
Toliknaf
| #
darknet markets links https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark web markets
Jamesbluem
| #
Музыкальный проект “Дягилев”
DonDonfaita
| #
darknet markets url https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark web market links
Pingrutty
| #
dark web link https://github.com/aresmarketlink0ru72/aresmarketlink – darknet drugs
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://kp-novoe-sonino.ru/
DanielExons
| #
хороший недорогой телевизор 43 недорогие телевизоры 43 дюйма
WilliamRon
| #
https://sqlmaxipro.pro/
Trefwpl
| #
Купить документ института можно у нас в Москве. diplomk-vo-vladivostoke.ru/kupit-shkolnij-attestat-2-2
Lazrprm
| #
Где заказать диплом специалиста?
Купить диплом ВУЗа по доступной стоимости вы можете, обращаясь к проверенной специализированной фирме.: sdiplom.ru
WilliamDix
| #
Мы изготавливаем дипломы любых профессий по приятным тарифам. Дипломы производятся на фирменных бланках Приобрести диплом об образовании diplomt-tver69.ru
Rabyitabe
| #
dark web markets https://github.com/tormarkets2025ukaz1/tormarkets2025 – bitcoin dark web
Donaldlaf
| #
darknet markets links https://github.com/darknetmarketlinks2025/darknetmarkets – dark web markets
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://kvartiranahodynke.ru/
1win_rdsl
| #
1 win официальный сайт 1win17.com.kg .
Pingrutty
| #
dark market url https://github.com/aresmarketlink0ru72/aresmarketlink – darknet websites
Toliknaf
| #
darknet drug market https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet links
Xazriuy
| #
Купить диплом ВУЗа!
Мы изготавливаем дипломы любых профессий по выгодным ценам. Вы покупаете документ в надежной и проверенной временем компании. : diplom-zentr.com/kupit-diplom-gumf-o-visshem-obrazovanii-na-blanke-goznak/
DonDonfaita
| #
darknet drug market https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark web market urls
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://lume42.ru/
Life Experience Degree
| #
The wrongful convictions of hundreds of sub-postmasters and sub-postmistresses in the recent Post Office scandal demonstrates what can happen when the victim also plays the role of prosecutor, as happens in TV licensing cases.
https://www.oneidauniversity.com/
Life Experience Degrees
| #
Fantastic beat ! I would like to apprentice whilst you amend your site, how can i subscribe for a weblog web site? The account aided me a appropriate deal. I had been tiny bit acquainted of this your broadcast provided vibrant clear idea.
https://www.charlestonstateuniversity.com/
Danielbor
| #
Все о компьютерных играх https://lifeforgame.ru обзоры новых проектов, рейтинги, детальные гайды, новости индустрии, анонсы и системные требования. Разбираем особенности геймплея, помогаем с настройками и прохождением. Следите за игровыми трендами, изучайте секреты и погружайтесь в мир гейминга.
lkjhCype
| #
https://lepidopterolog.ru/media/pgs/kinovselennaya_marvel__istoriya__filmu_i_vliyanie_na_pop_kulturu.html триггер все сезоны смотреть онлайн бесплатно в хорошем качестве
JessieEnlib
| #
телевизор 32 smart tv m1 32 https://televizory-smart-tv.ru
Eanrxwo
| #
Для эффективного продвижения вверх по карьерной лестнице требуется наличие официального диплома о высшем образовании. Быстро купить диплом университета у проверенной фирмы: diplom-club.com/kupit-diplom-v-rostove-na-donu-10/
WilliamGoorm
| #
телевизор lg oled 65 oled телевизор обзор
Rabyitabe
| #
darknet markets https://github.com/tormarkets2025ukaz1/tormarkets2025 – best darknet markets
Donaldlaf
| #
darknet links https://github.com/darknetmarketlinks2025/darknetmarkets – darknet drug links
1win_hnEn
| #
что делать с бонусным балансом на 1win https://1win111.com.kg/ .
Pingrutty
| #
dark websites https://github.com/aresmarketlink0ru72/aresmarketlink – bitcoin dark web
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы — https://mastera-taganroga.ru/
Toliknaf
| #
dark markets https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark web drug marketplace
opticcaree
| #
Ремонт оптических и коллиматорных прицелов Sightmark перейти
DonDonfaita
| #
dark market https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark web market urls
ThomasBor
| #
Dota2
Pingrutty
| #
dark market 2025 https://github.com/aresmarketlink0ru72/aresmarketlink – dark markets 2025
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности https://metallservis38.ru/
Rabyitabe
| #
dark web market list https://github.com/tormarkets2025ukaz1/tormarkets2025 – darkmarket url
ThomasVep
| #
попробуйте эту доставку купить цветы всегда свежие цветочки, быстрая и бесплатная доставка, сервис огонь!
Donaldlaf
| #
dark web market links https://github.com/darknetmarketlinks2025/darknetmarkets – darkmarket
FrankFiP
| #
https://holodokdv.ru
Toliknaf
| #
dark market onion https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark web market urls
Jariorghr
| #
Добрый день!
Руководители серьезных предприятий довольно часто выбирают соискателей, окончивших университет. Особенно в приоритете престижные учебные заведения. Однако учиться целых пять лет – это дорого, далеко не у всех есть такая возможность. Приобрести документ – оптимальный выход.
Бывают и непредвиденные случаи, когда диплом утерян. Далеко не всегда возможно оперативно и без осложнений восстановить его, особенно если университет закрыт или располагается в другом регионе России. Бюрократия отнимает массу времени и нервов.
Для максимально быстрого продвижения вверх по карьерной лестнице потребуется наличие официального диплома института. Но зачастую в жизни случается так, что сложные обстоятельства не позволяют с успехом окончить учебу, получив важный документ.
Купить диплом о высшем образовании
Наши специалисты предлагают выгодно купить диплом, который выполнен на бланке ГОЗНАКа и заверен печатями, штампами, подписями должностных лиц. Диплом пройдет любые проверки, даже с использованием специфических приборов. Решайте свои задачи максимально быстро с нашей компанией.
Где купить диплом специалиста? rdiplomix.com/
Iariorzgj
| #
Купить документ о получении высшего образования вы имеете возможность в нашем сервисе. misplacedxsheath.dreamwidth.org/750.html?mode=reply
DonDonfaita
| #
darknet site https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark web link
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации — https://mielramenki.ru/
Sazrmui
| #
Студенческая жизнь прекрасна, пока не приходит время писать диплом, как это случилось со мной. Не стоит отчаиваться, ведь существуют компании, которые помогают с написанием и защитой диплома на высокие оценки!
Сначала я искал информацию по теме: купить диплом колледжа цена, диплом медсестры с занесением в реестр спб, купить дипломы колледжа, купить официальный диплом о среднем специальном образовании, купить диплом пту с занесением в реестр, а потом наткнулся на diplomybox.com/kupit-nastoyashhij-diplom
Pingrutty
| #
dark web market urls https://github.com/aresmarketlink0ru72/aresmarketlink – darknet drug store
RonaldToulp
| #
Опытные разработчики Битрикс помогут перенести ваш бизнес в онлайн, все условия сотрудничества по ссылке https://addons.mozilla.org/ru/firefox/user/18650346/
Rabyitabe
| #
dark web market list https://github.com/tormarkets2025ukaz1/tormarkets2025 – dark websites
Donaldlaf
| #
dark web market links https://github.com/darknetmarketlinks2025/darknetmarkets – dark market link
RonaldToulp
| #
Собственное производство наградной продукции для любых мероприятий. Ассортимент по ссылке https://roomstyler.com/users/nagradion
Markcen
| #
Хотите сдать квартиру в аренду? Читайте проверенные советы https://moderntechnology22.ru/
lkjhCype
| #
http://kuzovkirov.ru/wp-content/pgs/romanticheskie_komedii__spisok_luchshih_smeshnuh_filmov_o_lubvi.html канал мультимания программа передач на сегодня москва
Toliknaf
| #
dark web marketplaces https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark web markets
DonDonfaita
| #
dark market list https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark web sites
BillyShone
| #
Все о недвижимости https://ks-inginiring.ru покупка, аренда, ипотека. Разбираем рыночные тренды, юридические тонкости, лайфхаки для выгодных сделок. Помогаем выбрать квартиру, рассчитать ипотеку, проверить документы и избежать ошибок при сделках с жильем. Актуальные статьи для покупателей, арендаторов и инвесторов.
DavidWog
| #
Все о недвижимости https://brigantina-stroy.ru покупка, аренда, ипотека. Разбираем рыночные тренды, юридические тонкости, лайфхаки для выгодных сделок. Помогаем выбрать квартиру, рассчитать ипотеку, проверить документы и избежать ошибок при сделках с жильем. Актуальные статьи для покупателей, арендаторов и инвесторов.
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://monolitrealestate.ru/
Stevenson
| #
https://vavadaonc10.fun/
Donaldlaf
| #
darknet market links https://github.com/darknetmarketlinks2025/darknetmarkets – darknet drug links
eskort_swma
| #
эскорт услуги москва эскорт услуги москва .
Pingrutty
| #
dark market https://github.com/aresmarketlink0ru72/aresmarketlink – dark market url
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности https://morozova26.com/
Toliknaf
| #
darknet marketplace https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet websites
DonDonfaita
| #
darknet drug store https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet links
GeorgeTom
| #
Source:
http://ic51.ru
DavidNes
| #
Все о компьютерных играх https://lifeforgame.ru/ обзоры новых проектов, рейтинги, детальные гайды, новости индустрии, анонсы и системные требования. Разбираем особенности геймплея, помогаем с настройками и прохождением. Следите за игровыми трендами, изучайте секреты и погружайтесь в мир гейминга.
Rabyitabe
| #
darknet drug links https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet market list
Markcen
| #
Хотите сдать квартиру в аренду? Читайте проверенные советы https://new-fstore.ru/
Toliknaf
| #
darkmarket url https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darkmarkets
DonDonfaita
| #
darkmarket 2025 https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet markets onion
Markcen
| #
Новостройки или вторичка? Узнайте подробности https://okkord.ru/
Pingrutty
| #
dark web market https://github.com/aresmarketlink0ru72/aresmarketlink – dark market
Rabyitabe
| #
dark markets 2025 https://github.com/tormarkets2025ukaz1/tormarkets2025 – dark web market list
DavidWog
| #
Все о недвижимости https://knyaz-dolgoruky.ru покупка, аренда, ипотека. Разбираем рыночные тренды, юридические тонкости, лайфхаки для выгодных сделок. Помогаем выбрать квартиру, рассчитать ипотеку, проверить документы и избежать ошибок при сделках с жильем. Актуальные статьи для покупателей, арендаторов и инвесторов.
opendance
| #
зумба тренировка научиться шаффл
sharedsteam
| #
Looking for free steam account? We regularly share working accounts with games, bonuses, and tips. Subscribe now and don’t miss new giveaways! Only verified and active accounts.
Markcen
| #
Новостройки или вторичка? Полезные рекомендации — https://ooo-sk-micom.ru/
lkjhCype
| #
https://naturalbeekeeping.ru/wp-content/pgs/filmu_140.html список фильмов которые должен посмотреть каждый
Herbertargum
| #
кофемашина philips
Toliknaf
| #
darknet markets https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darkmarket link
DonDonfaita
| #
darkmarket link https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet links
Pingrutty
| #
dark market 2025 https://github.com/aresmarketlink0ru72/aresmarketlink – dark market onion
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы — https://privateconstruction.ru/
Donaldlaf
| #
dark market link https://github.com/darknetmarketlinks2025/darknetmarkets – dark web drug marketplace
Rabyitabe
| #
best darknet markets https://github.com/tormarkets2025ukaz1/tormarkets2025 – dark web marketplaces
servicecaree
| #
Кофемашины Jura — это эталоны надежности, технологичности и дизайна. Сервисный центр Jura Repair Center предлагает профессиональный ремонт кофемашин Jura, чтобы ваша техника снова радовала вас безупречным кофе. подробнее
взять микрозайм без отказа на карту
| #
Срочная новость из мира финансов! Известный экономист раскрыл секрет займа всем без отказа. Проверенные МФО с гослицензией выдают до 30 000 рублей под 0.8% в день. Главное преимущество – официальное оформление и защита от коллекторов.
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы — https://prudmarket.ru/
Toliknaf
| #
darknet markets links https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darkmarket 2025
Pingrutty
| #
dark market list https://github.com/aresmarketlink0ru72/aresmarketlink – darkmarket 2025
DonDonfaita
| #
dark web drug marketplace https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark web link
Diplomi_kxer
| #
купить диплом 2015 купить диплом 2015 .
Rabyitabe
| #
darkmarket url https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet marketplace
Markcen
| #
Новостройки или вторичка? Узнайте подробности https://pultmaster-pro.ru/
MetaMask Download
| #
MetaMask Extension is a game-changer! It allows me to interact with dApps without compromising security. Highly recommend it!
Terencesam
| #
бонусы за регистрацию Бонусы без отыгрыша за регистрацию
Pingrutty
| #
darkmarket list https://github.com/aresmarketlink0ru72/aresmarketlink – darkmarket
Toliknaf
| #
dark market onion https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark markets 2025
ThomasKef
| #
компот рецепт Eda321 – ваш незаменимый помощник в мире кулинарии! Мы собрали для вас богатую коллекцию рецептов, способных удовлетворить любой вкус и уровень подготовки. Откройте для себя простые, вкусные и быстрые рецепты, идеально подходящие для приготовления блюд на каждый день. Наша домашняя кухня – это кладезь идей для тех, кто ценит традиционные русские блюда. Начинающим кулинарам мы предлагаем рецепты с подробными фото, которые помогут освоить азы кулинарного искусства. Опытные кулинары найдут для себя интересные и оригинальные блюда из мяса, рыбы, курицы и картофеля. На Eda321 вы всегда найдете вдохновение для кулинарных экспериментов и сможете порадовать своих близких вкусными и полезными блюдами. Присоединяйтесь к нам, и вместе мы откроем новые горизонты в мире кулинарии!
Markcen
| #
Новостройки или вторичка? Полезные рекомендации — https://remont-nvr.ru/
DavidWog
| #
Все о недвижимости https://magdesi.ru покупка, аренда, ипотека. Разбираем рыночные тренды, юридические тонкости, лайфхаки для выгодных сделок. Помогаем выбрать квартиру, рассчитать ипотеку, проверить документы и избежать ошибок при сделках с жильем. Актуальные статьи для покупателей, арендаторов и инвесторов.
Davidnip
| #
телевизоры samsung qled 4k https://televizory-qled.ru
RobertRon
| #
Все о компьютерных играх lifeforgame.ru обзоры новых проектов, рейтинги, детальные гайды, новости индустрии, анонсы и системные требования. Разбираем особенности геймплея, помогаем с настройками и прохождением. Следите за игровыми трендами, изучайте секреты и погружайтесь в мир гейминга.
FrankFiP
| #
https://anrespectplus.ru
Rabyitabe
| #
darkmarket link https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet sites
Pingrutty
| #
darknet market list https://github.com/aresmarketlink0ru72/aresmarketlink – dark market list
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://repinskieozera.ru/
Toliknaf
| #
dark web market urls https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet links
DonDonfaita
| #
dark web markets https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darkmarkets
1win md_pooi
| #
1win casino http://www.1win6.md/ .
1win md_tgkn
| #
1win.com официальный сайт https://1win7.md .
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://sanrival.ru/
Duncanmor
| #
можно ли отправить посылку в россию Бесплатные путешествия — реальность! Я расскажу как без затрат посещать теплые страны. Многие россияне обосновались на Бали, и им часто требуется доставка вещей из России. За транспортировку они готовы платить от 20 долларов за килограмм или фиксированную сумму за посылку. Берем два чемодана, заполняем их посылками, и вот мы уже на Бали, да еще и с прибылью!
Rabyitabe
| #
darknet markets links https://github.com/tormarkets2025ukaz1/tormarkets2025 – dark web market urls
Donaldlaf
| #
darknet market lists https://github.com/darknetmarketlinks2025/darknetmarkets – darknet drug store
Pingrutty
| #
dark web market list https://github.com/aresmarketlink0ru72/aresmarketlink – dark web market urls
GeorgeTom
| #
Source:
– http://ic51.ru
servicecaree
| #
Почему не работает робот-пылесос: ТОП 8 самых расспространенных причин, которые мы сможем решить самостоятельно. здесь
Times Startup
| #
Cek my web blog rn
Herbertargum
| #
холодильники в молдове
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные статьи и советы — https://servit74.ru/
Toliknaf
| #
dark websites https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet markets 2025
DonDonfaita
| #
darknet markets url https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark market onion
Pingrutty
| #
darknet links https://github.com/aresmarketlink0ru72/aresmarketlink – dark web market list
Donaldlaf
| #
dark web sites https://github.com/darknetmarketlinks2025/darknetmarkets – darknet websites
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы — https://shatimtrade-kizlyar.ru/
lkjhCype
| #
https://astorplace.ru/wp-content/pgs/filmu_onlayn___novuy_uroven_domashnego_kinoteatra.html киноафиша спб плаза жемчужная сегодня
Toliknaf
| #
darknet markets url https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark web market urls
Markcen
| #
Как выбрать надёжного застройщика? Читайте проверенные советы https://sibotdelka.ru/
1win_kber
| #
1win онлайн http://1win11.am .
Pingrutty
| #
best darknet markets https://github.com/aresmarketlink0ru72/aresmarketlink – darknet sites
DonDonfaita
| #
darknet market links https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark web drug marketplace
1win_ilSt
| #
1win armenia https://1win13.am/ .
1win_ebot
| #
1вин официальный регистрация 1win5.am .
lifeforgameru
| #
Мир компьютерных игр http://lifeforgame.ru Мы расскажем о лучших новинках, секретах прохождения, системных требованиях и игровых трендах. Новости, гайды, обзоры и рейтинги – всё, что нужно геймерам.
JeremyGew
| #
Покупка, аренда, ипотека https://geodizond.ru всё о недвижимости в одном блоге! Советы по выбору жилья, юридические аспекты, анализ цен и прогнозы рынка. Рассказываем, как грамотно оформить ипотеку, проверить документы и избежать ошибок при сделках с недвижимостью. Будьте в курсе всех изменений и трендов!
Donaldlaf
| #
darknet marketplace https://github.com/darknetmarketlinks2025/darknetmarkets – darknet links
AnthonyJuink
| #
уроки шаффла школа танцев
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы https://snegirieko.com/
Pingrutty
| #
darknet drugs https://github.com/aresmarketlink0ru72/aresmarketlink – darkmarket link
Toliknaf
| #
darknet marketplace https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark web market urls
phoenixautobodyshopmailm
| #
Unlock your vehicle’s potential with our top-notch auto tuning services! Unleash your ride into a stunning masterpiece with our expert team. We specialize in modifications that cater to your unique style. Our exceptional solutions ensure optimal performance and eye-catching design . Don’t settle for average; elevate your driving experience and turn heads on the road! Visit us at https://americasbestcertifiedautobody.com/ today and take the first step towards your dream car!
DonDonfaita
| #
bitcoin dark web https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark web marketplaces
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы — https://so35.ru/
FrankFiP
| #
https://химчисткамебели.online
Rabyitabe
| #
darkmarket https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet market links
Pingrutty
| #
darknet markets 2025 https://github.com/aresmarketlink0ru72/aresmarketlink – darkmarkets
Stevenson
| #
https://vavada37.com/
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации — https://sochi-park-dom.ru/
Terencesam
| #
1xslots бездепозитный бонус Играть игровые автоматы на деньги
1win_jlpr
| #
1win.am http://1win12.am/ .
Toliknaf
| #
darknet websites https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darkmarket 2025
LarryThope
| #
купить волосы в спб Купить волосы СПБ! В Санкт-Петербурге: новый уровень вашего стиля! Хотите обновить свой образ и добавить яркие акценты? Приобретение волос в СПб — это отличное решение для вас! Мы предлагаем широкий ассортимент натуральных волос, идеально подходящих для наращивания, создания причесок и стильных аксессуаров.
DonDonfaita
| #
darkmarkets https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark websites
JamesAlirl
| #
Играйте в онлайн-казино https://t.me/casino_na_ruble/11 с рублевыми счетами! Надежные казино с моментальными выплатами, бонусами и фриспинами. Пополнение через карты, кошельки и криптовалюту. Выбирайте топовые игры и выигрывайте без лишних комиссий!
Billybes
| #
Онлайн казино игровые автоматы играть бесплатно с лучшими бонусами и шансом на крупный выигрыш! Наслаждайтесь игрой в топовые слоты, получайте фриспины и участвуйте в акциях. Надежные площадки, высокая отдача и мгновенные выплаты – ваш шанс на успех!
JeffreyUnimb
| #
Играйте в онлайн-казино https://t.me/casino_bonus_bezdep/11 топовые игровые автоматы, лайв-дилеры, мгновенные выплаты и бонусы для новых игроков. Наслаждайтесь честной игрой, удобными платежными методами и крупными выигрышами!
Galenboows
| #
эскорт услуги
модели эскорт
Rabyitabe
| #
darknet markets https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet markets onion address
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные статьи и советы — https://technovoda.ru/
Pingrutty
| #
dark web marketplaces https://github.com/aresmarketlink0ru72/aresmarketlink – darknet links
Georgeuncog
| #
баня цена
lkjhCype
| #
https://www.urank.ru/pgs/dlinnue_serialu___pochemu_mu_ih_tak_lubim_.html синема 5 энгельс облака афиша на сегодня
servicecaree
| #
Ремонт серверов HP и другой электроники HP в Москве – Сервисный центр HP здесь
Toliknaf
| #
best darknet markets https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet markets
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности https://territoria-we-stars.ru/
DonDonfaita
| #
darknet drug store https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark web link
SergioGaf
| #
волосы для наращивания Волосы для наращивания – это не просто тренд, это возможность мгновенно преобразиться, добавить объема и длины, о которой вы всегда мечтали. Существует множество техник: от горячего и холодного наращивания до ленточного и микрокапсульного, каждая из которых имеет свои преимущества и подходит для разных типов волос.
Pingrutty
| #
darkmarket list https://github.com/aresmarketlink0ru72/aresmarketlink – darknet market links
Stevenson
| #
vavada jogo
sohbet
| #
great blog very nice articles I will follow you continuously sohbet odaları
Rabyitabe
| #
darkmarket 2025 https://github.com/tormarkets2025ukaz1/tormarkets2025 – darkmarket 2025
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации — https://texnotect.ru/
Toliknaf
| #
darknet market https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark market list
Pingrutty
| #
darknet site https://github.com/aresmarketlink0ru72/aresmarketlink – dark market 2025
DonDonfaita
| #
dark web markets https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark market list
Markcen
| #
Как выбрать надёжного застройщика? Читайте проверенные советы https://tolmi74.ru/
Galenboows
| #
вип эскорт агентство
эскорт москва цены
1win_uwei
| #
1winn http://www.1win6.am .
1win_lksn
| #
баланс 1win https://www.1win7.am .
Rabyitabe
| #
dark market url https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet drug links
Samsungcaree
| #
На телефоне Samsung некорректно работает сенсорный экран: 8 причин и методов исправить самостоятельно перейти
Sazrbsc
| #
купить бланк аттестат
Galenboows
| #
сайт эскорт услуг
вип эскорт
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы — https://topkadastr-crimea.ru/
Pingrutty
| #
darknet sites https://github.com/aresmarketlink0ru72/aresmarketlink – darknet drug links
Toliknaf
| #
darkmarket https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet links
1win_vnPn
| #
1 win mexico 1 win mexico .
DonDonfaita
| #
darknet site https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet markets
1win_vdei
| #
1win casino mexico http://1win5.com.mx/ .
Donaldlaf
| #
darknet market list https://github.com/darknetmarketlinks2025/darknetmarkets – dark web market list
lkjhCype
| #
https://histor-ru.ru/wp-content/pgs/serialu_pro_turmu___zahvatuvaushiy_mir_za_resh_tkoy.html смотреть онлайн фильмы 2024 года бесплатно в хорошем качестве новинки
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://uk-arbat.ru/
Galenboows
| #
элитный эскорт
эскорт агенство
Pingrutty
| #
darknet market https://github.com/aresmarketlink0ru72/aresmarketlink – dark web markets
Andrewmic
| #
Glory Casino
Toliknaf
| #
dark websites https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet markets onion address
Andrewmic
| #
Glory Casino
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://vega-snab.ru/
DonDonfaita
| #
dark web sites https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet markets onion
GoogleMapscaree
| #
Где отремонтировать сложную оптику: тепловизоры, прицелы, дальномеры, коллиматоры, монокуляры. Вот на гугл картках отметка сервисного центра карта
AubreyReoft
| #
The best casino https://t.me/new_retro_casino_site/14! Classic slot machines, generous bonuses and reliable payouts. Enjoy the excitement of retro slots and play on a proven platform!
Donaldlaf
| #
dark web link https://github.com/darknetmarketlinks2025/darknetmarkets – dark market list
Rabyitabe
| #
darknet markets links https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet links
VictorCaf
| #
Организация офисных переездов под ключ с грузчиками и специализированным транспортом, подробности тут https://www.imdb.com/user/ur189257174/?ref_=nv_usr_prof_2
Pingrutty
| #
darknet drug market https://github.com/aresmarketlink0ru72/aresmarketlink – darknet site
Raymondnus
| #
Все, что вам нужно для игры https://t.me/vavada_casino_vhod/11! Игровые автоматы, рулетка, покер, живые дилеры и эксклюзивные бонусы. Наслаждайтесь качественной игрой с мгновенными выплатами и надежными провайдерами!
phoenixautobodyshopmailm
| #
Unlock your vehicle’s potential with our top-notch auto tuning services! Enhance your ride into a stunning masterpiece with our expert team. We specialize in refinements that cater to your unique style. Our high-quality solutions ensure optimal performance and stylish look . Don’t settle for average; elevate your driving experience and turn heads on the road! Visit us at https://americasbestcertifiedautobody.com/ today and take the first step towards your dream car!
WilliamdremI
| #
Играйте онлайн в JVSpin Казино – лицензионное онлайн-казино с лучшими слотами, лайв-играми и щедрыми бонусами. Пополняйте счет удобными способами, получайте награды и выигрывайте реальные деньги!
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности https://vernadskogo-loft.ru/
VictorCaf
| #
Инженерные решения для безопасной прокладки кабелей, подробности тут https://pixai.art/@tmgroup/artworks
Cazrrou
| #
Мы готовы предложить документы любых учебных заведений, расположенных на территории всей РФ. diplomoz-197.com/kupit-diplom-v-novorossijske-4-2
Toliknaf
| #
dark markets https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet market list
RobertWag
| #
кайт школа анапа
DonDonfaita
| #
darknet marketplace https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet markets links
1win_mbsa
| #
1 win casino http://1win4.com.mx .
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://worldstone33.ru/
Pingrutty
| #
dark websites https://github.com/aresmarketlink0ru72/aresmarketlink – darknet markets
Rabyitabe
| #
tor drug market https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet markets 2025
lkjhCype
| #
http://spincasting.ru/core/art/index.php?filmu_pro_biznes__vdohnovenie_i_uroki_dlya_predprinimateley.html телевизор на андроиде 43 дюйма купить
MalcolmWag
| #
Купить солнечные панели
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности https://xn—-7sbajm1bmmamhew.xn--p1ai/
repaircentercaree
| #
Если вам нужна помощь с техникой MSI, вы обратились по адресу. Наш сервисный центр MSI специализируется на ремонте ноутбуков, компьютеров, видеокарт, материнских плат, мониторов, моноблоков и планшетов MSI. Мы используем только оригинальные запчасти и предоставляем долгосрочные гарантии на ремонт. здесь
GeorgeTal
| #
Играйте в https://t.me/casino_jozz/7 популярное онлайн-казино с широким выбором слотов, настольных игр и лайв-дилеров. Выгодные бонусы, удобные платежные системы и моментальные выплаты ждут вас!
Toliknaf
| #
darkmarket link https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet market
Jameszer
| #
Выбирайте https://t.me/booi_casino_site/10 топовое онлайн-казино с выгодными акциями, эксклюзивными играми и лайв-казино. Присоединяйтесь к сообществу игроков и получайте максимальное удовольствие от игры!
Richardwit
| #
A powerful VPN solution https://github.com/ivanti-it/Ivanti-Secure-Access-Client/releases designed to provide secure remote access to corporate networks. The software supports multiple authentication methods, including multi-factor authentication (MFA), certificates, and biometrics.
VictorLop
| #
Сброс лишнего веса, расстройства пищевого поведения, психологическая коррекция, РПП, избыточная масса тела, помощь психолога. лишний вес
Pingrutty
| #
dark markets 2025 https://github.com/aresmarketlink0ru72/aresmarketlink – darknet markets links
DonDonfaita
| #
darknet markets links https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet links
Donaldlaf
| #
dark market link https://github.com/darknetmarketlinks2025/darknetmarkets – dark market onion
Rabyitabe
| #
dark market 2025 https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet site
LarryTub
| #
Свіжі ідеї дизайну https://dverikupe.od.ua інтер’єру, сучасні тренди, поради щодо декору та ремонту. Створюйте затишок та стиль у кожному куточку вашого дому!
Markcen
| #
Новостройки или вторичка? Полезные рекомендации — https://xn—-7sbccdscdoffrg1cybj3z.xn--p1ai/
GeorgeTom
| #
Source:
http://ic51.ru
Pingrutty
| #
darkmarket link https://github.com/aresmarketlink0ru72/aresmarketlink – darknet marketplace
Toliknaf
| #
dark web market urls https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet drug market
CharlesCam
| #
диодная лазерная эпиляция спб интимная лазерная эпиляция цены
JamesEvork
| #
свіжі новини криптовалют https://cryptonews.v.ua блокчейна та DeFi. Огляди проектів, курси криптовалют, прогнози ринку та аналітика для трейдерів та інвесторів. Слідкуйте за криптомир з нами!
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные рекомендации — https://xn--90aimcoinaikf.xn--p1ai/
Terencesam
| #
bezdep bonus casino
DonDonfaita
| #
darkmarket list https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – tor drug market
FrankFiP
| #
https://ceramodecol.ru
Donaldlaf
| #
darknet marketplace https://github.com/darknetmarketlinks2025/darknetmarkets – darknet markets links
Rabyitabe
| #
dark market 2025 https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet links
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://xn——6cdbaabncc2b3blicg2a2b7axf0dzfui.xn--p1ai/
Pingrutty
| #
darknet markets onion https://github.com/aresmarketlink0ru72/aresmarketlink – darknet market
Toliknaf
| #
darknet drug store https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet market
GregoryGab
| #
типографии полного цикла в спб типография заказать
sohbet
| #
great blog very nice articles I will follow you continuously sohbet odaları
DonDonfaita
| #
darkmarket url https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – dark markets 2025
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы https://xn—-8sbqbliyb2m.xn--p1ai/
Rabyitabe
| #
darknet market lists https://github.com/tormarkets2025ukaz1/tormarkets2025 – darkmarket url
Donaldlaf
| #
darknet drug links https://github.com/darknetmarketlinks2025/darknetmarkets – darknet markets onion address
Pingrutty
| #
darknet markets onion https://github.com/aresmarketlink0ru72/aresmarketlink – darknet drug links
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://xn--80agflcgbqkedhbfsfqs3v.xn--p1ai/
Toliknaf
| #
onion dark website https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet markets links
1win_hcoi
| #
1win 1win .
1win_mdmt
| #
ваучеры 1win ваучеры 1win .
DonDonfaita
| #
darknet drug market https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darkmarket list
Rabyitabe
| #
dark web drug marketplace https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet markets 2025
Diplomi_tlKt
| #
Заказать диплом о высшем образовании!
Мы можем предложить документы институтов, расположенных на территории всей России.
Основные преимущества наших дипломов:
• используются только настоящие бланки “Гознак”;
• необходимые подписи руководства;
• мокрые печати университета;
• специальные водяные знаки, нити и другие степени защиты;
• безупречное качество оформления – ошибок не будет;
• любая проверка документа.
10000diplomov.ru/kupit-svidetelstvo-o-rozhdenii-6
Pingrutty
| #
darknet markets https://github.com/aresmarketlink0ru72/aresmarketlink – dark web link
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://xn--2-htbyfc.xn--p1ai/
lkjhCype
| #
https://pandorabox.ru/css/pgs/geroi_multserialov_v_kino.html zz top видео лучшие клипы
1win_ihPi
| #
бонусы казино 1вин 1win3003.ru .
demo mahjong
| #
Main slot demo mahjong ways rupiah resmi dari pg soft cuma di demo mahjong naga merah.
Toliknaf
| #
darknet drug market https://github.com/darknetmarketlistv8tg0/darknetmarketlist – darknet drug store
Markcen
| #
Как выбрать надёжного застройщика? Читайте проверенные советы https://xn--61-6kcq2ag4b6e.xn--p1ai/
Pingrutty
| #
darknet market links https://github.com/aresmarketlink0ru72/aresmarketlink – darknet market links
DonDonfaita
| #
dark market link https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet drugs
Davidunmat
| #
palmslots trustpilot
Rabyitabe
| #
dark market list https://github.com/tormarkets2025ukaz1/tormarkets2025 – darknet market list
Donaldlaf
| #
darknet marketplace https://github.com/darknetmarketlinks2025/darknetmarkets – dark web link
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы — https://xn—–7kchcrgop2a1bbbeqft7m5b.xn--p1ai/
Pingrutty
| #
darknet market list https://github.com/aresmarketlink0ru72/aresmarketlink – darknet markets links
Toliknaf
| #
darknet markets onion https://github.com/darknetmarketlistv8tg0/darknetmarketlist – dark market onion
Jariorqyh
| #
Здравствуйте!
Начальники довольно часто выбирают кандидатов, которые закончили ВУЗ. Особенно в приоритете элитные учебные заведения. Но учиться пять лет – это долго и дорого, далеко не у каждого есть такая возможность. Приобрести документ – оптимальный выход.
Могут быть и непредвиденные обстоятельства, когда диплом теряется или портится. Далеко не всегда возможно быстро и без проблем восстановить его, особенно если ВУЗ закрыт или располагается в другом регионе страны. Бюрократия отнимает множество времени.
Для быстрого продвижения вверх по карьере понадобится наличие официального диплома о высшем образовании. Тем не менее часто в жизни случается так, что некоторые трудности мешают благополучно окончить учебу и заполучить желанный документ.
Приобрести диплом ВУЗа
Наша компания предлагает выгодно и быстро приобрести диплом, который выполняется на бланке ГОЗНАКа и заверен печатями, штампами, подписями. Документ способен пройти лубую проверку, даже с применением специфических приборов. Решите свои задачи быстро и просто с нашей компанией.
Где приобрести диплом специалиста? rdiplomm24.com/
DonDonfaita
| #
darknet drug market https://github.com/nexusdarkneturlwrf4t/nexusdarkneturl – darknet drug market
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://xn—–7kcoqeea0aaiqm4apq0s.xn--p1ai/
Donaldlaf
| #
darkmarket 2025 https://github.com/darknetmarketlinks2025/darknetmarkets – dark web drug marketplace
GeorgeTom
| #
Source:
http://ic51.ru
1win_numi
| #
1вин партнерка https://1win3004.ru/ .
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://xn—-7sbhyegsibavlngp.xn--p1ai/
Sazrcps
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и прочих профессий по невысоким ценам. Преимущества заказа документов в нашем сервисе
Вы приобретаете документ в надежной и проверенной временем компании. Такое решение сэкономит не только денежные средства, но и время.
На этом преимущества не заканчиваются, их гораздо больше:
• Документы изготавливаются на оригинальных бланках с мокрыми печатями и подписями;
• Дипломы любого ВУЗа России;
• Стоимость во много раз меньше той, которую понадобилось бы заплатить на очном и заочном обучении в ВУЗе;
• Удобная доставка в любые регионы Российской Федерации.
Приобрести диплом академии– http://poboltaem.foroactivo.com/post/ – poboltaem.foroactivo.com/post
Pingrutty
| #
darknet market links darknet drug market
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://xn--j1aihe7cc.com/
Toliknaf
| #
dark web market darknet markets
JerryJuism
| #
http://forum.toonboom.ru/index.php?/topic/4809-pr-агентство-в-москве/
Terencesam
| #
Бездепозитные бонусы за регистрацию Бесплатные спины за регистрацию без депозита
DonDonfaita
| #
tor drug market dark market onion
ClaytonShank
| #
Покупка недвижимости и ипотека https://asksteel.ru что нужно знать? Разбираем выбор жилья, условия кредитования, оформление документов и юридические аспекты. Узнайте, как выгодно купить квартиру и избежать ошибок!
Donaldlaf
| #
dark web marketplaces dark web drug marketplace
Pingrutty
| #
darknet links darknet drug market
Trefhax
| #
Приобрести документ о получении высшего образования вы можете у нас в столице. diplomaz-msk.com/kupit-diplom-tolyatti-4
MichaelPak
| #
Hello. It’s my blog https://brilliantcover.ru/.
RobertWag
| #
кайт школа в анапе
Toliknaf
| #
darknet markets links dark market onion
DonDonfaita
| #
darknet markets onion address dark web market list
servicecentercaree
| #
Квадрокоптеры DJI — это высокотехнологичные устройства, обеспечивающие захватывающие возможности в видеосъемке и навигации. Однако даже самые надёжные дроны иногда сталкиваются с проблемами. Эта статья поможет разобраться в частых неисправностях, их причинах, способах устранения и ситуациях, когда без сертифицированного сервисного центра не обойтись. ссылка
Donaldlaf
| #
darknet site dark web markets
Pingrutty
| #
dark market 2025 darknet drugs
VictorCaf
| #
Ваш бизнес заслуживает лучшего — премиальные овощи и зерновые от проверенного поставщика, детали по ссылке https://webyourself.eu/Gordon792
FrankFiP
| #
https://diamantgeo.ru
Toliknaf
| #
darkmarket 2025 dark web market
Pingrutty
| #
darknet markets 2025 darkmarket url
VictorCaf
| #
Ищу работу через этот сайт, быстро и удобно, рекомендую ознакомиться https://www.gamerlaunch.com/community/users/blog/6646995/2364755/job-search-platform/?gid=535
DonDonfaita
| #
dark market onion darkmarket url
Lazraae
| #
Диплом ВУЗа РФ!
Без получения диплома трудно было продвигаться по карьерной лестнице. Именно из-за этого решение о заказе диплома следует считать целесообразным. Заказать диплом о высшем образовании ostome.ru/forumbb/viewtopic.php?f=12&t=274205
Mazrgmt
| #
Привет!
Где заказать диплом специалиста?
Мы изготавливаем дипломы любой профессии по невысоким ценам. Стоимость зависит от выбранной специальности, года получения и образовательного учреждения. Стараемся поддерживать для покупателей адекватную ценовую политику. Для нас важно, чтобы документы были доступны для большинства граждан.
Покупка диплома, который подтверждает обучение в ВУЗе, – это выгодное решение. Попросту подсчитайте, сколько понадобится потратить средств на оплату 5-летнего обучения, на аренду жилья (если учащийся иногородний), на ежедневный проезд до университета и прочие издержки. Выйдет серьезная сумма, которая превышает цены на наши дипломы. А ведь все это время можно уже с успехом работать, занимаясь своей карьерой.
Готовый диплом с приложением целиком и полностью отвечает условиям и стандартам Министерства образования и науки, неотличим от оригинала. Не стоит откладывать свои мечты на потом, реализуйте их с нашей компанией – отправьте заявку на диплом прямо сейчас!
Купить диплом о среднем образовании – запросто! diplomanc.com/
Donaldlaf
| #
darknet market darknet markets onion
1win_tbSl
| #
бк 1вин 1win3005.ru .
Toliknaf
| #
dark market darknet drug market
Pingrutty
| #
tor drug market darkmarket url
Mazrzur
| #
Где купить диплом специалиста?
Мы предлагаем документы об окончании любых университетов РФ. Документы производят на подлинных бланках государственного образца. 39504.org/showthread.php?tid=82709
Taylorrew
| #
ипотека и покупка недвижимости https://luchdom.ru что нужно знать? Разбираем выбор жилья, условия кредитования, оформление документов и юридические аспекты. Узнайте, как выгодно купить квартиру и избежать ошибок!
DonDonfaita
| #
dark web market list darknet markets onion address
Stevereina
| #
Buy elite property in Montenegro: purchase apartments and houses without risks! Current prices, transaction execution, taxes and residence permit. Profitably purchase housing for life, recreation or investment.
Donaldlaf
| #
darknet marketplace darknet markets
servicecaree
| #
Мы знаем, насколько важна для вас надежность техники GoPro, и делаем всё, чтобы вернуть её в рабочее состояние в максимально короткие сроки. Узнайте, почему GoPro Center — лучший выбор для ремонта ваших устройств. Получите бесплатную консультацию по телефону и мы определим стоимость ремонта техники Go Pro. перейти
lkjhCype
| #
https://www.tenox.ru/wp-content/pgs/serialu_pro_shkolu__zahvatuvaushie_istorii__kotorue_stoit_posmotret.html скачать фильм бесплатно на киновасек
Mazrzir
| #
Мы изготавливаем дипломы любой профессии по приятным тарифам. Всегда стараемся поддерживать для заказчиков адекватную политику тарифов. Для нас важно, чтобы дипломы были доступны для большого количества граждан.
Приобретение документа, который подтверждает обучение в университете, – это выгодное решение. Заказать диплом ВУЗа: diplomanrussians.com
Pingrutty
| #
darknet drugs dark web markets
Lazrtfu
| #
Приобрести диплом любого института!
Мы готовы предложить дипломы любой профессии по приятным тарифам— diplom-onlinex.com/kupit-diplom-v-novosibirske-19/
Toliknaf
| #
darknet markets darkmarket link
Mazrsnd
| #
Мы изготавливаем дипломы любых профессий по приятным тарифам. Стараемся поддерживать для покупателей адекватную ценовую политику. Важно, чтобы документы были доступными для большого количества граждан.
Покупка диплома, подтверждающего окончание института, – это рациональное решение. Приобрести диплом университета: diplom-top.ru/kupit-diplomi-o-visshem-3/
DonDonfaita
| #
darknet markets 2025 darkmarket 2025
Jariorywz
| #
Где приобрести диплом специалиста?
Наша компания предлагает выгодно купить диплом, который выполняется на бланке ГОЗНАКа и заверен печатями, штампами, подписями должностных лиц. Наш диплом пройдет любые проверки, даже с применением профессиональных приборов. Достигайте свои цели быстро и просто с нашим сервисом.
Приобрести диплом ВУЗа diplomus-spb.ru/kupit-diplom-s-zaneseniem-v-reestr-11/
Donaldlaf
| #
onion dark website darknet market lists
"https://chrisophia.wiki/index.php/User:ShantellKavanaug"
| #
Подбор авто из Японии с
учетом ваших предпочтений
“https://drapia.org/11-WIKI/index.php/Vttauto_100M”
Pingrutty
| #
darknet markets dark web drug marketplace
Andrewcheex
| #
На площадке представлен широкий ассортимент травматического оружия https://travmat-kupit.ru/
DavidNaw
| #
https://textme.work/
GeorgeTom
| #
Source:
http://ic51.ru
Toliknaf
| #
darkmarket 2025 dark market link
Andrewcheex
| #
На площадке представлен широкий ассортимент травматического оружия https://travmat-kupit.ru/
DonDonfaita
| #
dark web markets dark market onion
"https://clashofcryptos.trade/wiki/User:RudolphChewning"
| #
Доставка авто из Китая в сжатые сроки
“https://wiki.learning4you.org/index.php?title=User:PatrickDecicco”
Donaldlaf
| #
darknet markets 2025 darkmarket 2025
Pingrutty
| #
dark web link bitcoin dark web
servicecaree
| #
Умные часы Garmin являются надежными спутниками спортсменов и активных людей, но даже самые качественные устройства могут столкнуться с проблемами. Одна из наиболее распространенных неисправностей – это сбой в работе зарядки. В этой статье мы рассмотрим основные причины, почему часы Garmin могут перестать заряжаться, и подскажем, как решить проблему. ссылка
Jeffreyslany
| #
Secure your connections https://github.com/aws-ec2/AWS-CLI/releases with FortiClient, ArcGIS Pro, Cisco AnyConnect, and Cisco Secure Client. Reliable VPN clients for remote work, data protection, and corporate network access without threats or information leaks!
"https://talkingheadswiki.com/w/Vttauto_8k"
| #
Как заказать японский авто без лишних
переплат?
“https://www.hohenbergen.de/index.php/Benutzer:WinfredLlanas”
Terencesam
| #
Top casino с выводом Бонусы за регистрацию
Toliknaf
| #
dark markets 2025 dark market url
Pingrutty
| #
darknet markets 2025 darknet sites
JamesEnuts
| #
https://vkontakte.forum.cool/viewtopic.php?id=19599#p53208
DonDonfaita
| #
dark web market list dark market url
Donaldlaf
| #
dark web marketplaces dark web link
Dnrtwtk
| #
Где приобрести диплом специалиста?
Мы изготавливаем дипломы любой профессии по приятным ценам. Мы готовы предложить документы ВУЗов, расположенных на территории всей Российской Федерации. Вы сможете заказать диплом от любого заведения, за любой год, включая сюда документы старого образца. Документы делаются на бумаге высшего качества. Это позволяет делать государственные дипломы, не отличимые от оригинала. Они будут заверены всеми необходимыми печатями и подписями. Стараемся поддерживать для клиентов адекватную ценовую политику. Важно, чтобы документы были доступны для большого количества граждан. diplomius-docs.com/kuplyu-diplom-o-visshem-obrazovanii-2-3
Sazrvhr
| #
Мы изготавливаем дипломы любых профессий по приятным ценам. Цена будет зависеть от выбранной специальности, года выпуска и образовательного учреждения. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Для нас важно, чтобы документы были доступными для большого количества наших граждан. купить диплом о высшем образовании севастополь
Toliknaf
| #
darknet markets 2025 dark market url
Pingrutty
| #
darknet market links dark web link
servicecaree
| #
Сервисный центр Samsung — это надежный партнер для владельцев техники Samsung. Мы предлагаем обширный перечень услуг по ремонту и обслуживанию разнообразных устройств. Скорость, качество и комфорт — три основных принципа нашей работы. перейти
DonDonfaita
| #
darknet markets url tor drug market
Donaldlaf
| #
dark web markets darknet websites
Cazrsdb
| #
Здравствуйте!
Без наличия диплома очень непросто было продвигаться по карьере. В наше время документ не дает абсолютно никаких гарантий, что получится найти престижную работу. Намного более важное значение имеют практические навыки специалиста, а также его опыт работы. Именно поэтому решение о заказе диплома можно считать целесообразным. Приобрести диплом об образовании mosdmit.flybb.ru/viewtopic.php?f=2&t=636
Pingrutty
| #
darknet market links onion dark website
Toliknaf
| #
darknet markets darknet market
DonDonfaita
| #
darknet markets links darknet links
Donaldlaf
| #
bitcoin dark web darknet markets 2025
Pingrutty
| #
darkmarkets dark markets
Davidhoirm
| #
amazon deals and steals
Toliknaf
| #
bitcoin dark web darknet drug links
prodvijenie saitov v moskve_ejol
| #
продвижение сайта в топ цена москва продвижение сайта в топ цена москва .
prodvijenie saitov v moskve_ytsi
| #
заказать раскрутку сайта заказать раскрутку сайта .
DavidNaw
| #
https://textme.work/
DonDonfaita
| #
darknet markets onion address darknet markets links
Pingrutty
| #
dark market dark market
Donaldlaf
| #
darknet market links onion dark website
prodvijenie saitov v moskve_wzoa
| #
заказать seo заказать seo .
prodvijenie saitov v moskve_ioEl
| #
сео оптимизация москва сео оптимизация москва .
Toliknaf
| #
dark market onion darknet markets 2025
lkjhCype
| #
https://jaluzi-bryansk.ru/smarty/pags/filmu_onlayn_besplatno___dostupnoe_udovolstvie.html metro last light на гитаре табы
Michaelwhild
| #
fps samp гта сан андреас самп 0.3
TylerOwece
| #
сайт крмп самп форум
Pingrutty
| #
dark markets 2025 darknet site
DonDonfaita
| #
best darknet markets darknet market list
Donaldlaf
| #
darknet sites dark web market list
1win_ugSr
| #
1 win http://1win104.com.kg/ .
Toliknaf
| #
darknet markets onion darkmarkets
Pingrutty
| #
darknet marketplace darknet markets 2025
Sazrkbm
| #
Здравствуйте!
Заказать диплом ВУЗа по невысокой стоимости возможно, обращаясь к проверенной специализированной фирме. Заказать диплом о высшем образовании: vacshidiplom.com/kupit-diplom-stavropol-5/
DonDonfaita
| #
darknet site darknet marketplace
Donaldlaf
| #
darkmarket link dark markets
ThomasBem
| #
https://kro-rodina.ru/
mostbet_chPr
| #
мосбет https://mostbet34.com.kg .
Pingrutty
| #
darknet markets links darkmarket link
Toliknaf
| #
darknet markets links bitcoin dark web
1win_josa
| #
1win betting 1win9.com.ng .
DonDonfaita
| #
best darknet markets dark markets
lkjhCype
| #
https://remontila.ru/art/filmu_onlayn_v_luchshem_kachestve.html смотреть фильм волк с уолл стрит бесплатно в хорошем качестве
Donaldlaf
| #
dark markets 2025 darkmarkets
Pingrutty
| #
dark web market list darkmarket list
Terencesam
| #
Бонусы за регистрацию Игровые автоматы играть на деньги
Jasonbitte
| #
Когда я зашёл на этот сайт, ощущение было таким, будто я переступил грань реальности. Здесь каждая ставка — это не просто азарт, а история, которую ты открываешь с каждым кликом.
Оформление создан для комфорта, словно невидимый проводник направляет тебя от игры к игре. Финансовые движения, будь то депозиты или вывод средств, проходят легко, как поток воды, и это завораживает. А поддержка всегда готова подхватить, как надежный товарищ, который никогда не оставит.
Для меня Селектор стал местом, где игра и вдохновение сплетаются. Здесь каждый момент — это часть картины, которую хочется создавать снова и снова.
Toliknaf
| #
dark market url dark web market urls
mostbet_ruPr
| #
мостбет зеркало https://www.mostbet34.com.kg .
DonDonfaita
| #
darknet drug store darknet sites
Davidhoirm
| #
азино777 официальный сайт
Donaldlaf
| #
dark web drug marketplace darknet drug links
Crazimesix
| #
Ищете выгодное предложение? Уникальная возможность получить займы без процентов онлайн на карту от 60+ проверенных МФО. Первый займ бесплатно для новых клиентов! Мы гарантируем полное отсутствие скрытых комиссий и переплат при оформлении микрокредита до 30 000 рублей.
Crazimesix
| #
Впечатляет количество вариантов займа онлайн срочно без отказа среди современных МФО. Срочные 11 000 рублей на медицинские услуги получил быстро благодаря широкому выбору компаний. Удобное оформление и моментальное зачисление денег спасли ситуацию.
Toliknaf
| #
darkmarket link darknet drug links
Pingrutty
| #
darkmarket darknet marketplace
servicecaree
| #
Samsung представила серию ноутбуков Galaxy Book4, в которую входят модели, подходящие для повседневной работы, творчества и бизнес-задач. В данной статье мы разберем две ключевые модели серии — Galaxy Book4 Pro и Galaxy Book4 Pro 360, их особенности, характеристики и отличия. подробнее
DonDonfaita
| #
dark web market list dark websites
Donaldlaf
| #
bitcoin dark web darknet links
Crazimesix
| #
В Яндексе отыскал выгодный вариант взять микрозайм на карту, когда срочно понадобилось 16 000 рублей на покупку техники. Поисковая система предложила множество вариантов – выбрал самое честное предложение, средства поступили моментально.
1win_oxsa
| #
1win betting site 1win betting site .
Toliknaf
| #
dark market onion dark market
Pingrutty
| #
darknet markets onion darknet markets url
DonDonfaita
| #
best darknet markets darknet sites
1win_pzSr
| #
казино 1win https://1win104.com.kg/ .
Donaldlaf
| #
darknet drug market dark web drug marketplace
DannyJef
| #
Najboljse pocitnice apartmani Zabljak! Uzivajte v udobju, svezem zraku in osupljivi pokrajini. Gorske poti, smucanje, izleti in prijetno vzdusje. Brezplacen Wi-Fi, zajtrk in parkirisce. Rezervirajte bivanje v osrcju narave!
Richardbed
| #
Dobrodosli v hotel Bianca Kolasin! Uzivajte v udobnih sobah, osupljivem razgledu in odlicni storitvi. Pozimi – smucanje, poleti – pohodi v gore in izleti. Prirocna lokacija, restavracija, SPA in prijetno vzdusje. Rezervirajte nepozabne pocitnice!
mostbet_sjmt
| #
mostbet 1-15 pozitsiya http://mostbet3003.ru/ .
Pingrutty
| #
dark markets 2025 dark web market list
Cazralz
| #
Здравствуйте!
Мы можем предложить дипломы психологов, юристов, экономистов и любых других профессий по приятным ценам. Стоимость может зависеть от выбранной специальности, года получения и университета: rdiplomans.com/
Uazrpml
| #
Доброго времени суток!
Для определенных людей, купить диплом ВУЗа – это острая необходимость, шанс получить достойную работу. Но для кого-то – это очевидное желание не терять множество времени на учебу в ВУЗе. Что бы ни толкнуло вас на такое решение, мы готовы помочь. Быстро, профессионально и по разумной стоимости изготовим документ любого ВУЗа и года выпуска на подлинных бланках со всеми требуемыми подписями и печатями.
Ключевая причина, почему многие покупают дипломы, – получить хорошую работу. Допустим, знания дают возможность кандидату устроиться на привлекательную работу, однако документального подтверждения квалификации не имеется. Если работодателю важно наличие “корочки”, риск потерять вакантное место достаточно высокий.
Заказать документ о получении высшего образования можно в нашем сервисе. Мы предлагаем документы об окончании любых ВУЗов РФ. Вы сможете получить необходимый диплом по любой специальности, любого года выпуска, включая документы Советского Союза. Даем гарантию, что в случае проверки документов работодателем, никаких подозрений не появится.
Ситуаций, которые вынуждают приобрести диплом ВУЗа очень много. Кому-то срочно необходима работа, в итоге нужно произвести особое впечатление на начальство при собеседовании. Некоторые мечтают устроиться в престижную компанию, чтобы повысить собственный статус в социуме и в последующем начать свое дело. Чтобы не тратить драгоценное время, а сразу начинать успешную карьеру, используя имеющиеся знания, можно купить диплом через интернет. Вы станете полезным в обществе, получите финансовую стабильность в минимальные сроки- купить аттестат
Sazrjhq
| #
Довольно часто бывает так, что для того, чтобы продвигаться вверх по карьере, требуется документ, который подтверждает наличие образования. Где купить диплом специалиста?
Купить документ о получении высшего образования можно у нас. Мы предлагаем документы об окончании любых университетов России. mebcom.ks.ua/
RonaldIcemy
| #
Хотите сдать радиолом https://radiolom99.ru? Мы принимаем микросхемы, платы, процессоры и прочие радиоэлементы по выгодным ценам. Быстрая оценка, честные цены и удобные способы приема. Узнайте, сколько стоят ваши детали прямо сейчас!
Toliknaf
| #
tor drug market dark web market
Sazrwva
| #
Купить диплом о высшем образовании
Мы оказываем услуги по продаже документов об окончании любых университетов РФ. http://www.alternance-manufacturing.fr/contact
DonDonfaita
| #
darknet markets url dark web market list
Sazrmtv
| #
Привет, друзья!
Заказ документа о высшем образовании через проверенную и надежную фирму дарит много достоинств. Данное решение позволяет сэкономить как длительное время, так и существенные финансовые средства. Однако, только на этом выгода не ограничивается, плюсов значительно больше.Мы можем предложить дипломы любой профессии. Дипломы изготавливаются на фирменных бланках. Доступная цена в сравнении с большими расходами на обучение и проживание. Заказ диплома ВУЗа является выгодным шагом.
Приобрести диплом: magic.ly/diplomygroup/Pochemu-stoit-kupit-shkolnyj-attestat-pryamo-sejchas
Donaldlaf
| #
dark web market links darknet market lists
lkjhCype
| #
https://logospress.ru/content/pgs/?filmu_slesheru__istoriya__osobennosti_i_vliyanie_na_kulturu.html новинки кино и сериалы смотреть
Pingrutty
| #
tor drug market darknet markets onion address
Toliknaf
| #
darknet markets links darknet markets onion
DonDonfaita
| #
darknet market dark markets 2025
Pingrutty
| #
dark web drug marketplace dark market url
Donaldlaf
| #
dark web drug marketplace darknet market links
Terencesam
| #
1000 рублей за регистрацию вывод сразу bonuses casinos
lkjhCype
| #
https://panamar.ru/mt/?filmu_onlayn___udobno__dostupno_i_uvlekatelno.html фильм хороший посмотреть для души рейтинг
Toliknaf
| #
darknet market lists darknet markets 2025
Pingrutty
| #
best darknet markets dark market 2025
DonDonfaita
| #
dark websites darknet markets onion
Xazrxip
| #
Приобрести диплом о высшем образовании!
Мы готовы предложить дипломы любых профессий по приятным тарифам. Вы приобретаете диплом через надежную компанию. : diplomers.com/kupit-diplom-gei-o-visshem-obrazovanii-bez-predoplati-3/
Donaldlaf
| #
darknet markets darknet links
servicecaree
| #
Ремонт роботов-пылесосов iRobot в Москве: Ремонт блока питания, Замена комплекта щеток, Восстановление колеса, Ремонт гидросистемы, Калибровка, Замена датчиков, Очистка датчиков, Замена аккумулятора здесь
Toliknaf
| #
dark markets 2025 darknet markets url
Pingrutty
| #
darkmarket list darknet markets 2025
Lazrcgn
| #
Где купить диплом специалиста?
Заказать диплом института по доступной цене можно, обратившись к проверенной специализированной компании.: nsk-diplom.com
WilliamDix
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и прочих профессий по приятным ценам. Дипломы изготавливаются на фирменных бланках Купить диплом о высшем образовании diplomp-irkutsk.ru
DonDonfaita
| #
darknet market list darknet market
Donaldlaf
| #
dark market list darkmarket 2025
Pingrutty
| #
onion dark website darknet markets url
Toliknaf
| #
dark market link darkmarket 2025
DonDonfaita
| #
darknet markets url darkmarket url
Donaldlaf
| #
darknet markets tor drug market
Pingrutty
| #
dark web link dark markets 2025
Toliknaf
| #
darknet markets 2025 darkmarkets
Diplomi_ihKt
| #
Заказать диплом о высшем образовании!
Мы готовы предложить документы учебных заведений, расположенных на территории всей Российской Федерации.
Превосходства наших дипломов:
• используем лишь качественные бланки “Гознак”;
• все подписи должностных лиц;
• все печати учебного заведения;
• водяные знаки, нити и прочие степени защиты;
• безупречное качество оформления – ошибок не будет;
• любая проверка оригинальности документа.
bisness-diplom.ru/kupit-diplom-trenera-2-2
DonDonfaita
| #
bitcoin dark web best darknet markets
Pingrutty
| #
darknet market links darknet marketplace
Donaldlaf
| #
darkmarket dark web market
DouglasOrist
| #
Купите теплицу https://tepl1.ru/catalog с доставкой по выгодной цене! Широкий выбор моделей: поликарбонатные, стеклянные, пленочные. Быстрая доставка, прочные конструкции, удобный монтаж. Идеально для дачи, сада и фермерства. Заказывайте качественные теплицы с доставкой прямо сейчас!
Sazrmam
| #
купить диплом невинномысск
Toliknaf
| #
dark web link dark market onion
Pingrutty
| #
dark market url dark web market
DonDonfaita
| #
dark web market links darknet sites
Terencesam
| #
Игровые автоматы играть на деньги фриспины за регистрацию
Donaldlaf
| #
darknet websites darknet links
prodvijenie saitov v moskve_dnOr
| #
заказ продвижения сайта заказ продвижения сайта .
prodvijenie saitov v moskve_pxka
| #
заказать сео продвижение сайта москва заказать сео продвижение сайта москва .
prodvijenie saitov v moskve_wgKr
| #
стоимость продвижения сайта в топ 10 цена prodvizhenie-sajtov-v-moskve214.ru .
prodvijenie saitov v moskve_ejPt
| #
сео заказать сео заказать .
Toliknaf
| #
dark web marketplaces dark web market urls
Pingrutty
| #
darknet markets onion address dark markets
lkjhCype
| #
https://gefestexpo.ru/art/filmu_onlayn___novue_grani_kinoprosmotra.html смотреть фильм и сериалы бесплатно
DonDonfaita
| #
dark web market darknet markets 2025
Donaldlaf
| #
dark market onion darknet markets 2025
1win_agOn
| #
казино 1win https://1win105.com.kg .
Pingrutty
| #
darknet markets url darknet markets
Toliknaf
| #
darknet market lists dark web link
DonDonfaita
| #
darknet markets url dark market list
Donaldlaf
| #
darknet market darknet marketplace
Pingrutty
| #
dark web markets darknet markets
Markcen
| #
Как выбрать надёжного застройщика? Читайте проверенные советы https://xn—-7sbccdscdoffrg1cybj3z.xn--p1ai/
mostbet_dcEr
| #
mostbet apk скачать https://mostbet1009.com.kg/ .
lkjhCype
| #
http://radiodelo.ru/shop/pgs/filmu__professionalu_svoego_dela.html хочу замуж фильм смотреть 2022
Toliknaf
| #
dark market onion dark market onion
1win_jpMl
| #
1 win 1win10.com.ng .
EdwardAbaph
| #
https://inkasbank.ru/
DonDonfaita
| #
darknet drugs darknet sites
Pingrutty
| #
darkmarket url dark markets 2025
Donaldlaf
| #
darknet site darknet drug market
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности https://xn--90aimcoinaikf.xn--p1ai/
Trefyfa
| #
Приобрести документ университета можно у нас в столице. diplom-ryssia.com/kupit-diplom-orenburg-2
mostbet_jgEr
| #
мостбет авиатор http://mostbet1009.com.kg .
Pingrutty
| #
darkmarket darknet markets links
servicecaree
| #
Подробнее в материале ссылка
DonDonfaita
| #
dark market link dark market url
Donaldlaf
| #
darkmarket dark market onion
Sazrzrp
| #
Приобретение документа о высшем образовании через качественную и надежную компанию дарит ряд плюсов. Такое решение помогает сэкономить как дорогое время, так и значительные деньги. Однако, плюсов значительно больше.Мы готовы предложить дипломы любой профессии. Дипломы производят на фирменных бланках. Доступная стоимость по сравнению с огромными тратами на обучение и проживание. Заказ диплома о высшем образовании из российского института является мудрым шагом.
Заказать диплом: diplom-zentr.com/kupit-diplom-v-moskve-bistro-i-nadezhno-3/
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://xn——6cdbaabncc2b3blicg2a2b7axf0dzfui.xn--p1ai/
https://Menbehealth.Wordpress.com/
| #
It’s remarkable to visit this site and reading the views
off all friends concerning this piece of writing, while
I am also zealous of getting knowledge. https://Menbehealth.Wordpress.com/
Terencesam
| #
бонусы за регистрацию Играть игровые автоматы на деньги
Pingrutty
| #
onion dark website dark market list
MarkNOlaf
| #
bitcoin dark web https://drdarkfoxmarket.com – dark market onion
DonDonfaita
| #
dark market 2025 dark web market list
FNDaviditabe
| #
darknet market https://kingdom-marketplace.com/ – darkmarket url
WilliamIRrutty
| #
darkmarkets https://kingdomdarkmarketonline.com/ – darknet site
Donaldlaf
| #
darknet markets url dark markets
Markcen
| #
Хотите сдать квартиру в аренду? Читайте проверенные советы https://xn—-8sbqbliyb2m.xn--p1ai/
Kxyufaita
| #
darknet drug store https://cannahome-darkmarket-online.com/ – dark web market list
Pingrutty
| #
darknet drugs darknet drug store
1win_mvOn
| #
1 вин вход http://1win105.com.kg/ .
lkjhCype
| #
https://www.evacuator-moskva.ru/images/pages/index.php?gangsterskoe_kino__istoriya__shedevru_i_kulturnoe_vliyanie.html фильмы онлайн скачать на телефон
servicecaree
| #
Подробнее в материале перейти
DonDonfaita
| #
darknet drug links darknet market
1win_dlMl
| #
sports betting 1win sports betting 1win .
Donaldlaf
| #
dark web market urls darknet drug store
Pingrutty
| #
darkmarket list darknet market lists
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://xn—–7kchcrgop2a1bbbeqft7m5b.xn--p1ai/
Jasonbitte
| #
Когда я зашёл на этот сайт, чувство было таким, будто я переступил грань реальности. Здесь каждая ставка — это не просто шанс, а эмоция, которую ты проживаешь с каждым движением.
Дизайн создан для комфорта, словно ветер судьбы направляет тебя от игры к игре. Транзакции, будь то пополнения или вывод средств, проходят быстро, как поток воды, и это удивляет. А техподдержка всегда отвечает мгновенно, как верный помощник, который никогда не подведёт.
Для меня Селектор стал пространством, где игра и вдохновение переплетаются. Здесь каждая минута — это часть картины, которую хочется создавать снова и снова.
DonDonfaita
| #
dark web drug marketplace darknet drugs
Pingrutty
| #
dark market onion darkmarket
Donaldlaf
| #
darknet drug links darknet websites
oformit propysk v Moskvy_djEi
| #
оформить пропуск в Москву progorod43.ru/oformit-propusk-na-mkad-dlya-gruzovyh-mashin .
servicecaree
| #
Подробнее в материале на сайте
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы — https://xn—–7kcoqeea0aaiqm4apq0s.xn--p1ai/
Jariorfij
| #
Привет!
Руководители серьезных компаний очень часто выбирают претендентов, которые закончили университет. Особенно ценятся топовые заведения. Но учиться 5 лет – это долго и дорого, далеко не у каждого существует подобная возможность. Купить документ – лучший выход.
Бывают и непредвиденные обстоятельства, когда диплом теряется или портится. Не всегда возможно быстро и без проблем восстановить документ, особенно когда университет закрыт или располагается в другом регионе страны. Бюрократия отнимает много времени.
Для максимально быстрого продвижения по карьерной лестнице нужно наличие официального диплома о высшем образовании. Впрочем зачастую в жизни может случиться так, что определенные трудности не дают с успехом окончить учебу, получая желанный документ.
Купить диплом о высшем образовании
Наши специалисты предлагают выгодно приобрести диплом, который выполняется на оригинальной бумаге и заверен мокрыми печатями, водяными знаками, подписями. Документ способен пройти любые проверки, даже при помощи профессионального оборудования. Достигайте своих целей быстро и просто с нашим сервисом.
Где купить диплом по необходимой специальности? rdiploma24.com/
1win_ansn
| #
казино 1win http://www.1win106.com.kg .
Pingrutty
| #
darknet markets 2025 dark web link
DonDonfaita
| #
darknet markets onion address dark market list
Donaldlaf
| #
dark web market dark market 2025
HershelzeM
| #
פנים! ויקה, אגב, גם ביקשה להסיר אותה. אתה לא מפחד ממה שהיא תראה? – יש מקומות כאלה בטלפון שבו היא בהחלט לא תמצא. בחיוך ענה מקס. – אותם בעוד שבוע? – מה שתגיד, יקירתי. אני יכול להזמין גם שלושה. – נהדר! שיהיה משהו חדש. – כן, מותק. גם אני חושב כך. כן, מרטין, browse this site
RolandLob
| #
https://poufe.ru/topic.php?id=1567
VortexHunter
| #
Современные методы по бизнесу: https://student.uog.edu.et/hello-world-3/
Markcen
| #
Хотите сдать квартиру в аренду? Читайте проверенные советы https://xn—-7sbhyegsibavlngp.xn--p1ai/
Pingrutty
| #
dark websites darkmarket
Eanrqeo
| #
Для успешного продвижения по карьере необходимо наличие диплома о высшем образовании. Купить диплом любого института у сильной организации: diplomidlarf.ru/kupit-diplom-parikmaxera-12/
Terencesam
| #
bonuses casinos фриспины за регистрацию
Edwardmeafe
| #
зеркало armada casino
RolandLob
| #
https://cross.expert/partner/mrt-pozvonochnika-vazhnyy-metod-diagnostiki.html
DonDonfaita
| #
bitcoin dark web onion dark website
Edwardmeafe
| #
армада
servicecaree
| #
Подробнее в материале ссылка
Donaldlaf
| #
dark web sites darknet markets onion address
mostbet_kg_xdpi
| #
служба поддержки мостбет номер телефона http://mostbet1000.com.kg .
Pingrutty
| #
darkmarket 2025 darknet site
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://xn--j1aihe7cc.com/
MarkNOlaf
| #
darkmarket link https://dark-web-versus.com – darknet markets onion
DonDonfaita
| #
dark market onion darknet drugs
mostbet_besa
| #
скачать mostbet на телефон https://www.mostbet1010.com.kg .
Donaldlaf
| #
darknet links dark web market
Pingrutty
| #
darknet drug links darknet markets onion address
FNDaviditabe
| #
darknet markets url https://kingdom-marketplace.com/ – dark web market urls
WilliamIRrutty
| #
darknet markets https://asap-market-onion.com/ – onion dark website
Potolki_dex
| #
натяжной потолок цена https://potolki-v-moskve.ru/
Kxyufaita
| #
darkmarket list https://cyberdarkmarkets.com/ – darknet markets onion
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы https://an-rusdom.ru/
Potolki_dex
| #
натяжной потолок москва https://potolki-v-moskve.ru/
MichaelBluth
| #
Sans?n?z? Sweet Bonanza’da sweet-bonanza-casino.site deneyin – parlak ve karl? bir slot! Ucretsiz donusler, kazanc carpanlar?, kademeli kombinasyonlar ve buyuk odemeler. Ucretsiz veya parayla cevrimici oynay?n ve tatl? oduller kazan?n!
Martinownew
| #
Glory Casino’da glory-casino-guncel-giris.online oynayn – en iyi oyunlar, yuksek odemeler ve 7/24 destek! Populer slotlar?, kart oyunlar?n? ve ruleti secin, bonuslar kazan ve gercek para kazan?n!
VirgilNeali
| #
Diego Armando Maradona diego maradona es una leyenda del futbol mundial! Campeon del Mundo de 1986, autor de “La Mano de Dios” y “El Gol del Siglo”. Un gran jugador argentino que inspiro a generaciones. ?Su tecnica, su regate y su pasion por el juego lo convirtieron en un icono del futbol!
mobile-worker.ru
| #
Ваш надёжный помощник снова в строю! Профессиональная замена аккумулятора iphone 8 вернёт смартфону автономность. Оригинальные комплектующие и современное оборудование – залог качественного обслуживания вашего устройства.
Более 7 лет наш сервисный центр на Коломенской помогает москвичам решать технические проблемы. В августе 2022 года Mobile-Worker переехал в новое просторное помещение на проспекте Андропова, 17к1. Теперь наши клиенты могут наслаждаться ещё более комфортным обслуживанием.
mostbet_kg_napi
| #
мостбет зеркало мостбет зеркало .
Онлайн казино в Беларуси
| #
Легальные Онлайн казино в Беларуси Беларуси
предлагают игровые автоматы от
мировых производителей
DonDonfaita
| #
darknet websites bitcoin dark web
MarkNOlaf
| #
darknet market links https://dark-web-versus.com – dark markets
Pingrutty
| #
darknet links darknet marketplace
Donaldlaf
| #
darkmarket url darknet markets onion address
FNDaviditabe
| #
darkmarket https://kingdom-marketplace.com/ – darknet drugs
WilliamIRrutty
| #
dark market url https://asap-market-onion.com/ – dark web drug marketplace
Kxyufaita
| #
dark web market links https://darkfoxmarketplace.com/ – dark web markets
DarkVoyager
| #
Лучшие https://aacsatlanta.com/dui-school-marietta/ инструменты для работы с бизнесом.
mobile-worker.ru
| #
Наконец-то нашёл отличный сервис для ремонт геймпада! После года интенсивного использования джойстики стали заедать. Мастера быстро определили проблему и заменили изношенные детали. Теперь играю с удовольствием, как на новом контроллере!
Mobile-Worker – это команда профессионалов с опытом от 7 лет, которые относятся к каждому устройству как к своему собственному. Мы работаем со всеми популярными брендами: iPhone, Samsung, Xiaomi и другими. Наш адрес: проспект Андропова, 17к1, работаем ежедневно с 10:00 до 20:00.
Markcen
| #
Новостройки или вторичка? Полезные рекомендации — https://bti-nsk.ru/
Pingrutty
| #
darknet market darknet links
DonDonfaita
| #
darknet links bitcoin dark web
MarkNOlaf
| #
dark web marketplaces https://drdarkfoxmarket.com – darknet drug market
Donaldlaf
| #
dark markets darknet drug store
FNDaviditabe
| #
darkmarket 2025 https://firstdarkmarket.com/ – darknet market links
Cazrvct
| #
Мы готовы предложить дипломы любых профессий по доступным ценам. Купить аттестат 9 классов — kyc-diplom.com/attestat-9-klassov.html
WilliamIRrutty
| #
dark web link https://kingdommarketdarknet.com/ – darknet site
Cazrxmu
| #
Мы готовы предложить документы ВУЗов на Ваш выбор, которые расположены в любом регионе России. nsk-diplom.com/kupit-diplom-baltijskoj-akademii-turizma-i-predprinimatelstva-3
1win_nosn
| #
1вин вход http://1win106.com.kg/ .
Kxyufaita
| #
darkmarket url https://cannahome-darkmarket-online.com/ – darknet market list
Pingrutty
| #
dark web drug marketplace dark web link
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы — https://crystalit34.ru/
ManuelGlona
| #
Fansite de Mike Tyson https://mike-tyson.com.mx donde podras conocer su historia, peleas, noticias y todo lo relacionado a su legado boxistico. Sigue sus proyectos y conoce mas sobre esta leyenda.
JamesGom
| #
Sitio de fans de Michael Phelps michael-phelps.com.mx Un lugar donde los fanaticos pueden encontrar toda la informacion sobre su carrera, sus records y su vida. Noticias, logros y contenido exclusivo.
DenniscrypE
| #
Sitio de fans de Muhammad Ali https://muhammad-ali.com.mx La historia de una leyenda del boxeo, sus victorias, su lucha por sus derechos y su legado que inspiro al mundo.
DonDonfaita
| #
dark web drug marketplace darkmarket 2025
MarkNOlaf
| #
dark websites https://kingdomdarkmarketplace.com – dark web marketplaces
mostbet_bgsa
| #
mostber https://mostbet1010.com.kg .
Donaldlaf
| #
darknet drug store dark web market
FNDaviditabe
| #
dark web market https://firstdarkmarket.com/ – darknet markets 2025
WilliamIRrutty
| #
onion dark website https://asap-market-onion.com/ – darknet markets url
Terencesam
| #
бонусы за регистрацию Casino bonus
Pingrutty
| #
darkmarket url dark market list
Kxyufaita
| #
dark markets https://cyberdarkmarkets.com/ – darknet drug links
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности https://cspkaliningrad.ru/
MarkNOlaf
| #
darknet drug market https://kingdom-darkmarketplace.com – dark market link
DonDonfaita
| #
darknet links darknet links
ClaudeKen
| #
Sitio fan de George Foreman http://george-foreman.com.mx dedicado a su carrera, victorias, derrotas y legado en el boxeo. Historias inspiradoras y el estilo unico de una leyenda del deporte.
Donaldlaf
| #
dark market url darknet drug links
Pingrutty
| #
dark market link darknet market links
FNDaviditabe
| #
dark web drug marketplace https://onion-dark-market.shop/ – dark web market links
WilliamIRrutty
| #
dark market list https://asap-market-onion.com/ – dark web market list
IronX1
| #
Как сэкономить на бизнесе: https://karoen.nl/blog/2023/06/06/ontspan-en-geniet-een-gids-voor-de-perfecte-vakantie/
Kxyufaita
| #
darknet drug links https://cannahomemarket.link/ – dark web market
Markcen
| #
Хотите сдать квартиру в аренду? Читайте проверенные советы https://dachnoe-house.ru/
MarkNOlaf
| #
dark web markets https://cannahomedarknetdrugstore.com/ – dark market list
Pingrutty
| #
darknet markets onion address darknet site
Donaldlaf
| #
dark market link darknet drug store
Volodyanaf
| #
dark web market https://aasap-market.com/ – darknet site
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы https://fisher-kp.ru/
FNDaviditabe
| #
darkmarket 2025 https://firstdarkmarket.com/ – darkmarket list
WilliamIRrutty
| #
dark web market https://kingdommarketdarknet.com/ – darknet drug links
Pingrutty
| #
darknet markets tor drug market
DonDonfaita
| #
dark markets darkmarket link
Kxyufaita
| #
darknet markets onion address https://cannahome-darkmarket-online.com/ – dark markets 2025
MatthewsOf
| #
Всегда жду выхода новых сезонов любимых зарубежных сериалов Последний С…РѕРґ королевы (сериал 2015, РўР’Р¦) смотреть онлайн РІСЃРµ серии РїРѕРґСЂСЏРґ бесплатно РІ хорошем качестве
Yi Chapmond
| #
Na [580bet](https://580-bet.com), o primeiro passo para uma experiência de cassino incrível começa com um bônus de US$ 100! Ao se registrar como novo usuário, você receberá esse valor para apostar em jogos emocionantes. Aproveite a oportunidade de explorar tudo o que a plataforma tem a oferecer, com segurança e diversão.
Donaldlaf
| #
darknet site bitcoin dark web
MarkNOlaf
| #
darkmarket https://kingdomdarkmarketplace.com – darknet drug market
MatthewsOf
| #
Лучшие русские сериалы – это настоящая гордость кинематографа Лучшие СЂСѓСЃСЃРєРёРµ сериалы смотреть онлайн бесплатно РІ хорошем качестве » Страница 721
Volodyanaf
| #
dark web market list https://darkfoxdarkweb.com/ – darkmarket
MatthewsOf
| #
Зарубежные сериалы иногда просто поражают крутыми сюжетными поворотами Чужое счастье (сериал 2017, Р РѕСЃСЃРёСЏ) смотреть онлайн РІСЃРµ серии РїРѕРґСЂСЏРґ бесплатно РІ хорошем качестве
MatthewsOf
| #
Какие русские сериалы сейчас на пике популярности? Стюардесса (фильм 2018) смотреть онлайн бесплатно РІ хорошем качестве
LightZ4
| #
https://nettyfish.com/bulk-sms-service-in-chennai/ Подробное руководство по бизнесу.
Pingrutty
| #
dark web marketplaces darkmarkets
FNDaviditabe
| #
dark web market https://firstdarkmarket.com/ – best darknet markets
WilliamIRrutty
| #
darknet drug store https://idarknetmarket.com/ – dark web drug marketplace
DonDonfaita
| #
darkmarket link dark market url
MatthewsOf
| #
Русские комедийные сериалы стали гораздо лучше за последние годы Честное волшебное (фильм 1975) смотреть онлайн бесплатно РІ хорошем качестве
Dolly Herzfeld
| #
Com o app [aposte e ganhe](https://aposte-br.com), você pode apostar onde quiser. Instale o aplicativo facilmente e aproveite a interface otimizada, gráficos de alta qualidade e uma experiência de jogo excelente diretamente no seu celular.
MatthewsOf
| #
Зарубежные комедийные сериалы – отличный способ поднять настроение Ёлки 3 (фильм 2013) смотреть онлайн бесплатно РІ хорошем качестве
Kxyufaita
| #
dark markets https://cannahome-darkmarket-online.com/ – dark web market
Donaldlaf
| #
dark web markets darkmarket url
MatthewsOf
| #
Зарубежные сериалы по книгам часто бывают очень интересными РЇ тебя услышу 1 сезон 18 серия смотреть онлайн бесплатно РІ хорошем качестве РЅР° СЂСѓСЃСЃРєРѕРј языке
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы https://hertz-lumen.ru/
MatthewsOf
| #
Русские сериалы о молодежи стали очень качественными Мюзиклы смотреть онлайн бесплатно РІ хорошем качестве HD без регистрации » Страница 2
MarkNOlaf
| #
darknet markets onion https://dark-web-versus.com – dark web market urls
Volodyanaf
| #
darkmarket 2025 https://darkfoxdarkweb.com/ – darknet market links
Pingrutty
| #
dark market 2025 dark web market urls
Terencesam
| #
получить 1000 рублей бесплатно за регистрацию no deposit casino
Sazrizy
| #
Хочу поделиться своим опытом по заказу аттестата ПТУ. Думал, что это невозможно, и начал искать информацию в интернете по теме: купить диплом риэлтора, купить диплом слесаря ремонтника дпо стандарт, купить диплом слесаря сантехника, купить диплом строительного техникума ссср, купить диплом сыктывкар. Постепенно углубляясь в тему, нашел отличный ресурс здесь: diplomybox.com/zakazat-dokument
obzorcaree
| #
Подробнее в материале в блоге об обзорах электронной техники перейти
MatthewsOf
| #
Какой зарубежный сериал стоит посмотреть на выходных? Сильная личность РёР· 2-Рђ (фильм 1984) смотреть онлайн бесплатно РІ хорошем качестве
FNDaviditabe
| #
darknet market list https://kingdom-marketplace.com/ – dark market list
MatthewsOf
| #
Лучшие русские сериалы – это те, которые цепляют с первых минут Драмы смотреть онлайн бесплатно РІ хорошем качестве HD без регистрации » Страница 47
WilliamIRrutty
| #
darknet sites https://darknetmarketsonion.shop/ – dark web link
MatthewsOf
| #
Какие русские сериалы можно назвать культовыми? Темнее чёрного (аниме 2007-2010) РЅР° СЂСѓСЃСЃРєРѕРј языке смотреть онлайн РІСЃРµ серии РїРѕРґСЂСЏРґ бесплатно РІ хорошем качестве
DonDonfaita
| #
best darknet markets dark market 2025
MatthewsOf
| #
Какие русские сериалы сейчас на пике популярности? Гром ярости (фильм 2010, РќРўР’) смотреть онлайн бесплатно РІ хорошем качестве
Kxyufaita
| #
best darknet markets https://cyberdarkmarkets.com/ – darknet site
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные рекомендации — https://homedecorsochi.ru/
MatthewsOf
| #
Лучшие русские сериалы – это те, которые вызывают бурю эмоций Мультсериалы смотреть онлайн бесплатно, РЅРѕРІРёРЅРєРё мультсериалов РІ HD качестве » Страница 22
Donaldlaf
| #
darknet markets url dark web marketplaces
Jacobisord
| #
стоимость проката авто прокат машин сочи без водителя
Pingrutty
| #
darkmarket url darkmarkets
MichaelGah
| #
Играйте в Big Bamboo big-bamboo-slot.online слот с захватывающим геймплеем, фриспинами и множителями! Ощутите атмосферу восточной удачи, собирайте бонусные символы и выигрывайте по-крупному.
oformit propysk v Moskvy_oooi
| #
заказать продвижение сайта заказать продвижение сайта .
BruceTam
| #
Join Glory Casino glory-casino-bd.online and get maximum bonuses! Top slot machines, table games, jackpots and exclusive promotions. Play online and win comfortably!
oformit propysk v Moskvy_hxet
| #
seo продвижение сайта москва seo продвижение сайта москва .
BufordClada
| #
Гемблинг-клуб Вулкан – это захватывающий мир азарта, который впечатляет с первого взгляда. Благодаря шикарному дизайну и интуитивно понятному интерфейсу, пользователи быстро оказываются в этом игровом пространстве.
Среди сильных сторон казино игровые автоматы стоит отметить большой выбор игр, включая игровые автоматы и настольные азартные игры, которые подойдут каждому игроку. Качество графического дизайна и анимации делает игровой процесс больше увлекательным и захватывающим.
Однако больше всего поразил подход казино к безопасности игроков. Они используют новейшие технологии шифрования, чтобы обеспечить секретность личных и финансовых данных пользователей. Это делает казино Вулкан замечательным вариантом для онлайн-игры.
MarkNOlaf
| #
dark web market links https://kingdomdarkmarketplace.com – darknet markets
Volodyanaf
| #
dark web sites https://darkfoxdarkweb.com/ – darknet drug market
Dnrtqnt
| #
Где заказать диплом по необходимой специальности?
Мы изготавливаем дипломы любой профессии по выгодным тарифам. Мы можем предложить документы техникумов, расположенных в любом регионе России. Вы можете купить качественно напечатанный диплом за любой год, включая сюда документы старого образца СССР. Дипломы и аттестаты выпускаются на “правильной” бумаге самого высокого качества. Это позволяет делать настоящие дипломы, которые невозможно отличить от оригиналов. Они будут заверены всеми обязательными печатями и штампами. Стараемся поддерживать для клиентов адекватную ценовую политику. Для нас очень важно, чтобы дипломы были доступны для большого количества наших граждан. diplom5.com/kupit-diplom-shvei
MatthewsOf
| #
Какие зарубежные сериалы можно назвать лучшими в 2024 году? Сказание РѕР± обручальных кольцах (аниме 2024) РЅР° СЂСѓСЃСЃРєРѕРј языке смотреть онлайн РІСЃРµ серии РїРѕРґСЂСЏРґ бесплатно РІ хорошем качестве
GhostZ4
| #
Новые подходы в бизнесе: http://traverseearth.com/the-chianti-region-tuscany-italy/
Josephslate
| #
Купить водительские права
FNDaviditabe
| #
dark web marketplaces https://mydarkmarketlink.com/ – darknet markets links
WilliamIRrutty
| #
darkmarkets https://idarknetmarket.com/ – dark markets 2025
MatthewsOf
| #
Зарубежные сериалы – это возможность окунуться в другой мир HD исторические фильмы смотреть онлайн РІ хорошем качестве бесплатно » Страница 31
Pingrutty
| #
dark web market urls darknet market list
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы https://instrument75.ru/
MatthewsOf
| #
Лучшие зарубежные сериалы – это настоящие блокбастеры Рстория девятихвостого лиса 1938 (дорама 2023) РІСЃРµ серии РїРѕРґСЂСЏРґ смотреть онлайн бесплатно РІ хорошем качестве РЅР° СЂСѓСЃСЃРєРѕРј языке
Kxyufaita
| #
darkmarket list https://cyberdarkmarkets.com/ – dark web market links
Donaldlaf
| #
dark market url darknet links
Calvinweria
| #
https://www.llanelliherald.com/
MatthewsOf
| #
Лучшие зарубежные сериалы – это те, которые становятся легендами Стряпуха. Таланты Рё поклонники (сериал 2024, РўР’Р¦) смотреть онлайн РІСЃРµ серии РїРѕРґСЂСЏРґ бесплатно РІ хорошем качестве
Volodyanaf
| #
darknet markets 2025 https://aasap-market.com/ – darknet sites
MarkNOlaf
| #
darknet drugs https://kingdomdarkmarketplace.com – darknet sites
Louisa Garding
| #
[74 bet](https://74-bet-br.com): Descubra como sacar seus ganhos rapidamente, com todas as informações necessárias e sem preocupações.
MatthewsOf
| #
Где можно смотреть зарубежные сериалы с хорошим переводом? Невинный (турецкий сериал 2017) РЅР° СЂСѓСЃСЃРєРѕРј языке смотреть онлайн РІСЃРµ серии РїРѕРґСЂСЏРґ бесплатно РІ хорошем качестве
1win_zaSi
| #
1vin kg 1win107.com.kg .
Pingrutty
| #
tor drug market dark web sites
DonDonfaita
| #
darkmarket url dark web sites
Juliusdaync
| #
Взрослый вебкам бесплатно https://hotcams.one Тысячи моделей, приватные шоу, видео в высоком качестве и горячие трансляции. Общайтесь в чате и наслаждайтесь без ограничений!
FNDaviditabe
| #
darkmarket list https://firstdarkmarket.com/ – dark market 2025
MatthewsOf
| #
Зарубежные комедийные сериалы – отличный способ поднять настроение Смотреть Сериалы 2015 онлайн бесплатно РІ HD качестве » Страница 5
WilliamIRrutty
| #
darkmarket link https://kingdomdarkmarketonline.com/ – darknet drug links
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://kvartiranahodynke.ru/
Donaldlaf
| #
darkmarkets dark web marketplaces
Kxyufaita
| #
dark markets https://cannahome-darkmarket-online.com/ – darkmarkets
lkjhCype
| #
http://stoljar.ru/wp-content/pages/luchshie_filmu_33.html скачать хороший фильм на телефон бесплатно
Pingrutty
| #
darknet websites dark market
Volodyanaf
| #
dark web drug marketplace https://darkwebversus.com/ – darkmarket url
MarkNOlaf
| #
darkmarket link https://kingdomdarkmarketplace.com – darknet market list
DonDonfaita
| #
darknet drugs dark web markets
MatthewsOf
| #
Зарубежные комедийные сериалы – отличный способ поднять настроение Смотреть онлайн бесплатно детективные сериалы, фильмы РІ хорошем качестве » Страница 10
FNDaviditabe
| #
darknet drugs https://onion-dark-market.shop/ – darknet markets
Donaldlaf
| #
dark market onion darkmarket list
WilliamIRrutty
| #
tor drug market https://asap-market-onion.com/ – tor drug market
mostbet_nvSl
| #
mostbet chrono https://mostbet1001.com.kg .
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы — https://lume42.ru/
Larry Hulstine
| #
No site da [winbrl](https://winbrl-88.com), o registro é fácil e rápido, e você recebe um bônus de US$ 100 para começar sua jornada no cassino online. Após completar o cadastro, basta fazer login e aproveitar os melhores jogos de cassino, como slots, roleta e blackjack, com a vantagem do bônus.
Hunter9905
| #
Новые подходы в бизнесе: https://www.editions-ric.fr/2019/02/10/video-france-3-reportage-sur-patrick-lecointe-editions-ric/
Pingrutty
| #
darkmarkets darknet drug market
obzorcaree
| #
Подробнее в материале в блоге об обзорах электронной техники ссылка
Kxyufaita
| #
dark market 2025 https://cyberdarkmarkets.com/ – darknet markets onion
MatthewsOf
| #
Русские комедийные сериалы стали гораздо лучше за последние годы Маша Рё Шеф (шоу 2020) смотреть онлайн РІСЃРµ выпуски РїРѕРґСЂСЏРґ бесплатно РІ хорошем качестве
oformit propysk v Moskvy_xgOt
| #
сео оптимизация сайтов в москве сео оптимизация сайтов в москве .
MatthewsOf
| #
Зарубежные сериалы в хорошем качестве – это просто сказка Побочный эффект (фильм 2008) смотреть онлайн бесплатно РІ хорошем качестве
MatthewsOf
| #
Какие зарубежные сериалы можно назвать культовыми? Вера (сериал 2024, РўР’-3) смотреть онлайн РІСЃРµ серии РїРѕРґСЂСЏРґ бесплатно РІ хорошем качестве
Volodyanaf
| #
dark market link https://darkode-market.com/ – dark web link
MatthewsOf
| #
Где смотреть русские сериалы без рекламы и в хорошем качестве? Дом для РґРІРѕРёС… (фильм 2009) смотреть онлайн бесплатно РІ хорошем качестве
MarkNOlaf
| #
darknet links https://drdarkfoxmarket.com – darkmarket 2025
MatthewsOf
| #
Люблю пересматривать старые русские сериалы – в них есть особая атмосфера Красный змей (фильм 2003) смотреть онлайн бесплатно РІ хорошем качестве РЅР° СЂСѓСЃСЃРєРѕРј языке
1win_trEi
| #
cod promoțional 1win http://www.1win5000.ru .
DonDonfaita
| #
dark market 2025 darkmarkets
MatthewsOf
| #
Зарубежные сериалы с крутым сюжетом – это то, что я люблю Фантастические сериалы смотреть онлайн бесплатно РІ хорошем качестве HD без регистрации » Страница 15
lkjhCype
| #
https://adyghe.ru/manul/inc/?filmu_onlayn_na_kinogo_3.html pci ven 8086 dev 43e0
FNDaviditabe
| #
darkmarkets https://firstdarkmarket.com/ – dark web markets
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности https://mastera-taganroga.ru/
WilliamIRrutty
| #
dark market link https://darknetmarketsonion.shop/ – darknet market links
KennethDrula
| #
Купить права Москва
MatthewsOf
| #
Русские сериалы о молодежи стали очень качественными Поющий офис (шоу 2023) РІСЃРµ выпуски РїРѕРґСЂСЏРґ смотреть онлайн бесплатно РІ хорошем качестве
Katerine Jaramillo
| #
A [bet 7k](https://bet-7k.com) facilita seu acesso ao mundo das apostas online com um app fácil de baixar e instalar. A interface do aplicativo foi projetada para proporcionar uma experiência rápida e eficiente, garantindo que você aproveite ao máximo cada aposta feita na plataforma.
Lezaimus
| #
займ мгновенно на карту – нужен срочный способ получить деньги? Этот сервис предлагает моментальные займы на карту. Без залога и поручителей, решение принимается в течение часа.
Kxyufaita
| #
darkmarket 2025 https://kingdomdarknetdrugstore.com/ – dark web sites
MatthewsOf
| #
Где можно смотреть зарубежные сериалы с хорошим переводом? Путь Рє сердцу мужчины смотреть онлайн бесплатно РІ хорошем качестве РЅР° СЂСѓСЃСЃРєРѕРј языке
Davidhip
| #
крым отдых в крыму
MatthewsOf
| #
Русские сериалы приятно удивляют качеством и интересными сюжетами Тайны дворцовых переворотов (сериал 2000) смотреть онлайн РІСЃРµ серии РїРѕРґСЂСЏРґ бесплатно РІ хорошем качестве
MatthewsOf
| #
Лучшие русские сериалы – это настоящая гордость кинематографа Смотреть онлайн бесплатно детективные сериалы, фильмы РІ хорошем качестве » Страница 118
Volodyanaf
| #
darknet markets onion address https://darkwebversus.com/ – darknet market
MarkNOlaf
| #
dark web link https://kingdom-darkmarketplace.com – darkmarket list
MatthewsOf
| #
Русские исторические сериалы заслуживают внимания Падение Берлина (сериал 1949) смотреть онлайн РІСЃРµ серии РїРѕРґСЂСЏРґ бесплатно РІ хорошем качестве
mostbet_esSl
| #
мостбет скачать бесплатно мостбет скачать бесплатно .
obzorcaree
| #
Подробнее в материале в блоге об обзорах электронной техники перейти
Terencesam
| #
bonuses casinos 1000 рублей за регистрацию вывод сразу без вложений
FNDaviditabe
| #
dark web link https://darkmarketdarkfox.com/ – dark web market links
Markcen
| #
Покупка квартиры без рисков? Полезные статьи и советы — https://moderntechnology22.ru/
WilliamIRrutty
| #
darknet drug market https://idarknetmarket.com/ – dark web sites
Terry Berdes
| #
[bingo](https://bingo-br.com): Atendimento ao cliente que resolve suas dúvidas com rapidez, a qualquer momento.
MatthewsOf
| #
Лучшие зарубежные сериалы – это те, которые становятся легендами РўРќРў сериалы смотреть онлайн РЅРѕРІРёРЅРєРё РІСЃРµ серии РїРѕРґСЂСЏРґ РІ хорошем качестве HD без регистрации
Lezaimus
| #
взять кредит на карту без отказа срочно – не бойтесь отказов! Этот сервис работает с любыми клиентами. Простая регистрация, моментальное одобрение. Деньги на карту без проверок.
SteelSlayer
| #
Какие изменения в бизнесе? https://www.tandem.edu.co/grados-2022-1/
Kxyufaita
| #
darknet markets links https://cannahomemarket.link/ – dark markets 2025
Volodyanaf
| #
dark markets https://darkmarketpremium.com/ – darknet market
Zoe Franeo
| #
Bônus de US$ 100 ao se cadastrar na [doce](https://doce-br.com) – cadastre-se já!
MarkNOlaf
| #
darkmarket https://kingdom-darkmarketplace.com – dark market 2025
JamesMeerb
| #
набрать безопасно
MatthewsOf
| #
Всегда жду выхода новых сезонов любимых зарубежных сериалов Мелодрамы смотреть онлайн бесплатно РІ хорошем качестве HD » Страница 276
MatthewsOf
| #
Зарубежные сериалы бывают настолько захватывающими, что невозможно оторваться Сериалы онлайн смотреть бесплатно, РЅРѕРІРёРЅРєРё сериалов РІ HD » Страница 37
MatthewsOf
| #
Какие зарубежные сериалы можно назвать лучшими в 2024 году? Западня (сериал 2024, РўР’Р¦) смотреть онлайн РІСЃРµ серии РїРѕРґСЂСЏРґ бесплатно РІ хорошем качестве
JamesMeerb
| #
набрать дешево
MatthewsOf
| #
Лучшие русские сериалы – это те, которые заставляют задуматься Сериалы онлайн смотреть бесплатно, РЅРѕРІРёРЅРєРё сериалов РІ HD » Страница 25
Markcen
| #
Как выбрать надёжного застройщика? Читайте проверенные советы https://monolitrealestate.ru/
FNDaviditabe
| #
darknet markets url https://kingdom-marketplace.com/ – darknet markets onion address
WilliamIRrutty
| #
bitcoin dark web https://asap-market-onion.com/ – darkmarket list
Sazrchx
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и других профессий по выгодным тарифам. Стоимость зависит от выбранной специальности, года выпуска и образовательного учреждения. Всегда стараемся поддерживать для покупателей адекватную ценовую политику. Для нас очень важно, чтобы дипломы были доступными для большого количества наших граждан. купить диплом москва высокого
MatthewsOf
| #
Какой зарубежный сериал стоит начать смотреть прямо сейчас? Версия (сериал 2015, Р РѕСЃСЃРёСЏ) смотреть онлайн РІСЃРµ серии РїРѕРґСЂСЏРґ бесплатно РІ хорошем качестве РЅР° СЂСѓСЃСЃРєРѕРј языке
1win_biEi
| #
1win md 1win md .
1win_ztSi
| #
1вин вход http://1win107.com.kg/ .
Kxyufaita
| #
darkmarket list https://cannahomemarket.link/ – darknet market links
MatthewsOf
| #
Люблю сериалы, в которых много неожиданных поворотов Марьина роща (сериал 2014, Р РѕСЃСЃРёСЏ) 1, 2 сезон смотреть онлайн РІСЃРµ серии РїРѕРґСЂСЏРґ бесплатно РІ хорошем качестве
Volodyanaf
| #
dark market list https://aasap-market.com/ – best darknet markets
CharlesSpolf
| #
Witamy w Slottica! Nasza platforma to harmonijne połączenie innowacji, różnorodności i najwyższych standardów bezpieczeństwa, zaprojektowane z myślą o wymagających graczach z Polski. Oferujemy nie tylko szeroki wybór gier – od dynamicznych automatów, przez strategiczne gry stołowe, po ekskluzywne sesje z live dealerami – ale także kompleksowe rozwiązania dostosowane do Twoich preferencji. Dzięki zaawansowanej technologii mobilnej, ciesz się płynnym dostępem do ulubionych rozgrywek na dowolnym urządzeniu, gdziekolwiek jesteś. Slottica Casino współpracuje wyłącznie z uznanymi dostawcami oprogramowania, gwarantując najwyższą jakość grafiki, uczciwość mechaniki i regularne aktualizacje biblioteki gier.
MarkNOlaf
| #
darknet marketplace https://drdarkfoxmarket.com – best darknet markets
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные статьи и советы — https://new-fstore.ru/
MatthewsOf
| #
Лучшие зарубежные сериалы – это те, которые заставляют думать Шахта фильм смотреть онлайн бесплатно РІ хорошем качестве РЅР° СЂСѓСЃСЃРєРѕРј языке
FNDaviditabe
| #
dark web drug marketplace https://firstdarkmarket.com/ – darkmarket url
WilliamIRrutty
| #
onion dark website https://kingdomdarkmarketonline.com/ – darknet links
Travisbub
| #
іменини соломії церковний календар
Janean Steely
| #
Como se registrar na [bet 7k](https://bet-7k.com) e receber seu bônus de US$ 100!
Kxyufaita
| #
darknet sites https://cannahomemarket.link/ – darkmarkets
Storm_Master
| #
Какой https://www.tandem.edu.co/halloween/ вариант предпочесть в бизнесе?
MatthewsOf
| #
Какой зарубежный сериал стоит посмотреть на выходных? РћРґРёРЅ настоящий день (фильм 2022) смотреть онлайн бесплатно РІ хорошем качестве
Volodyanaf
| #
dark web market links https://darkode-market.com/ – darknet market
MatthewsOf
| #
Где можно найти подборку лучших зарубежных сериалов всех времен? Семейные сериалы смотреть онлайн бесплатно РІ хорошем качестве HD 720 без регистрации » Страница 9
Carisa Botero
| #
Com o aplicativo [leao](https://leao-88.com), você tem uma experiência de apostas sem igual. Baixe o app de maneira simples, instale e desfrute da interface otimizada, com navegação rápida e fluída, proporcionando a melhor jogabilidade a qualquer hora e lugar.
MarkNOlaf
| #
dark web market list https://drdarkfoxmarket.com – dark web market list
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности https://okkord.ru/
Jewell Guy
| #
Ao se registrar como novo usuário na [luck 2](https://luck-2.com), você não só tem acesso a uma das melhores plataformas de cassino online, mas também recebe um bônus de US$ 100 para começar a jogar. Aproveite essa chance de ouro para aumentar suas chances de ganhar nas slots e outros jogos de cassino populares.
FNDaviditabe
| #
darkmarket https://firstdarkmarket.com/ – darknet marketplace
WilliamIRrutty
| #
darknet market list https://asap-market-onion.com/ – dark web drug marketplace
Debera Kirksey
| #
Bônus de US$ 100 para novos usuários: Como se registrar na [john bet](https://john-bet.com)!
prodvijenie saitov v moskve_dcOa
| #
продвижение веб сайтов москва продвижение веб сайтов москва .
MatthewsOf
| #
Зарубежные сериалы – это отличный способ подтянуть иностранный язык Криминальные сериалы смотреть онлайн бесплатно в хорошем качестве HD без регистрации » Страница 25
Kxyufaita
| #
darknet market https://cyberdarkmarkets.com/ – darknet drugs
CharlesSpolf
| #
Witamy w Slottica! Nasza platforma to harmonijne połączenie innowacji, różnorodności i najwyższych standardów bezpieczeństwa, zaprojektowane z myślą o wymagających graczach z Polski. Oferujemy nie tylko szeroki wybór gier – od dynamicznych automatów, przez strategiczne gry stołowe, po ekskluzywne sesje z live dealerami – ale także kompleksowe rozwiązania dostosowane do Twoich preferencji. Dzięki zaawansowanej technologii mobilnej, ciesz się płynnym dostępem do ulubionych rozgrywek na dowolnym urządzeniu, gdziekolwiek jesteś. Slottica Vladimir współpracuje wyłącznie z uznanymi dostawcami oprogramowania, gwarantując najwyższą jakość grafiki, uczciwość mechaniki i regularne aktualizacje biblioteki gier.
Volodyanaf
| #
darkmarket link https://darkmarketpremium.com/ – darkmarket 2025
lifeautobodymailm
| #
Experience the convergence of artistry and technology at Life Auto Body. We are proficient in revitalizing your vehicle into a unique gem . Our proficient team provides exceptional auto tuning packages . Feel the rush of driving a perfectly tuned car crafted to your taste. Visit us at https://lifeautobody.com/ and start your car’s transformation journey with us!
lkjhCype
| #
http://electrotrade.biz/images/pages/?filmu_pro_samoletu___zahvatuvaushiy_mir_v_nebe.html скачать фильм на ноутбук без торрента
MarkNOlaf
| #
dark web market urls https://cannahomedarknetdrugstore.com/ – darknet markets 2025
MatthewsOf
| #
Лучшие сериалы – это те, которые захватывают с первых серий Жуковский (фильм 1950) смотреть онлайн бесплатно в хорошем качестве
MatthewsOf
| #
Зарубежные сериалы позволяют увидеть другие культуры и традиции Казнить нельзя помиловать (сериал 2017, НТВ) 1-12 серия смотреть онлайн бесплатно в хорошем качестве
Markcen
| #
Хотите сдать квартиру в аренду? Читайте проверенные советы https://privateconstruction.ru/
MatthewsOf
| #
Зарубежные сериалы бывают настолько захватывающими, что невозможно оторваться Один на всех 1, 2 сезон (сериал 2012, Россия) смотреть онлайн все серии подряд бесплатно в хорошем качестве
FNDaviditabe
| #
darkmarkets https://onion-dark-market.shop/ – darknet markets onion address
Terencesam
| #
Игровые автоматы на деньги реальные Casino top
MetaMask Download
| #
MetaMask Chrome is essential for crypto users. It simplifies transactions, provides robust security, and integrates perfectly with DeFi.
WilliamIRrutty
| #
darknet market links https://asap-market-onion.com/ – darknet market lists
Kxyufaita
| #
dark web market links https://cannahome-darkmarket-online.com/ – dark websites
Volodyanaf
| #
darknet market links https://darkfoxdarkweb.com/ – darkmarket url
Blue60
| #
Сравнительный анализ бизнеса: https://www.tandem.edu.co/presentacion-the-space/
Devin Clampitt
| #
A [bet7](https://bet7-88.com) oferece uma oferta imperdível para novos jogadores! Ao se registrar no site, você recebe US$ 100 para apostar em diversos jogos de cassino. Essa é uma excelente oportunidade para aproveitar ao máximo sua experiência de apostas online sem riscos iniciais.
MatthewsOf
| #
Где найти список лучших русских сериалов всех времен? HD военные сериалы смотреть онлайн бесплатно в хорошем качестве » Страница 7
MarkNOlaf
| #
darkmarket link https://cannahomedarknetdrugstore.com/ – darkmarket url
Andree Henifin
| #
A [pk55](https://pk55-88.com) é a escolha ideal para quem busca diversão e segurança. Com uma ampla variedade de jogos populares e um sistema justo, você pode apostar sem preocupações. Além disso, a plataforma oferece bônus exclusivos que garantem mais chances de vitória e recompensas para todos.
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://prudmarket.ru/
MatthewsOf
| #
Какие русские сериалы стоит посмотреть в 2024 году? Сквозь чёрное стекло фильм смотреть онлайн бесплатно в хорошем качестве на русском языке
Potolki_dex
| #
Хорошо сочетаются с разными стилями: классика, модерн, минимализм – https://potolki-v-moskve.ru/
FNDaviditabe
| #
darknet market https://kingdom-marketplace.com/ – darkmarket
Rosalia Verrone
| #
Ao se registrar na [8800 bet](https://8800-bet.com), você ganha US$ 100 de bônus! O processo é simples e o login é imediato. Depois de criar sua conta, você pode começar a jogar em jogos como roleta, blackjack e slots, tudo com a vantagem do bônus de boas-vindas. Registre-se já!
WilliamIRrutty
| #
darknet markets url https://kingdomdarkmarketonline.com/ – dark web link
AlfredoBlape
| #
Looking for high-quality slots to play? AbeBet Casino offers the best online slots for all players!
At AbeBet abe-casino.com/tr/games/demo/pragmatic_play_starlight_princess_1000, you’ll find slot hits from leading providers.
Register now to get newbie gifts.
Dive into a world of winnings with AbeBet!
MatthewsOf
| #
Где можно посмотреть новый сезон моего любимого зарубежного сериала? Пустыня (сериал 2019, НТВ) смотреть онлайн все серии подряд бесплатно в хорошем качестве
Timothyvop
| #
казино онлайн
Kxyufaita
| #
onion dark website https://cyberdarkmarkets.com/ – dark market url
Volodyanaf
| #
dark web drug marketplace https://darkode-market.com/ – darkmarket list
Jariornma
| #
Где заказать диплом по нужной специальности?
Наша компания предлагает быстро и выгодно приобрести диплом, который выполняется на оригинальной бумаге и заверен мокрыми печатями, штампами, подписями. Документ способен пройти лубую проверку, даже при использовании специального оборудования. Решите свои задачи быстро с нашей компанией.
Приобрести диплом о высшем образовании kupitediplom.ru/kupit-attestati-za-9-13/
Lazravr
| #
Быстро и просто приобрести диплом ВУЗа!
Мы изготавливаем дипломы любых профессий по приятным тарифам— asxdiploman.com/kupit-diplom-s-zaneseniem-v-reestr-12/
Iariorqdb
| #
Заказать документ университета можно в нашей компании в столице. http://www.festivalcinepamplona.com/2020/10/29/hello-world
MatthewsOf
| #
Зарубежные сериалы в хорошем качестве – это просто сказка Урга: Территория любви (фильм 1991) смотреть онлайн бесплатно в хорошем качестве
MatthewsOf
| #
Русские исторические сериалы заслуживают внимания Так бывает (фильм 2007) смотреть онлайн бесплатно в хорошем качестве
lkjhCype
| #
http://arbaletspb.ru/img/pgs/?luchshie_filmu_dlya_prosmotra.html сколько будет 7 от 2000
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности https://pultmaster-pro.ru/
MarkNOlaf
| #
darknet site https://cannahomedarknetdrugstore.com/ – dark websites
CharlesPUSLY
| #
Официальный веб-сайт казино Рояль предлагает стильную обстановку для гемблеров. С его изысканным дизайном и элитным выбором игр, Рояль является превосходным местом для тех, кто обожает качественное азартное развлечение. Исследуйте широкий спектр игровых автоматов и настольных игр на основном веб-сайте royalcasino-slots.com/ru/.
Казино Рояль предоставляет высокое качество клиентов и поддержку. Внимательная команда поддержки в наличии круглосуточно, готова реагировать на любые возникающие вопросы и исправить проблемы, обеспечивая беспрерывную игру и удовлетворение.
MatthewsOf
| #
Русские сериалы в жанре драмы всегда вызывают сильные эмоции Солнце, море и любовь (сериал 2023, Домашний) смотреть онлайн все серии подряд бесплатно в хорошем качестве
DavidCooma
| #
прокат дорогого авто аренда машины адлер цены
1win_vgpt
| #
cod promoțional 1win 1win5001.ru .
Georgianne Triplette
| #
Na [bet 4](https://bet-4-br.com), você encontra uma vasta seleção de jogos justos e bônus exclusivos!
MatthewsOf
| #
Русские сериалы становятся все популярнее за рубежом НТВ сериалы смотреть онлайн новинки все серии подряд в хорошем качестве HD без регистрации » Страница 29
FNDaviditabe
| #
darknet sites https://onion-dark-market.shop/ – darknet drug links
KennethDrula
| #
Купить водительские права официально
mostbet_cgei
| #
скачать мостбет официальный сайт скачать мостбет официальный сайт .
WilliamIRrutty
| #
onion dark website https://asap-market-onion.com/ – darknet links
obzorcaree
| #
Подробнее в материале в блоге об обзорах электронной техники диджитал гаджет ссылка
Cazrmbj
| #
Приобрести диплом о высшем образовании поспособствуем. Часто задаваемые вопросы – diplomybox.com/voprosy-i-otvety
MatthewsOf
| #
Люблю зарубежные сериалы за атмосферу и крутых персонажей Помнить (фильм 2022) смотреть онлайн бесплатно в хорошем качестве на русском языке
mostbet_ygSi
| #
motsbet http://www.mostbet1002.com.kg .
Volodyanaf
| #
dark web market https://darkfoxdarkweb.com/ – darknet drug store
Kxyufaita
| #
dark web market list https://cannahomemarket.link/ – dark web link
Frankglync
| #
casebattle
MatthewsOf
| #
Лучшие зарубежные сериалы – это настоящие блокбастеры Сериалы 2019 года смотреть онлайн бесплатно, лучшие Сериалы 2019 » Страница 23
MarkNOlaf
| #
dark web market links https://dark-web-versus.com – darknet site
WolfStriker
| #
Практические советы для https://mejorsintlc.cl/litio-e-hidrogeno-verde-en-la-maleta-del-presidente-boric/ работы с бизнесом.
Williamvop
| #
На площадке представлен широкий ассортимент травматического оружия https://travmat-kupit.ru/
MatthewsOf
| #
Где найти подборку русских сериалов с высоким рейтингом? Движение вверх (фильм 2017) смотреть онлайн бесплатно в хорошем качестве на русском языке
MatthewsOf
| #
Русские сериалы про врачей стали очень популярны Нарушение правил 1 сезон 4 серия смотреть онлайн бесплатно в хорошем качестве на русском языке
FNDaviditabe
| #
dark web markets https://mydarkmarketlink.com/ – darknet websites
ThomasFette
| #
томми ли
WilliamIRrutty
| #
tor drug market https://kingdomdarkmarketonline.com/ – darknet markets onion
Cazraxz
| #
Здравствуйте!
Без ВУЗа очень непросто было продвигаться вверх по карьере. В наше время этот документ не дает гарантий, что получится получить хорошо оплачиваемую работу. Куда более важное значение имеют навыки и знания специалиста, а также его постоянный опыт. По этой причине решение о покупке диплома можно считать мудрым и рациональным. Заказать диплом любого университета milanoregola.copiny.com/question/details/id/1052647
MatthewsOf
| #
Зарубежные сериалы – это возможность окунуться в другой мир Волчий остров (фильм 2012) смотреть онлайн бесплатно в хорошем качестве
MatthewsOf
| #
Какие зарубежные сериалы стоит посмотреть в жанре фэнтези? HD триллеры смотреть онлайн бесплатно в хорошем качестве » Страница 3
Volodyanaf
| #
dark web market urls https://aasap-market.com/ – darknet drug market
Kxyufaita
| #
darknet markets links https://cyberdarkmarkets.com/ – darknet drug market
MatthewsOf
| #
Люблю сериалы, в которых много неожиданных поворотов Веселые Расплюевские дни (фильм 1966) смотреть онлайн бесплатно в хорошем качестве
1win_cypt
| #
pariuri sportive moldova http://www.1win5001.ru .
MatthewsOf
| #
Люблю сериалы с историческим сюжетом, особенно русские Платье цвета моря (сериал 2023, Россия) смотреть онлайн бесплатно все серии подряд в хорошем качестве
mostbet_zyei
| #
mostbet chrono mostbet1003.com.kg .
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы — https://repinskieozera.ru/
MarkNOlaf
| #
darknet markets https://dark-web-versus.com – dark web marketplaces
Jamesfrapy
| #
армада казино 2025
mostbet_vySi
| #
мостбет скачать казино http://mostbet1002.com.kg/ .
MatthewsOf
| #
Лучшие русские сериалы – это те, которые заставляют переживать за героев Внутри (турецкий сериал 2016) на русском языке смотреть онлайн все серии подряд бесплатно в хорошем качестве
FrankFiP
| #
https://ramregion.ru
Donaldlaf
| #
darkmarket list dark market link
Pingrutty
| #
darknet market list dark web sites
FNDaviditabe
| #
darkmarket list https://onion-dark-market.shop/ – darkmarket
Toliknaf
| #
dark markets 2025 darknet marketplace
WilliamIRrutty
| #
darknet markets https://idarknetmarket.com/ – darkmarkets
Rabyitabe
| #
darknet markets onion dark website
Volodyanaf
| #
darknet sites https://darkmarketpremium.com/ – darknet drugs
Kxyufaita
| #
bitcoin dark web https://cannahome-darkmarket-online.com/ – dark web market links
MatthewsOf
| #
Русские сериалы в жанре драмы всегда вызывают сильные эмоции Чары любви (сериал 2024, Домашний) смотреть онлайн бесплатно все серии подряд в хорошем качестве
obzorcaree
| #
Подробнее в обозревательном журнале чек-ноутбук ссылка
MatthewsOf
| #
Лучшие зарубежные сериалы – это те, которые заставляют думать Случайная любовь (турецкий сериал 2021) на русском языке смотреть онлайн все серии подряд бесплатно в хорошем качестве
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://shatimtrade-kizlyar.ru/
MatthewsOf
| #
Где можно скачать сериалы в хорошем качестве? Хроники призрачного племени (фильм 2015) на русском языке смотреть онлайн бесплатно в хорошем качестве
lkjhCype
| #
http://monetoss.ru/news/molodezghnue_serialu__pochemu_ih_tak_lubyat_.html тнт телепрограмма на сегодня тольятти
MarkNOlaf
| #
darknet market list https://kingdom-darkmarketplace.com – darknet site
DonDonfaita
| #
darknet market lists darknet market list
MatthewsOf
| #
Люблю зарубежные сериалы за оригинальные сценарии и качественную съемку Жемчужная свадьба 1 сезон 4 серия смотреть онлайн бесплатно в хорошем качестве на русском языке
Pingrutty
| #
dark web markets dark websites
MatthewsOf
| #
Какой зарубежный сериал стоит посмотреть на выходных? Спрячь меня (турецкий сериал 2023, Домашний) на русском языке смотреть онлайн все серии подряд бесплатно в хорошем качестве
Fang2203
| #
Хочу понять в https://epiczo.com/?p=254 бизнесе. Куда зайти?
MatthewsOf
| #
Где можно смотреть зарубежные сериалы бесплатно и без регистрации? Скоряк 2 сезон 30 серия смотреть онлайн бесплатно в хорошем качестве на русском языке
Donaldlaf
| #
dark market list darknet markets
FNDaviditabe
| #
darknet site https://mydarkmarketlink.com/ – dark market onion
Toliknaf
| #
darknet markets dark web markets
WilliamIRrutty
| #
darknet market list https://idarknetmarket.com/ – dark market 2025
MatthewsOf
| #
Какие русские сериалы можно посоветовать для просмотра? Семья Зацепиных (фильм 1977) смотреть онлайн бесплатно все серии подряд в хорошем качестве
Volodyanaf
| #
dark web drug marketplace https://darkfoxdarkweb.com/ – darknet drug market
Rabyitabe
| #
dark web drug marketplace darknet drugs
Terencesam
| #
Игровые автоматы с бонусом за регистрацию без депозита no deposit casino
Kxyufaita
| #
darknet markets https://cannahomemarket.link/ – darknet drug store
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://sibotdelka.ru/
MatthewsOf
| #
Зарубежные сериалы про детективов всегда на высоте Не бойся, я с тобой! (фильм 1981) смотреть онлайн бесплатно в хорошем качестве на русском языке
JasonIsole
| #
https://textme.work/
MarkNOlaf
| #
dark market list https://cannahomedarknetdrugstore.com/ – darkmarket list
Pingrutty
| #
darknet markets onion address dark web link
MatthewsOf
| #
Лучшие русские сериалы – это те, которые вызывают бурю эмоций Комедии смотреть онлайн бесплатно в хорошем качестве HD без регистрации » Страница 144
mikrozaim
| #
Микрозайм онлайн на карту без проверок срочно микрозайм онлайн на карту без проверок срочно
MatthewsOf
| #
Какие зарубежные сериалы можно посмотреть в жанре фантастики? Смотреть Сериалы 2015 онлайн бесплатно в HD качестве » Страница 7
GeorgeTom
| #
Source:
– badmintonplayer.ru
DonDonfaita
| #
darknet drug market dark web drug marketplace
vzyatzaim
| #
Займы интернет оформление займа онлайн
avtoplastics
| #
онлайн-магазин автозапчастей https://avtoplastics.ru
MatthewsOf
| #
Зарубежные сериалы – это возможность окунуться в другой мир Охотничий (фильм 2010) на русском языке смотреть онлайн бесплатно в хорошем качестве
digitalcaree
| #
Подробнее в обозревательном журнале new digital tech здесь
MatthewsOf
| #
Зарубежные сериалы – это всегда высокий уровень съемок и крутые спецэффекты Ангел-полицейский (аниме 1989) на русском языке смотреть онлайн все серии подряд бесплатно в хорошем качестве
RicardoDitle
| #
Glory Casino
FNDaviditabe
| #
dark web markets https://firstdarkmarket.com/ – dark markets 2025
Donaldlaf
| #
dark markets darknet markets onion
WilliamIRrutty
| #
darknet markets onion address https://darknetmarketsonion.shop/ – darknet markets
MatthewsOf
| #
Смотреть сериалы в хорошем качестве – настоящее удовольствие Сериалы 2023 года смотреть онлайн бесплатно, лучшие фильмы 2023, в хорошем качестве » Страница 35
Toliknaf
| #
dark market link dark market link
Volodyanaf
| #
darknet markets https://darkmarketpremium.com/ – darkmarket
Kxyufaita
| #
dark websites https://cannahome-darkmarket-online.com/ – darknet drugs
Rabyitabe
| #
darkmarket 2025 dark market 2025
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://snegirieko.com/
MatthewsOf
| #
Ищу зарубежный сериал, который можно смотреть без остановки Дом с чёрными котами (сериал 2018) смотреть онлайн все серии подряд бесплатно в хорошем качестве
Pingrutty
| #
dark markets 2025 darknet sites
MarkNOlaf
| #
tor drug market https://kingdom-darkmarketplace.com – dark web market list
DonDonfaita
| #
darknet markets darknet markets
Andrewwhone
| #
pokerdom
durmitor
| #
prirodni biser Durmitor park Jedinstveni planinski pejzazi, bistra jezera, kanjoni i guste sume. Idealno mjesto za planinarenje, rafting, skijanje i rekreaciju na otvorenom. Otkrijte nacionalni park pod zastitom UNESCO-a!
nuevoscasinosonlineespana
| #
codigo promocional de olimpo bet nuevoscasinosonlineespana.com
FNDaviditabe
| #
darknet markets https://mydarkmarketlink.com/ – darknet drug links
Andrewwhone
| #
joycasino бонус код
Donaldlaf
| #
dark market url darknet links
WilliamIRrutty
| #
darknet market list https://darknetmarketsonion.shop/ – onion dark website
Volodyanaf
| #
darkmarkets https://darkfoxdarkweb.com/ – dark web market
MatthewsOf
| #
Русские сериалы про реальную жизнь всегда интересны Мелодрамы смотреть онлайн бесплатно в хорошем качестве HD » Страница 34
Toliknaf
| #
darknet market lists dark markets 2025
mtsnovosibirsksig
| #
провайдер мтс
https://digitalbarker.com/employer/gertrude2406
мтс интернет
Kxyufaita
| #
darkmarket 2025 https://kingdomdarknetdrugstore.com/ – darknet markets
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://so35.ru/
Pingrutty
| #
darknet markets dark market url
Blade7928
| #
https://dmpublicidad.com.ar/974-2/ Пример из практики: Бизнес.
Trefujx
| #
Заказать документ университета вы сможете в нашей компании. diplom-club.com/kupit-diplom-krasnoyarsk-4
MatthewsOf
| #
Всегда ищу лучшие русские сериалы, чтобы насладиться интересными историями Татьянина ночь (сериал 2014) смотреть онлайн все серии подряд бесплатно в хорошем качестве
MarkNOlaf
| #
tor drug market https://drdarkfoxmarket.com – darknet market links
Tamojennii predstavitel Moskva_vmki
| #
таможенный брокер импорт https://tamozhennyj-predstavitel-moskva12.ru/ .
Tamojennii broker Moskva_wcKr
| #
компания таможенный брокер компания таможенный брокер .
prodvijenie saitov v moskve_oxOa
| #
продвижение сайта онлайн продвижение сайта онлайн .
DonDonfaita
| #
darkmarket link darkmarket url
1win_omPn
| #
win 1 http://cah.forum24.ru/?1-19-0-00000716-000-0-0-1741702224/ .
Sazrweg
| #
купить диплом петербург одно
FNDaviditabe
| #
bitcoin dark web https://onion-dark-market.shop/ – darkmarket link
WilliamIRrutty
| #
onion dark website https://darknetmarketsonion.shop/ – darkmarket list
mostbet_dppt
| #
скачат мостбет http://dubna.myqip.ru/?1-18-0-00000145-000-0-0-1741708632 .
Volodyanaf
| #
dark web market https://darkode-market.com/ – darkmarket 2025
Donaldlaf
| #
dark web market links dark web sites
Pingrutty
| #
darknet markets links darknet drug market
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://sochi-park-dom.ru/
Toliknaf
| #
darknet market darknet market list
MatthewsOf
| #
Лучшие русские сериалы – это те, которые заставляют задуматься Гордость мира боевых искусств (дорама 2023) на русском языке смотреть онлайн все серии подряд бесплатно в хорошем качестве
Kxyufaita
| #
tor drug market https://kingdomdarknetdrugstore.com/ – dark web sites
proektoroffcaree
| #
Подробнее в обозревательном журнале proektorOFF на сайте
Rabyitabe
| #
dark web markets dark websites
1win_eaKl
| #
1win.online https://aktivnoe.forum24.ru/?1-2-0-00000100-000-0-0-1741701286 .
MarkNOlaf
| #
dark market https://kingdomdarkmarketplace.com – darknet links
Mazrtdv
| #
Где купить диплом специалиста?
Мы оказываем услуги по изготовлению и продаже документов об окончании любых университетов РФ. Документы производят на оригинальных бланках. lightsdemons.phorum.pl/viewtopic.php?p=70808#70808
DonDonfaita
| #
dark web link dark web market urls
Pingrutty
| #
darknet markets onion darkmarkets
FNDaviditabe
| #
darkmarket https://onion-dark-market.shop/ – onion dark website
Volodyanaf
| #
darknet markets url https://darkmarketpremium.com/ – darknet markets links
mtsnovosibirsksig
| #
мтс подключение
https://www.listandrelax.com/profile/johnniechaves
мтс подключить
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности https://technovoda.ru/
Donaldlaf
| #
dark market best darknet markets
1win_wcPn
| #
1win https://cah.forum24.ru/?1-19-0-00000716-000-0-0-1741702224/ .
Kxyufaita
| #
dark web market urls https://cannahomemarket.link/ – darknet markets onion
Toliknaf
| #
tor drug market darkmarket list
mostbet_bzpt
| #
мостбет промокод http://dubna.myqip.ru/?1-18-0-00000145-000-0-0-1741708632/ .
Rabyitabe
| #
dark market darknet market list
МФО новые
| #
Когда экстренно нужны деньги, а банки не одобряют, альтернатива есть — быстрые микрозаймы. Теперь взять кредит может каждый, ключевое условие — паспорт. Наша группа экспертов собрала список из десятков малоизвестные займы на карту онлайн которые работают официально и выдают суммы до 30 тысяч гарантированно.
Даже если вам отказывали в финансирование, в этом списке шанс почти 100%! Тарифы выгодные, подписание договора моментальное. Без звонков, изучения кредитной истории и справок. Такие займы — лучшее решение, когда средства нужны здесь и сейчас.
MarkNOlaf
| #
darknet websites https://dark-web-versus.com – darkmarket link
Pingrutty
| #
dark web sites dark markets 2025
Terencesam
| #
Игровые автоматы играть на деньги 1000 рублей за регистрацию вывод сразу
DonDonfaita
| #
dark web market list dark web market urls
FNDaviditabe
| #
dark web marketplaces https://firstdarkmarket.com/ – darknet links
Volodyanaf
| #
dark market 2025 https://darkode-market.com/ – tor drug market
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы https://territoria-we-stars.ru/
WilliamIRrutty
| #
darknet marketplace https://darknetmarketsonion.shop/ – darkmarket list
МФО новые
| #
Когда срочно нужны средства, а финансовые организации отказывают, решение есть — быстрые микрозаймы. Теперь оформить займ может все, главное условие — паспорт. Наша специалисты собрала базу из более 20 самые новые займы которые легально действуют и предоставляют небольшие кредиты гарантированно.
Если раньше вам отказывали в кредит, в этом списке шанс максимальный! Тарифы низкие, подписание договора моментальное. Никаких звонков, проверок и справок. Такие займы — оптимальный вариант, когда финансы нужны здесь и сейчас.
Donaldlaf
| #
dark websites darkmarket link
1win_mqKl
| #
1wln http://www.aktivnoe.forum24.ru/?1-2-0-00000100-000-0-0-1741701286 .
Kxyufaita
| #
darknet sites https://kingdomdarknetdrugstore.com/ – dark web markets
Michaelabaks
| #
Это лучший способ вернуть себе полноценную улыбку без ущерба для соседних зубов https://dentalaesthetics.ru/
Sazrzbr
| #
Приветствую!
Приобрести диплом ВУЗа по доступной стоимости можно, обращаясь к проверенной специализированной компании. Купить диплом: diplomt-v-chelyabinske.ru/kupit-diplom-kursk-5/
Toliknaf
| #
bitcoin dark web darknet drugs
MatthewsOf
| #
Всегда жду выхода новых сезонов любимых зарубежных сериалов Драмы смотреть онлайн бесплатно в хорошем качестве HD без регистрации » Страница 20
Rabyitabe
| #
tor drug market darknet markets 2025
Lesha Hipple
| #
Venha para a [f12bet](https://f12–bet.com) e aproveite os jogos de cassino justos com bônus imperdíveis!
MarkNOlaf
| #
dark market onion https://kingdom-darkmarketplace.com – darknet market
Lesha Hipple
| #
A [bet7](https://bet-7-br.com) oferece um bônus de US$ 100 para novos jogadores assim que se registram no site. Não perca tempo e aproveite essa chance de começar a sua experiência no cassino com um bônus que vai dar aquele empurrão inicial nas suas apostas. Faça seu cadastro agora mesmo!
Pingrutty
| #
darknet market list bitcoin dark web
mtsnovosibirsksig
| #
мтс тарифы на интернет
https://empleo.infosernt.com/employer/shanna1183/
мтс тв новосибирск
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы https://texnotect.ru/
Volodyanaf
| #
darkmarket link https://darkfoxdarkweb.com/ – dark market 2025
FNDaviditabe
| #
darkmarket link https://onion-dark-market.shop/ – dark web market links
DonDonfaita
| #
darknet markets links darknet drug store
WilliamIRrutty
| #
darknet markets links https://kingdomdarkmarketonline.com/ – dark market onion
Michaelabaks
| #
Имплантация позволяет избавиться от неудобств, связанных с ношением съемных протезов: этапы имплантации зубов
MatthewsOf
| #
Зарубежные сериалы по книгам часто бывают очень интересными Эластико смотреть онлайн бесплатно в хорошем качестве на русском языке
RedZ3
| #
Где смотреть статистику о бизнесе? https://shop.platinumwellness.net/hello-world/
MatthewsOf
| #
Русские сериалы про криминал всегда интересные и захватывающие Одноклассники.ru: НаCLICKай удачу фильм смотреть онлайн бесплатно в хорошем качестве на русском языке
Lazrmfg
| #
Диплом ВУЗа России!
Без ВУЗа достаточно сложно было продвигаться по карьерной лестнице. Именно из-за этого решение о покупке диплома стоит считать мудрым и рациональным. Приобрести диплом института deves.flybb.ru/viewtopic.php?f=2&t=1168
Kxyufaita
| #
darknet websites https://cannahomemarket.link/ – dark web drug marketplace
MatthewsOf
| #
Зарубежные сериалы по книгам часто бывают очень интересными Бродяга (дорама 2019) на русском языке смотреть онлайн бесплатно все серии подряд в хорошем качестве
Toliknaf
| #
dark web marketplaces darknet links
MatthewsOf
| #
Смотреть сериалы в хорошем качестве – настоящее удовольствие Жила-была девочка фильм смотреть онлайн бесплатно в хорошем качестве на русском языке
Pingrutty
| #
darkmarkets darkmarket list
Rabyitabe
| #
dark web marketplaces darknet market
MarkNOlaf
| #
bitcoin dark web https://drdarkfoxmarket.com – darknet markets
MatthewsOf
| #
Какие русские сериалы стоит посмотреть в 2024 году? Одноклассники.ru: НаCLICKай удачу фильм смотреть онлайн бесплатно в хорошем качестве на русском языке
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://tolmi74.ru/
lkjhCype
| #
https://ourmind.ru//wp-content/news/filmu_pro_budushee__vzglyad_skvoz_prizmu_vremeni.html фильмы фул hd 1080 смотреть онлайн бесплатно
Volodyanaf
| #
darknet markets url https://darkode-market.com/ – dark web markets
MatthewsOf
| #
Где можно смотреть сериалы без регистрации и смс? Мелодрамы смотреть онлайн бесплатно в хорошем качестве HD » Страница 51
FNDaviditabe
| #
darkmarket link https://onion-dark-market.shop/ – dark market url
WilliamIRrutty
| #
dark websites https://darknetmarketsonion.shop/ – dark markets
DonDonfaita
| #
dark web drug marketplace dark web market list
Diplomi_bwKt
| #
Приобрести диплом любого университета!
Мы можем предложить документы институтов, которые расположены на территории всей РФ.
Превосходства наших дипломов:
• используем настоящие бланки “Гознака”;
• необходимые подписи руководства;
• мокрые печати учебного заведения;
• специальные водяные знаки, нити и прочие степени защиты;
• идеальное заполнение и оформление – ошибок не бывает;
• любая проверка документов.
magazin-diplomov.ru/kupit-diplom-v-bryanske-3
Kxyufaita
| #
dark market link https://cannahomemarket.link/ – dark web link
Donaldlaf
| #
best darknet markets darknet drug links
Pingrutty
| #
dark web sites darknet market
Toliknaf
| #
dark markets dark web market urls
ufc_ar
| #
everything about the world https://t.me/ufc_ar of UFC in Arabic! Latest news, tournament schedules, fighter ratings and exclusive analytical materials. Follow the best fights and stay up to date with all the events in the world of mixed martial arts!
MarkNOlaf
| #
dark markets 2025 https://kingdom-darkmarketplace.com – darknet drug links
Rabyitabe
| #
darknet markets links darknet market
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы https://topkadastr-crimea.ru/
MasonBed
| #
steam desktop authenticator github
Volodyanaf
| #
darknet market links https://aasap-market.com/ – dark web link
FNDaviditabe
| #
dark web link https://darkmarketdarkfox.com/ – onion dark website
Jamesbop
| #
Looking for best online casino games? MasalBet is the place to find favorite games from renowned providers.
At https://denizliotoekspertiz.com, you’ll find newbie gifts and a user-friendly interface.
Sign up now and get amazing bonuses.
Play true excitement with Masal Bet!
WilliamIRrutty
| #
darknet market list https://kingdommarketdarknet.com/ – dark web marketplaces
Jamesbop
| #
Игровая платформа 7k — это современная платформа для игр с ставками с огромным выбором развлечений на любой вкус. Здесь можно найти лучшие слот-машины, настольные игры и реальные игры с дилерами с опытными дилерами. Интерфейс пространный и простой в навигации, что делает игру удобной и увлекательной. На 7k740.casino предусмотрены щедрые бонусы для игроков. В целом, ресурс предоставляет отличный опыт для каждого игрока.
DonDonfaita
| #
dark web drug marketplace darknet links
JosephElari
| #
https://msk.sweet-escort.co/
Pingrutty
| #
dark market 2025 dark market link
Pioneer4200
| #
Пытаюсь разобраться в http://vitanuova.ru/u-severnoj-korei-est-svoj-planshet/ бизнесе. Что посоветуете?
Kxyufaita
| #
darknet market list https://darkfoxmarketplace.com/ – darknet markets onion address
mostbet_noKr
| #
мосбет https://www.aktivnoe.forum24.ru/?1-8-0-00000260-000-0-0-1741701879 .
lkjhCype
| #
https://yarkompany.ru/language/pages/?volshebnue_skazki___okna_v_mir_chudes.html фильмы с высоким рейтингом зарубежные смотреть онлайн
Donaldlaf
| #
darknet markets onion darknet market lists
1win_gsPn
| #
1 win официальный https://aktivnoe.forum24.ru/?1-8-0-00000259-000-0-0-1741701621/ .
Mazrpro
| #
Добрый день!
Где заказать диплом по актуальной специальности?
Мы изготавливаем дипломы любых профессий по приятным тарифам. Стоимость может зависеть от определенной специальности, года выпуска и ВУЗа. Стараемся поддерживать для заказчиков адекватную политику тарифов. Для нас очень важно, чтобы дипломы были доступны для большого количества наших граждан.
Покупка документа, подтверждающего окончание института, – это выгодное решение. Просто посчитайте, сколько понадобится потратить средств на ежемесячную оплату 5 лет обучения, на аренду квартиры (если учащийся из другого города), на проезд до ВУЗа и многие другие издержки. Получится приличная сумма, которая превышает цены на наши документы. А ведь все это время можно уже с успехом работать, развивая собственные навыки на практике.
Готовый диплом с приложением отвечает стандартам, неотличим от оригинала. Не откладывайте свои мечты и цели на потом, реализуйте их с нашей компанией – отправьте простую заявку на изготовление документа сегодня!
Диплом о высшем образовании – легко! diplomanruss.com/
Toliknaf
| #
darknet links darknet markets url
MarkNOlaf
| #
darknet markets https://cannahomedarknetdrugstore.com/ – darknet site
DonaldTob
| #
Сертификат происхождения СТ-1 подтверждает, что товар был произведен в определенной стране, что важно для таможенного оформления и применения тарифных преференций. Протокол испытания, в свою очередь, документирует результаты лабораторных исследований продукции, подтверждая соответствие определенным стандартам качества и безопасности. Сертификат соответствия на продукцию удостоверяет, что товар соответствует требованиям технических регламентов или национальных стандартов. Свидетельство о государственной регистрации продукции требуется для товаров, подлежащих санитарно-эпидемиологическому надзору, и подтверждает их безопасность для здоровья человека. Декларация ТР ТС – это документ, которым производитель или импортер подтверждает соответствие продукции требованиям технических регламентов Таможенного союза. Нотификация ФСБ требуется для ввоза или вывоза продукции, содержащей шифровальные (криптографические) средства. Декларация Минсвязи, в свою очередь, необходима для оборудования связи, подтверждая его соответствие установленным требованиям в сфере связи и телекоммуникаций. Все эти документы играют важную роль в обеспечении качества, безопасности и законности продукции, обращающейся на рынке. сертификат происхождения СТ-1
Markcen
| #
Покупка квартиры без рисков? Полезные статьи и советы — https://uk-arbat.ru/
Rabyitabe
| #
darknet drug store darknet drug store
ThomasMaw
| #
иногда лучше доверить seo профессионалу, чтобы избежать ошибок https://seo-tver.ru/
MasonBed
| #
скачать steam desktop authenticator github
mostbet_hnKa
| #
mostbet промокод mostbet промокод .
Volodyanaf
| #
best darknet markets https://darkmarketpremium.com/ – tor drug market
Danielgen
| #
ProxyLib for Startups
FNDaviditabe
| #
darknet site https://onion-dark-market.shop/ – tor drug market
WilliamIRrutty
| #
darkmarkets https://idarknetmarket.com/ – darknet websites
Pingrutty
| #
darknet markets onion address darkmarket url
kiprus
| #
аренда авто в сиде прокат машин сочи без водителя
Terencesam
| #
Бонусы без отыгрыша за регистрацию фриспины без депозита с выводом за регистрацию
DonDonfaita
| #
bitcoin dark web dark market onion
Kxyufaita
| #
darknet market links https://cannahomemarket.link/ – onion dark website
DenverPek
| #
“Witryna pia, 12 sierpnia 2018 to katastrofalna klapa! Laduje sie z opoznieniem, pliki zawsze znikaja, a pomoc kontaktuje sie bardzo wolno.
Interfejs jest nieintuicyjny, funkcje uruchamiaja sie z bledami, a produkt obrazow pozostawia wiele do zyczenia.
Zarejestrowalem sie na konto premium, kwota zostaly zabrane natychmiast, ale dostep do przywilejow nie otworzyl sie. To po prostu oszustwo!
Nie polecam przyjaciolom, zmarnujesz zasoby i oszczednosci.”
Donaldlaf
| #
darkmarkets darkmarkets
Toliknaf
| #
dark market onion dark web marketplaces
MarkNOlaf
| #
dark web sites https://kingdom-darkmarketplace.com – dark web link
Zulma Bywater
| #
https://api.lioleo.edu.vn/public/images/course/4.png.php?id=link-pg-bet
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://vernadskogo-loft.ru/
lkjhCype
| #
https://school33-perm.ru/media/pgs/?hitu_amediateki.html фильмы с высокими рейтингами смотреть онлайн бесплатно в хорошем качестве
MasonBed
| #
steam authenticator
mostbet_jnKr
| #
мостбет скачать мостбет скачать .
Billy Ishak
| #
https://lioleo.edu.vn/creatorcourse02/4.png.php?id=joker-bet-21
Rabyitabe
| #
darknet markets 2025 darknet links
Coreen Giarusso
| #
https://lioleo.edu.vn/daily01-huyen/4.png.php?id=bet-roulette
1win_gjPn
| #
1win бк https://aktivnoe.forum24.ru/?1-8-0-00000259-000-0-0-1741701621/ .
Volodyanaf
| #
darknet drug market https://darkode-market.com/ – darknet drug market
Leoma Witthuhn
| #
https://lioleo.edu.vn/daily01-huyen/4.png.php?id=bet-city
Pingrutty
| #
darknet markets onion darknet drug links
FNDaviditabe
| #
darknet markets onion https://firstdarkmarket.com/ – bitcoin dark web
mtsnovosibirsksig
| #
мтс подключить новосибирск
https://smlord.com/employer/efrain2784/
мтс подключение новосибирск
Prodvizhen_mror
| #
Как продвигать сайт успешно, изучите.
Продвижение сайтов: от теории к практике, знания.
Узнайте, как продвигать сайт, которые повысит.
Будущее онлайн-продвижения, которые изменят ваш бизнес.
Углубляемся в продвижение сайтов, доступные каждому.
Продвижение сайтов в 2025 году, которые нужно освоить.
Лучшие агентства по SEO, на которые можно положиться.
14 ошибок при продвижении сайтов, чтобы не потерять трафик.
Доступные методы SEO, без лишних затрат.
Топовые инструменты для анализа, что упростят вашу работу.
Как анализировать успех продвижения сайта, чтобы добиться результата.
Как контент влияет на трафик, не забывайте.
Успех в локальном SEO, стратегии, которые работают.
Психология пользователей и SEO, приемы, которые сработают.
Как оптимизировать сайт для мобильных, может стать вашим преимуществом.
Как выбрать метод продвижения, на основе фактов.
Значение ссылочного продвижения, чтобы повысить авторитет.
Ежегодные тренды в SEO, изучите подробнее.
Сила SMM в SEO, вовлекайте пользователей.
Как правильно оптимизировать, используйте для продвижения.
продвижение сайта в гугл продвижение сайта в гугл .
WilliamIRrutty
| #
darknet markets onion address https://asap-market-onion.com/ – dark web market list
Tuyet Measheaw
| #
https://lioleo.edu.vn/christmas-section/4.png.php?id=bet-365
Jeannie Forss
| #
https://api.lioleo.edu.vn/public/images/course/4.png.php?id=bet-1000
pinco kazino _jjmt
| #
pinko casino pinko casino .
mostbet_peKa
| #
мостбет скачать cah.forum24.ru/?1-19-0-00000715-000-0-0-1741702061 .
ufc-ar
| #
Follow UFC fights with https://t.me/s/ufc_ar full tournament schedule, fight results, fighter ratings and analytics. Exclusive news, reviews and interviews with top MMA athletes – all in one place!
DonDonfaita
| #
darknet drug store darknet market links
pinco kazino _xzSn
| #
pinko casino pinko casino .
casino-reyting
| #
Рейтинг казино лицензионные https://t.me/casino_list_reyting/
Kxyufaita
| #
darknet links https://kingdomdarknetdrugstore.com/ – dark market url
DanielRup
| #
Уже много лет мы предоставляем услуги по ремонту и обслуживанию автомобилей Citroen, обеспечивая надежность и безопасность вашего автомобиля. Наши специалисты проводят диагностику, замену запчастей и техническое обслуживание как для владельцев легковых автомобилей, так и для корпоративных клиентов, гарантируя высокий уровень сервиса и индивидуальный подход к каждому автомобилю https://citroen-avtoservise.ru/
JamesPat
| #
задумываетесь об улучшении позиций вашего сайта в поисковой выдаче? возможно, вам будет полезен этот материал https://1st-seo.ru/
Phantom_Warden
| #
Лучшие инструменты для работы с бизнесом: http://www.pureatz.com/recipes/penne-with-shrimp-and-asparagus/
Donaldlaf
| #
darknet drug market darkmarkets
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://worldstone33.ru/
MarkNOlaf
| #
dark markets 2025 https://kingdomdarkmarketplace.com – darkmarket
Toliknaf
| #
darknet drug links dark market
Davidpeark
| #
Zajrzalem na strone 1496 bridge convention, i bez owijania w bawelne, calkowicie zawiedziony. Oprawa wizualna wyglada przestarzale, czesto pojawiaja sie problemy podczas otwierania, co utrudnia sprawnym dzialaniu. Nie wspominajac o tym, ze prezentacja informacji nie jest najlepsza. Support praktycznie nie istnieje, co sprawia, ze sytuacja staje sie jeszcze bardziej uciazliwa. Dostepne gry, jesli w ogole mozna je tak nazwac, sa calkowicie nieciekawe i nawet czesto sie zawieszaja. Nikomu tracic swojego czasu na te strone, jesli chcecie przyjemnej gry.
Lavina Mantano
| #
https://lioleo.edu.vn/app-dang-ky-thanh-cong/4.png.php?id=bet-slot-apk
Pingrutty
| #
dark web drug marketplace dark markets
Rabyitabe
| #
dark web marketplaces dark market onion
Volodyanaf
| #
darknet sites https://aasap-market.com/ – darknet markets onion
Malorie Parrot
| #
https://api.lioleo.edu.vn/public/images/course/4.png.php?id=bet-pemda
FNDaviditabe
| #
darknet links https://darkmarketdarkfox.com/ – darknet market links
WilliamIRrutty
| #
darkmarket url https://darknetmarketsonion.shop/ – darknet market
Selina Wasielewski
| #
https://lioleo.edu.vn/creatorcourse01/4.png.php?id=guci-bet
DonDonfaita
| #
best darknet markets darkmarkets
Kxyufaita
| #
darknet drug links https://cyberdarkmarkets.com/ – darknet drug store
Lazroqw
| #
Где заказать диплом по необходимой специальности?
Приобрести диплом института по невысокой цене возможно, обращаясь к проверенной специализированной фирме.: magazin-diplomov.ru
lkjhCype
| #
https://allboiler.ru/news/poznavatelnoe_kino__uchimsya__razvivaemsya_i_vdohnovlyaemsya.html fc zenit ru официальный сайт
WilliamDix
| #
Мы изготавливаем дипломы любой профессии по приятным тарифам. Дипломы производятся на подлинных бланках государственного образца Заказать диплом о высшем образовании diplomt-tver69.ru
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://komproekt-spb.ru/
Donaldlaf
| #
dark market onion darknet markets url
MarkNOlaf
| #
darknet marketplace https://dark-web-versus.com – dark market 2025
Pingrutty
| #
dark markets dark web market
Toliknaf
| #
darknet drugs dark web market
mtsnovosibirsksig
| #
провайдер мтс
https://bcde.ru/employer/autumn8982/
мтс подключить
Volodyanaf
| #
darknet markets onion address https://darkode-market.com/ – bitcoin dark web
Rabyitabe
| #
best darknet markets darknet sites
FNDaviditabe
| #
darkmarket url https://darkmarketdarkfox.com/ – dark markets
WilliamIRrutty
| #
dark web link https://asap-market-onion.com/ – darknet links
бнанс Створити акаунт
| #
I don’t think the title of your article matches the content lol. Just kidding, mainly because I had some doubts after reading the article.
Kxyufaita
| #
dark markets 2025 https://kingdomdarknetdrugstore.com/ – onion dark website
Cyndy Wadkins
| #
https://lioleo.edu.vn/app-dang-ky-thanh-cong/4.png.php?id=golf-bet
DonDonfaita
| #
tor drug market darknet marketplace
Mazrkun
| #
Где купить диплом по необходимой специальности?
Готовый диплом со всеми печатями и подписями 100% отвечает запросам и стандартам Министерства образования и науки, неотличим от оригинала. Не откладывайте свои мечты и задачи на потом, реализуйте их с нами – отправляйте быструю заявку на диплом уже сегодня! Диплом о среднем образовании – не проблема! diplom-top.ru/kupit-diplom-medrabotnika-bistro-i-nadezhno/
Pingrutty
| #
dark market list dark web market list
Markcen
| #
Новостройки или вторичка? Узнайте подробности https://konsta-ovk.ru/
Lindsay Warlick
| #
https://lioleo.edu.vn/creatorcourse01/4.png.php?id=qq1221-bet
MarkNOlaf
| #
dark web markets https://drdarkfoxmarket.com – darknet drug links
Donaldlaf
| #
darknet drug store darkmarket list
lkjhCype
| #
https://clockfase.com/auth/elmn/?filmu_na_kinogo___luchshiy_vubor_dlya_lubiteley_kino.html скачать фильм на телефон без регистрации
Toliknaf
| #
darknet websites darkmarkets
Volodyanaf
| #
darkmarket url https://darkode-market.com/ – dark market link
Dark555
| #
Авторитетное мнение о бизнесе: https://mattaarquitectos.es/portfolio/avenida-reina-victoria-8-madrid/main-31/
Rabyitabe
| #
darknet markets links darknet markets onion
FNDaviditabe
| #
dark web market list https://onion-dark-market.shop/ – darkmarket url
WilliamIRrutty
| #
darknet market list https://asap-market-onion.com/ – best darknet markets
Sazrdev
| #
Очень часто случается так, что для продвижения по карьерной лестнице, требуется документ, который подтверждает наличие профильного образования. Где заказать диплом специалиста?
Купить документ института вы имеете возможность у нас. Мы предлагаем документы об окончании любых университетов РФ. bagira-school.com.ua/
Xazrojr
| #
Приобрести диплом о высшем образовании!
Мы изготавливаем дипломы любых профессий по приятным ценам. Вы покупаете документ через надежную и проверенную временем компанию. : diplomc-v-ufe.ru/kupit-diplom-veip-o-visshem-obrazovanii-na-blanke-goznak/
Terencesam
| #
Top casino с выводом 1000 рублей за регистрацию вывод сразу
Christian Fonteneau
| #
https://api.lioleo.edu.vn/public/images/skills/4.png.php?id=jet88-bet
Kxyufaita
| #
darknet site https://cannahome-darkmarket-online.com/ – dark market url
Pingrutty
| #
dark web market links dark web drug marketplace
Dnrtqro
| #
Где купить диплом специалиста?
Мы изготавливаем дипломы любой профессии по невысоким тарифам. Мы готовы предложить документы ВУЗов, расположенных в любом регионе Российской Федерации. Можно купить диплом за любой год, указав необходимую специальность и хорошие оценки за все дисциплины. Документы выпускаются на “правильной” бумаге высшего качества. Это дает возможности делать настоящие дипломы, не отличимые от оригинала. Документы будут заверены необходимыми печатями и подписями. Стараемся поддерживать для заказчиков адекватную политику тарифов. Для нас важно, чтобы документы были доступны для большого количества граждан. kupitediplom0029.ru/kupit-diplom-rossiya-2-3
DonDonfaita
| #
dark web sites dark web sites
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://monsalvat.ru/
mtsnovosibirsksig
| #
провайдер мтс
https://cannabisjobs.solutions/companies/daniella5036/
мтс тарифы новосибирск
MarkNOlaf
| #
darknet markets onion https://cannahomedarknetdrugstore.com/ – darknet links
Sazrkkf
| #
Приобрести диплом о высшем образовании !
Покупка диплома любого ВУЗа России у нас является надежным делом, потому что документ будет заноситься в реестр. Печать выполняется на фирменных бланках ГОЗНАКа. Купить диплом об образовании arenadiplom24.online/vuzy/uralskij-gosudarstvennyj-universitet-putej-soobshcheniya
Donaldlaf
| #
dark web market darkmarket link
Volodyanaf
| #
darknet drug store https://darkmarketpremium.com/ – darkmarket
Toliknaf
| #
darknet links darknet drugs
Cazrrzs
| #
Приобрести диплом университета по невысокой цене вы сможете, обратившись к проверенной специализированной фирме. Мы можем предложить документы учебных заведений, расположенных в любом регионе РФ. diplomers.com/kak-kupit-diplom
GeorgeTom
| #
Source:
– eurekatomsk.ru
FNDaviditabe
| #
darknet site https://kingdom-marketplace.com/ – dark web market urls
BryanNeisM
| #
Играйте в Mostbet Casino mostbet spas extreme лицензированное онлайн-казино с огромным выбором игр! Получайте бонусы, выигрывайте в лучших слотах и наслаждайтесь быстрыми выплатами. Удобные способы пополнения счета!
Rabyitabe
| #
darkmarket url dark web markets
Diplomi_dnKt
| #
Приобрести диплом университета!
Мы готовы предложить документы университетов, расположенных на территории всей РФ.
diplom-onlinex.com/magistr-kupit-diplom-2
Sazrfll
| #
Заказать диплом университета по невысокой цене возможно, обращаясь к надежной специализированной компании. Мы оказываем услуги по производству и продаже документов об окончании любых университетов России. Приобрести диплом университета– diploma-groups24.ru/kupit-diplom-v-gorode/cherepovec.html
WilliamIRrutty
| #
dark web link https://kingdomdarkmarketonline.com/ – darknet market
Pingrutty
| #
dark web sites darknet market links
Anjelica Mac
| #
https://lioleo.edu.vn/app-dang-ky-thanh-cong/4.png.php?id=ppg-bet
Kxyufaita
| #
dark web market urls https://darkfoxmarketplace.com/ – darkmarket 2025
lkjhCype
| #
https://taktikiipraktiki.ru/news/serialu_pro_derevnu__prostota__iskrennost_i_osobaya_atmosfera.html смотреть фильм гримм все серии
Markcen
| #
Как выбрать надёжного застройщика? Читайте проверенные советы https://chelstg.ru/
Niki Rhea
| #
https://lioleo.edu.vn/christmas-section/4.png.php?id=bet-hoki-77
Anthony_Inads
| #
накрутка лайков в ВК на пост бесплатно
DonDonfaita
| #
darknet markets darknet site
Jamesenved
| #
Автополив участка — идеальное решение для вашего сада и дачи! Наша ландшафтная студия предлагает профессиональный монтаж автоматической системы орошения, которая гарантирует эффективный полив и уход за вашим газоном и растениями. Забудьте о рутинном поливе! Мы проведем проектирование, установку и настройку оборудования под ваши нужды. С нашим сервисом вы сможете наслаждаться уютом загородного участка без забот. Обеспечьте своим растениям качественный уход с помощью автополива https://1landshaftniy-dizayn.ru/
Charlesnag
| #
Выбираем недвижимость в Киеве https://automat.kiev.ua как оценить район, новостройки и вторичный рынок? Анализ цен, проверка застройщика, юридическая безопасность сделки. Полезные советы для покупки квартиры без ошибок и переплат!
Anthony_Inads
| #
накрутка лайков в ВК онлайн бесплатно
Jamespeege
| #
Строительный онлайн журнал https://kero.com.ua ваш источник актуальной информации о строительстве и ремонте! Советы, обзоры материалов, технологии, тренды и новости отрасли. Узнайте о современных методах строительства, инженерных решениях и дизайне интерьера!
MarkNOlaf
| #
bitcoin dark web https://kingdomdarkmarketplace.com – best darknet markets
Volodyanaf
| #
darknet markets links https://darkode-market.com/ – dark websites
Donaldlaf
| #
darkmarket link darkmarket list
FNDaviditabe
| #
dark web marketplaces https://firstdarkmarket.com/ – darkmarket link
Toliknaf
| #
dark web marketplaces tor drug market
WilliamIRrutty
| #
darkmarkets https://darknetmarketsonion.shop/ – bitcoin dark web
Pingrutty
| #
dark web market list darkmarket
Rabyitabe
| #
dark market link darknet markets links
Rickie Litteral
| #
https://lioleo.edu.vn/creatorcourse02/4.png.php?id=win-win-bet
Kxyufaita
| #
darknet markets onion address https://cyberdarkmarkets.com/ – darknet market list
VortexV5
| #
Подкаст о https://schule-am-volkspark.de/plusminus-umfrage бизнесе.
Michaelimami
| #
frumzi.gr.com
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности https://rentalsea.ru/
DanielRak
| #
Аркада казино, или казино аркада, – это относительно новое направление в индустрии азартных игр, которое стремится сочетать элементы классических аркадных игр с традиционными казино-играми. Этот гибридный формат нацелен на привлечение более молодой аудитории, выросшей на видеоиграх, и тех, кто ищет более интерактивный и захватывающий опыт, нежели стандартные слоты и настольные игры. аркада казино бонусы
1win_seml
| #
1win. com http://1win112.com.kg/ .
MarkNOlaf
| #
dark market url https://cannahomedarknetdrugstore.com/ – dark web markets
DonDonfaita
| #
darknet markets 2025 darknet market links
1win_ajkn
| #
1вин сайт https://www.1win113.com.kg .
Volodyanaf
| #
dark web market list https://darkmarketpremium.com/ – darknet websites
Jamesenved
| #
Автополив участка – это идеальное решение для тех, кто ценит время и хочет эффективно ухаживать за своим садом и газоном. Наша ландшафтная студия предлагает полный спектр услуг по проектированию, установке и обслуживанию автоматических систем орошения. С помощью современного оборудования вы сможете легко и быстро полить ваш дачный участок, теплицу или парк. Наши специалисты проведут пуско-наладочные работы, настроят систему под ваши нужды и обеспечат качественное обслуживание. Не упустите возможность упростить уход за вашим озеленением с помощью автополива: сайт
1win_qypi
| #
1 win https://1win713.ru .
Donaldlaf
| #
darknet site darkmarket
Pingrutty
| #
dark market link dark web market urls
FNDaviditabe
| #
darknet market https://onion-dark-market.shop/ – dark web marketplaces
Toliknaf
| #
darknet drug links darknet drug market
WilliamIRrutty
| #
darknet marketplace https://kingdomdarkmarketonline.com/ – onion dark website
Rabyitabe
| #
darkmarkets dark market link
Michaelsoifs
| #
Glory Casino
Kxyufaita
| #
dark market 2025 https://cyberdarkmarkets.com/ – dark market link
Gianna Hardridge
| #
https://lioleo.edu.vn/christmas-section/4.png.php?id=8364-bet
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://ramregion.ru/
Michaelsoifs
| #
Glory Casino
MarkNOlaf
| #
dark markets https://cannahomedarknetdrugstore.com/ – darkmarket link
Volodyanaf
| #
darknet markets 2025 https://darkfoxdarkweb.com/ – tor drug market
DonDonfaita
| #
darknet drugs dark web drug marketplace
Pingrutty
| #
darkmarkets dark web market urls
1win_njml
| #
1 вин вход в личный кабинет http://1win112.com.kg/ .
1win_uakn
| #
1win официальный сайт вход 1win113.com.kg .
FNDaviditabe
| #
dark web link https://firstdarkmarket.com/ – dark web market links
Donaldlaf
| #
dark web market urls darknet markets
WilliamIRrutty
| #
dark web market https://kingdomdarkmarketonline.com/ – darkmarkets
Terencesam
| #
спины за регистрацию без депозита с выводом Играть игровые автоматы на деньги
Toliknaf
| #
darknet drug market dark web market links
1win_ippi
| #
1win зайти 1win зайти .
Michaelsoifs
| #
Glory Casino
Kxyufaita
| #
darknet markets onion address https://cannahome-darkmarket-online.com/ – darkmarket 2025
Rabyitabe
| #
darknet market darknet marketplace
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности https://imcongroup.ru/
Kartu Poker
| #
thanks everything will change after you post this blog
MarkNOlaf
| #
dark market 2025 https://kingdom-darkmarketplace.com – darkmarket link
AlphaV1
| #
Как не сдаться при изучении бизнеса: https://aroagardenbar.com.br/hello-world-2/
Molly Arbogust
| #
https://lioleo.edu.vn/christmas-section/4.png.php?id=win-bet-88
Pingrutty
| #
dark web markets darknet markets links
DonDonfaita
| #
darknet sites dark markets
Michaelsoifs
| #
Glory Casino
mtsekaterinburgsig
| #
мтс екатеринбург
https://timviec24h.com.vn/companies/24inetrnet-domashnij-ekb-3/
мтс тв
Michaelsoifs
| #
Glory Casino
FNDaviditabe
| #
tor drug market https://darkmarketdarkfox.com/ – darkmarket
Michaelimami
| #
https://frumzi.gr.com/
Michaelsoifs
| #
Glory Casino
Michaelsoifs
| #
Glory Casino
Donaldlaf
| #
dark web link darknet markets url
WilliamIRrutty
| #
dark web market https://kingdommarketdarknet.com/ – darknet market list
Michaelsoifs
| #
Glory Casino
Toliknaf
| #
darknet marketplace dark web market links
Kxyufaita
| #
darknet markets https://kingdomdarknetdrugstore.com/ – dark markets
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://realsfera.ru/
Rabyitabe
| #
darknet markets onion address tor drug market
Michaelsoifs
| #
Glory Casino
Pingrutty
| #
darknet market links dark market list
Volodyanaf
| #
dark markets 2025 https://darkwebversus.com/ – darknet market list
DonDonfaita
| #
tor drug market dark markets
FNDaviditabe
| #
darknet markets https://kingdom-marketplace.com/ – darknet markets 2025
webpage
| #
Incredible points. Outstanding arguments. Keep up the amazing
effort.
WilliamIRrutty
| #
tor drug market https://darknetmarketsonion.shop/ – dark web market
Donaldlaf
| #
dark market onion dark market
prodvijenie saitov v moskve_qlpt
| #
сео сайта prodvizhenie-sajtov-v-moskve221.ru .
prodvijenie saitov v moskve_nvKl
| #
заказать продвижение сайтов заказать продвижение сайтов .
WilliamDog
| #
Всё для мужчин https://kompanion.com.ua в одном месте! Спорт, мода, авто, гаджеты, здоровье и лайфхаки. Читай полезные советы, следи за трендами и будь лучшей версией себя.
HenryTut
| #
Новости шоу-бизнеса https://mediateam.com.ua мода, красота и афиша – всё в одном месте! Узнавайте о главных событиях, свежих трендах и топовых мероприятиях. Будьте в курсе всего, что происходит в мире стиля и развлечений!
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации — https://talisman-65.ru/
Kxyufaita
| #
dark markets https://cannahome-darkmarket-online.com/ – tor drug market
Aaronaleft
| #
Все автомобильные новости https://nmiu.org.ua в одном месте! Новые модели, обзоры, сравнения, тест-драйвы, технологии и автоиндустрия. Следите за тенденциями и оставайтесь в курсе событий автопрома!
Pingrutty
| #
dark market list dark market link
Rabyitabe
| #
onion dark website darknet drugs
MarkNOlaf
| #
darknet links https://dark-web-versus.com – tor drug market
mtsekaterinburgsig
| #
мтс домашний интернет екатеринбург
https://stepstage.fr/employer/24inetrnet-domashnij-ekb/
мтс подключить
GeorgeTom
| #
Source:
https://eurekatomsk.ru/
Sazrmtn
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и любых других профессий по выгодным тарифам. Цена зависит от определенной специальности, года получения и образовательного учреждения. Стараемся поддерживать для клиентов адекватную политику цен. Важно, чтобы дипломы были доступны для большого количества граждан. купить диплом форум
Blue_Reaper
| #
Что лучше выбрать в бизнесе? https://kubalitours.com/family-friendly-resorts-in-coastal-kenya/
Cazrjlx
| #
Привет!
Мы изготавливаем дипломы любой профессии по приятным тарифам. Стоимость будет зависеть от определенной специальности, года получения и университета: rdiploman.com/
Uazrpsw
| #
Здравствуйте!
Для некоторых людей, заказать диплом о высшем образовании – это необходимость, возможность получить отличную работу. Но для кого-то – это желание не терять огромное количество времени на учебу в университете. С какой бы целью вам это не потребовалось, мы готовы помочь. Быстро, профессионально и выгодно сделаем диплом нового или старого образца на подлинных бланках со всеми печатями.
Главная причина, почему многие люди прибегают к покупке документов, – получить определенную работу. Например, навыки и опыт дают возможность человеку устроиться на работу, однако документального подтверждения квалификации нет. Если работодателю важно наличие “корочек”, риск потерять вакантное место очень высокий.
Приобрести документ ВУЗа можно в нашей компании в Москве. Мы предлагаем документы об окончании любых ВУЗов РФ. Вы сможете получить диплом по любой специальности, любого года выпуска, в том числе документы старого образца. Даем 100% гарантию, что в случае проверки документа работодателем, подозрений не возникнет.
Ситуаций, которые вынуждают заказать диплом ВУЗа очень много. Кому-то очень срочно требуется работа, в итоге необходимо произвести особое впечатление на начальство на протяжении собеседования. Некоторые хотят устроиться в престижную компанию, чтобы повысить собственный статус в обществе и в последующем начать собственное дело. Чтобы не тратить драгоценное время, а сразу начать удачную карьеру, используя имеющиеся знания, можно приобрести диплом в интернете. Вы сможете быть полезным в социуме, обретете финансовую стабильность максимально быстро и просто- купить диплом техникума
DonDonfaita
| #
darknet markets onion address darknet marketplace
FNDaviditabe
| #
dark web market list https://kingdom-marketplace.com/ – dark web market urls
WilliamIRrutty
| #
dark market link https://asap-market-onion.com/ – dark market 2025
Donaldlaf
| #
darknet websites darknet markets onion
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://brigantina-stroy.ru/
Pingrutty
| #
dark web market urls darknet markets onion
Kxyufaita
| #
dark web market https://cannahomemarket.link/ – darknet drug market
Toliknaf
| #
darkmarket url darknet site
Franksaumn
| #
Актуальные новости бизнеса https://rentwell.in.ua экономики и недвижимости! Аналитика, тренды, инвестиции, рынок недвижимости и финансовые прогнозы. Будьте в курсе главных событий и принимайте взвешенные решения!
JamesLep
| #
Выбор недвижимости в Киеве https://odinden.com.ua на что обратить внимание? Анализ районов, проверка застройщиков, сравнение цен на новостройки и вторичное жилье.
Franciswealk
| #
Лучший женский портал https://rosetti.com.ua Стиль, красота, здоровье, семья, карьера и саморазвитие. Узнавайте о новых трендах, читайте полезные советы и вдохновляйтесь идеями для счастливой жизни!
MarkNOlaf
| #
dark market url https://drdarkfoxmarket.com – darkmarket link
Volodyanaf
| #
dark web drug marketplace https://darkwebversus.com/ – best darknet markets
Rabyitabe
| #
darkmarket link darkmarket link
Terencesam
| #
bezdep Играть игровые автоматы на деньги
Odellnus
| #
https://cross.expert/partner/mrt-pozvonochnika-vazhnyy-metod-diagnostiki.html
DonDonfaita
| #
dark web link dark web market urls
FNDaviditabe
| #
darknet markets https://kingdom-marketplace.com/ – dark websites
lkjhCype
| #
http://dentaone.ru/wp-content/pages/uzghasu_na_kinogo___parad_igolok_dlya_nervov_i_fantaziy.html кинопремьеры смотреть онлайн бесплатно в хорошем качестве
WilliamIRrutty
| #
darknet drug market https://darknetmarketsonion.shop/ – darkmarket list
Pingrutty
| #
dark market list dark web market urls
mostbet_yemt
| #
mostbet игры https://mostbet1011.com.kg/ .
mostbet_ufMr
| #
мрстбет http://www.mostbet1004.com.kg .
Markcen
| #
Хотите сдать квартиру в аренду? Читайте проверенные советы https://kazhdogopravo.ru/
Kxyufaita
| #
dark market list https://cyberdarkmarkets.com/ – dark market 2025
Donaldlaf
| #
dark web marketplaces tor drug market
1win_ng_ifSi
| #
1 win nigeria 1 win nigeria .
mtsekaterinburgsig
| #
мтс тарифы на интернет
https://www.workinternational-df.com/employer/24inetrnet-domashnij-ekb-3/
сайт мтс екатеринбург
Michaelsoifs
| #
Glory Casino
Toliknaf
| #
darknet markets links onion dark website
Stephenloole
| #
Современный женский портал https://womanlife.kyiv.ua мода, стиль, красота, здоровье, отношения и карьера. Читайте актуальные статьи, находите полезные советы и вдохновляйтесь на новые достижения!
Volodyanaf
| #
darknet site https://darkmarketpremium.com/ – dark websites
MarkNOlaf
| #
darknet site https://kingdom-darkmarketplace.com – darknet sites
HaroldKnita
| #
Все для женщин https://timelady.kyiv.ua в одном месте! Новости моды, бьюти-советы, отношения, карьера, психология и лайфхаки. Читайте статьи, находите вдохновение и будьте уверены в себе каждый день!
Jordanmip
| #
Детский портал о здоровье https://run.org.ua полезная информация для заботливых родителей! Советы педиатров, питание, развитие, вакцинация, профилактика болезней и здоровый образ жизни. Всё, что нужно знать о здоровье вашего ребенка!
Michaelsoifs
| #
Glory Casino
Rabyitabe
| #
dark websites bitcoin dark web
Peternig
| #
The cryptocurrency market remains a whirlwind of activity, with Bitcoin price predictions oscillating wildly amidst global economic uncertainty. Ethereum’s market analysis reveals a struggle to maintain momentum, weighed down by network congestion and scaling challenges, even as the Merge’s long-term potential remains undeniable. Investors are keenly eyeing top altcoins, searching for the next breakout star, while bracing themselves for potential market corrections. Blockchain adoption updates
Charlescon
| #
накрутка дешево
Pingrutty
| #
darknet drug market darkmarket url
FNDaviditabe
| #
darknet markets onion https://darkmarketdarkfox.com/ – darkmarket
DonDonfaita
| #
best darknet markets dark market url
WilliamIRrutty
| #
dark web sites https://darknetmarketsonion.shop/ – darknet markets 2025
frumzi_Wob
| #
https://frumzi.xn--qxam/
RedV3
| #
Повышение эффективности через бизнес: https://asesinosasueldo.org/busco-un-sicario/.
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://1domovoy.ru/
Hubertfoend
| #
https://salda.ws/f/topic.php?f=12&t=81373
Kxyufaita
| #
darkmarket https://darkfoxmarketplace.com/ – darknet market list
lkjhCype
| #
https://rumol.ru/wp-includes/wli/smotret_serialu_onlayn___udobstvo_i_vubor_dlya_kazghdogo.html фильмы для просмотра на вечер
Donaldlaf
| #
darknet markets 2025 dark web link
sohbet
| #
great blog very nice articles I will follow you continuously sohbet odaları
Volodyanaf
| #
darknet market links https://darkmarketpremium.com/ – onion dark website
mostbet_ygmt
| #
mostbet kg скачать на андроид mostbet kg скачать на андроид .
MarkNOlaf
| #
darknet market list https://kingdom-darkmarketplace.com – darknet drugs
mostbet_omMr
| #
mostbet скачать https://mostbet1004.com.kg/ .
localtorontomovers
| #
toronto moving and storage company movers toronto
LandonOveta
| #
Всё для женщин https://allwoman.kyiv.ua в одном месте! Советы по стилю, уходу за собой, психологии, семье, карьере и саморазвитию. Узнавайте о новинках моды, секретах успешных женщин и будьте на шаг впереди!
Jimmyjab
| #
Современный женский сайт https://dama.kyiv.ua красота, стиль, здоровье, семья и карьера. Читайте актуальные статьи, находите полезные советы и вдохновляйтесь на новые достижения!
Rabyitabe
| #
dark market list best darknet markets
1win_ng_ntSi
| #
1win online games https://1win11.com.ng/ .
Pingrutty
| #
darknet sites dark web marketplaces
binance註冊獎金
| #
Can you be more specific about the content of your article? After reading it, I still have some doubts. Hope you can help me.
FNDaviditabe
| #
darknet market lists https://kingdom-marketplace.com/ – dark market list
mtsekaterinburgsig
| #
мтс подключить
http://modiyil.com/profile/will2470972029
мтс подключение
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности https://anrespectplus.ru/
WilliamIRrutty
| #
darknet drugs https://kingdomdarkmarketonline.com/ – best darknet markets
DonDonfaita
| #
dark market 2025 onion dark website
Kxyufaita
| #
darkmarket 2025 https://cannahome-darkmarket-online.com/ – darknet drug market
Volodyanaf
| #
darknet drugs https://darkode-market.com/ – darknet sites
Donaldlaf
| #
onion dark website darknet marketplace
MarkNOlaf
| #
dark market onion https://drdarkfoxmarket.com – darknet marketplace
Toliknaf
| #
darkmarkets darknet markets 2025
Pingrutty
| #
dark web market dark web link
Rabyitabe
| #
dark web market best darknet markets
FNDaviditabe
| #
darknet markets 2025 https://onion-dark-market.shop/ – darkmarkets
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://17metrov.ru/
WilliamIRrutty
| #
darkmarket 2025 https://asap-market-onion.com/ – darknet markets 2025
DonDonfaita
| #
tor drug market dark web link
Michaelles
| #
Женский онлайн-журнал https://krasotka.kyiv.ua мода, красота, здоровье, семья и карьера. Полезные статьи, тренды, лайфхаки и вдохновение для современной женщины. Читайте, развивайтесь и наслаждайтесь жизнью!
Moshebuh
| #
Онлайн-журнал для женщин https://otnoshenia.net которые стремятся к лучшему! Советы по стилю и макияжу, секреты счастливых отношений, здоровое питание, психология и идеи для отдыха.
Danieljoymn
| #
Лучший сайт для женщин https://model.kyiv.ua секреты красоты, стильные образы, отношения, карьера, лайфхаки и вдохновение. Оставайтесь в курсе трендов, читайте экспертные советы и развивайтесь вместе с нами!
Kxyufaita
| #
dark web market https://darkfoxmarketplace.com/ – darknet drugs
lkjhCype
| #
https://rosohrancult.ru/wp-includes/pages/filmu_142.html пастбище на лесной поляне 5 букв сканворд
Volodyanaf
| #
dark web sites https://darkode-market.com/ – darknet markets onion
DragonV3
| #
Что нельзя делать с бизнесом? https://seointellect.com/2023/04/09/boost-your-online-presence-our-top-digital-marketing/
ArthurElelp
| #
https://wiki.lovettcreations.org/index.php/Voditel_71V
MarkNOlaf
| #
darknet site https://cannahomedarknetdrugstore.com/ – dark web market list
Pingrutty
| #
dark market darkmarket list
ArthurElelp
| #
https://dptotti.fic.edu.uy/mediawiki/index.php/Voditel_52o
Toliknaf
| #
darknet market list darknet drugs
Rabyitabe
| #
darknet sites dark web market urls
mtsekaterinburgsig
| #
мтс подключение екатеринбург
https://skillsdiscover.com/profile/anitaalcock302
мтс тарифы на интернет
FrankFiP
| #
https://akvilon152.ru
ArthurElelp
| #
http://kousokuwiki.org/wiki/%E5%88%A9%E7%94%A8%E8%80%85:CortezSpyer5372
Markcen
| #
Новостройки или вторичка? Полезные рекомендации — https://ubkrent.ru/
GeorgeTom
| #
Source:
https://eurekatomsk.ru/
FNDaviditabe
| #
darkmarket list https://mydarkmarketlink.com/ – dark market link
Sazrlye
| #
Мы изготавливаем дипломы любой профессии по приятным тарифам. Преимущества покупки документов в нашем сервисе
Вы заказываете диплом через надежную фирму. Такое решение позволит вам сохранить не только массу средств, но и ваше драгоценное время.
Плюсов намного больше:
• Дипломы изготавливаются на подлинных бланках с мокрыми печатями и подписями;
• Дипломы любого ВУЗа России;
• Цена в разы ниже той, которую пришлось бы платить на очном обучении в университете;
• Доставка как по Москве, так и в другие регионы Российской Федерации.
Заказать диплом института– http://powerofmony.copiny.com/question/details/id/1061041/ – powerofmony.copiny.com/question/details/id/1061041
ArthurElelp
| #
https://techuswiki.xyz/index.php/Voditel_39A
WilliamIRrutty
| #
darknet market links https://idarknetmarket.com/ – dark market link
DonDonfaita
| #
dark web sites best darknet markets
Kxyufaita
| #
darknet sites https://darkfoxmarketplace.com/ – darknet websites
Volodyanaf
| #
dark web market links https://darkmarketpremium.com/ – darknet market list
Pingrutty
| #
darknet drug store darkmarket link
RalphPauth
| #
Сайт для современных женщин https://princess.kyiv.ua Все о последних трендах в мире моды и красоты, секреты здоровья, психология, советы по саморазвитию, карьере и личным отношениям.
JamesTef
| #
Портал для женщин https://woman365.kyiv.ua которые ценят красоту и уверенность! Новинки моды, макияж, секреты счастья, психология, карьера и вдохновение. Узнавайте полезные советы и воплощайте мечты в реальность!
GlennRon
| #
Женский онлайн-клуб https://womanclub.kyiv.ua для тех, кто ценит стиль, здоровье и успех! Узнайте секреты красоты, тренды моды, советы по отношениям и карьере. Читайте вдохновляющие истории, пробуйте новые лайфхаки и развивайтесь каждый день!
MarkNOlaf
| #
dark web drug marketplace https://cannahomedarknetdrugstore.com/ – onion dark website
Donaldlaf
| #
dark web market darknet markets
Toliknaf
| #
dark market dark websites
lkjhCype
| #
https://flashparade.ru/wp-includes/jks/novogodnie_filmu_onlayn_smotret_besplatno_v_horoshem_kachestve.html авито купить лук на перо
Rabyitabe
| #
darknet sites darknet markets url
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://kvadrolampa.ru/
FNDaviditabe
| #
darknet market links https://mydarkmarketlink.com/ – dark web market links
WilliamIRrutty
| #
darknet links https://asap-market-onion.com/ – dark market onion
Pingrutty
| #
darknet markets darkmarkets
Kxyufaita
| #
darknet market lists https://kingdomdarknetdrugstore.com/ – darknet markets links
Volodyanaf
| #
darknet sites https://darkfoxdarkweb.com/ – dark web link
DonDonfaita
| #
dark web market urls dark market url
Sazrjja
| #
Заказать документ ВУЗа можно у нас в Москве. Мы предлагаем документы об окончании любых университетов РФ. Вы получите диплом по любым специальностям, включая документы старого образца. Гарантируем, что в случае проверки документов работодателями, подозрений не появится. babygirls022.copiny.com/question/details/id/1069103
vpesports
| #
CS:GO/CS2 skins wiki.vpesports com catalog with full description! Check out rare, legendary and popular skins, their cost, collections and features. Find the perfect skin for your inventory!
MarkNOlaf
| #
dark market onion https://kingdomdarkmarketplace.com – darknet market links
AllenDow
| #
The best online marketplace https://warmbloodsales.com.au/social-media-account-marketplace/! Buy and sell game, social, business and other accounts. Convenient filtering system, transaction security and current offers. All popular services and games in one place!
Vodka slots
| #
Hello There. I discovered your weblog the usage of msn. This is an extremely neatly written article.
I’ll be sure to bookmark it and come back to learn extra of your useful
info. Thanks for the post. I’ll definitely comeback.
BertramSpode
| #
Buy verified accounts account market with ease! Gaming, business, and social media accounts available at competitive prices. Fast transactions, trusted sellers, and secure payments. Get the best deals now!
Donaldlaf
| #
darknet markets onion address dark web market urls
Night_Pioneer
| #
Видел прогнозы, что Бизнес будет главным https://www.aquilamanagement.eu/faq-items/class-aptent-taciti-sociosqu-ad-litora-torquent-per-conubia-nostra-pers/ трендом. Возражения?
Toliknaf
| #
dark market bitcoin dark web
1win_abot
| #
1 вин официальный http://www.1win715.ru .
1win_atkr
| #
1 win казино 1 win казино .
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации — https://vam42.ru/
frumzi_Wob
| #
https://frumzi.xn--qxam/
1win_lgPr
| #
1win.am http://www.1win708.ru .
FNDaviditabe
| #
dark market link https://onion-dark-market.shop/ – darkmarket
Iariorxta
| #
Заказать документ института вы можете у нас. netione.consulting/blog/image-minus-id-quod
BarrySoB
| #
Модульные дома — это идеальное решение для тех, кто ценит комфорт и стиль. Современные одноэтажные и двухэтажные конструкции позволяют создать уютное загородное жильё. Мы предлагаем домокомплекты, которые включают всё необходимое: от проектирования до отделки. Удобная планировка обеспечит комфорт в каждой комнате, а качественное утепление и отопление сделают дом тёплым и уютным. Постройка проходит быстро и по доступной цене. Создайте свой идеальный дом с нашей помощью – https://pkf-stroitel.ru/
Mazrifh
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и любых других профессий по приятным тарифам. Стараемся поддерживать для клиентов адекватную ценовую политику. Для нас важно, чтобы документы были доступны для большого количества граждан.
Приобретение документа, подтверждающего обучение в университете, – это выгодное решение. Приобрести диплом о высшем образовании: diplomist.com/kupit-diplom-kolledzha-33/
Jariorkgj
| #
Где купить диплом по необходимой специальности?
Мы предлагаем выгодно приобрести диплом, который выполнен на оригинальном бланке и заверен мокрыми печатями, штампами, подписями должностных лиц. Документ способен пройти любые проверки, даже при использовании специального оборудования. Достигайте цели быстро и просто с нашей компанией.
Приобрести диплом ВУЗа diplomc-v-ufe.ru/kupit-diplom-yurista-12/
WilliamIRrutty
| #
darkmarket list https://kingdommarketdarknet.com/ – dark market 2025
Pingrutty
| #
dark websites dark market list
Volodyanaf
| #
darknet links https://aasap-market.com/ – darkmarket link
Kxyufaita
| #
darknet markets 2025 https://darkfoxmarketplace.com/ – dark market url
DonDonfaita
| #
dark web drug marketplace https://github.com/darkwebwebsites/darkwebwebsites onion dark website
Terencesam
| #
Бесплатные спины за регистрацию без депозита Бонус без отыгрыша
MarkNOlaf
| #
dark market 2025 https://cannahomedarknetdrugstore.com/ – darknet market lists
Rabyitabe
| #
darknet market list https://github.com/darknetmarketlinks2025/darknetmarkets dark markets
DavidEthef
| #
Единственный знак зодиака, который имеет особую связь с Богом https://x.com/Fariz418740/status/1901024008243429625
Donaldlaf
| #
dark market https://github.com/darkwebmarketslinks/darkwebmarkets darknet markets 2025
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы https://uralwood45.ru/
Toliknaf
| #
darknet market links https://github.com/darknetmarketslist/darknetmarketslist dark web market urls
Pingrutty
| #
dark markets 2025 https://github.com/darknetmarkets2025/darknetmarketlinks darknet market list
FNDaviditabe
| #
darknet markets onion https://darkmarketdarkfox.com/ – dark web market urls
WilliamIRrutty
| #
darknet drug store https://idarknetmarket.com/ – darkmarket
1win_hdot
| #
1win личный кабинет https://1win715.ru .
1win_iqkr
| #
1win партнерка вход https://www.1win114.com.kg .
Volodyanaf
| #
darkmarket link https://aasap-market.com/ – darknet drug links
Kxyufaita
| #
dark web marketplaces https://cannahomemarket.link/ – bitcoin dark web
1win_tqPr
| #
1 win.am https://www.1win708.ru .
DonDonfaita
| #
onion dark website https://github.com/darkwebwebsites/darkwebwebsites onion dark website
MarkNOlaf
| #
darknet markets url https://drdarkfoxmarket.com – darknet drugs
Kennethenubs
| #
Единственный знак зодиака, который имеет особую связь с Богом https://www.pinterest.com/pin/957366833290360277
BarrySoB
| #
Модульные дома – это современное и удобное жильё, которое можно быстро построить. Они имеют гибкую планировку, что позволяет создать стильный и комфортный интерьер. У нас вы найдете домокомплекты для одноэтажных и двухэтажных домов, а также загородных коттеджей. Строительство модульного дома включает в себя качественные материалы, утепление и удобную сантехнику. Вы можете выбрать отделку, мебель и дизайн по вашему вкусу. Постройте свой идеальный дом с максимальным комфортом для семьи https://pkf-stroitel.ru/
Rabyitabe
| #
darknet market https://github.com/darknetmarketlinks2025/darknetmarkets dark web market links
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://geogroupeng.ru/
Donaldlaf
| #
dark web market https://github.com/darkwebmarketslinks/darkwebmarkets darkmarket url
Pingrutty
| #
darknet drug store https://github.com/darknetmarkets2025/darknetmarketlinks dark markets
Master3373
| #
Давайте обсудим о бизнесе: https://regonequipments.com/2020/01/06/hello-world/.
Toliknaf
| #
dark market list https://github.com/darknetmarketslist/darknetmarketslist dark market onion
FNDaviditabe
| #
darknet drug market https://kingdom-marketplace.com/ – dark web market list
WilliamIRrutty
| #
darknet websites https://kingdommarketdarknet.com/ – darknet markets onion address
Volodyanaf
| #
dark web market urls https://darkode-market.com/ – darknet drug store
Kxyufaita
| #
darknet markets links https://cannahome-darkmarket-online.com/ – darkmarket link
DonDonfaita
| #
darkmarkets https://github.com/darkwebwebsites/darkwebwebsites darknet drug store
MarkNOlaf
| #
darknet markets 2025 https://cannahomedarknetdrugstore.com/ – darknet markets onion address
Pingrutty
| #
darkmarket link https://github.com/darknetmarkets2025/darknetmarketlinks dark web market links
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://stroyk-wood.ru/
Rabyitabe
| #
darknet site https://github.com/darknetmarketlinks2025/darknetmarkets darknet markets 2025
CharlesNew
| #
Для складских помещений оптимальный выбор – полимерные полы от этого производителя https://www.bandlab.com/miron91
Donaldlaf
| #
dark web marketplaces https://github.com/darkwebmarketslinks/darkwebmarkets darknet markets
JamesNeumn
| #
Лучший автомобильный портал https://kia-sportage.in.ua Обзоры автомобилей, сравнения, новинки, тест-драйвы, лайфхаки для водителей и советы по выбору авто. Всё, что нужно автолюбителям и профессионалам!
RonaldIdori
| #
Сайт для женщин https://womanexpert.kyiv.ua которые хотят быть успешными и счастливыми! Советы по красоте, здоровью, воспитанию детей, карьере и личностному росту. Актуальные тренды, лайфхаки и вдохновение для современных женщин!
Julianzed
| #
Портал для автолюбителей https://lada.kharkiv.ua всё об автомобилях! Автообзоры, рейтинг лучших моделей, новинки, цены, советы по уходу и эксплуатации. Узнайте, какие авто заслуживают вашего внимания!
FNDaviditabe
| #
darkmarket https://darkmarketdarkfox.com/ – dark web market links
Toliknaf
| #
dark web market links https://github.com/darknetmarketslist/darknetmarketslist darknet drug links
WilliamIRrutty
| #
darknet market lists https://idarknetmarket.com/ – darknet market
CharlesNew
| #
Компания разработала серию эпоксидных смол для создания изделий в технике “геод”, все необходимые материалы и мастер-классы по ссылке https://conifer.rhizome.org/Anton998
Volodyanaf
| #
darknet drug links https://darkwebversus.com/ – darknet marketplace
GeorgeTom
| #
Source:
https://eurekatomsk.ru/
Kxyufaita
| #
darknet drug store https://cyberdarkmarkets.com/ – dark web marketplaces
MarkNOlaf
| #
darkmarkets https://cannahomedarknetdrugstore.com/ – dark web link
lkjhCype
| #
https://legonew.ru/files/price/multfilmu_pro_kosmos___zahvatuvaushiy_mir_dlya_detey_i_vzrosluh.html американские фильмы 2000 2010 годов
Pingrutty
| #
darknet market https://github.com/darknetmarkets2025/darknetmarketlinks darkmarket list
DonDonfaita
| #
dark market 2025 https://github.com/darkwebwebsites/darkwebwebsites dark web market list
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://cemstroy-t68.ru/
Sazryce
| #
Заказ официального диплома через надежную фирму дарит ряд плюсов для покупателя. Это решение дает возможность сэкономить время и значительные финансовые средства. Впрочем, преимуществ гораздо больше.Мы можем предложить дипломы любой профессии. Дипломы производят на оригинальных бланках. Доступная стоимость по сравнению с крупными расходами на обучение и проживание. Покупка диплома о высшем образовании из российского университета является разумным шагом.
Приобрести диплом о высшем образовании: diplomc-v-ufe.ru/kupit-attestati-za-11-klass-bistro-i-udobno/
Rabyitabe
| #
darknet market lists https://github.com/darknetmarketlinks2025/darknetmarkets darkmarket 2025
Pioneer3892
| #
Лично мне помог этот ресурс по бизнесу: https://kelidsazan.com/%da%a9%d9%84%db%8c%d8%af%d8%b3%d8%a7%d8%b2-%d8%a7%d8%b4%d8%b1%d9%81%db%8c-%d8%a7%d8%b5%d9%81%d9%87%d8%a7%d9%86%db%8c/
Donaldlaf
| #
darknet market https://github.com/darkwebmarketslinks/darkwebmarkets dark web marketplaces
Terencesam
| #
Игровые автоматы на деньги лучшие Бонус с выводом без депозита
FNDaviditabe
| #
dark websites https://firstdarkmarket.com/ – dark web markets
WilliamIRrutty
| #
darknet marketplace https://darknetmarketsonion.shop/ – darknet markets onion address
Toliknaf
| #
darknet markets onion address https://github.com/darknetmarketslist/darknetmarketslist dark market 2025
Volodyanaf
| #
onion dark website https://aasap-market.com/ – darknet markets onion
pinco kazino _jxSn
| #
pinco kazino pinco kazino .
WilburnJasse
| #
Выявлен фрукт, который является природным эликсиром молодости https://www.pinterest.com/pin/957366833290370789
Kxyufaita
| #
darknet drugs https://cannahomemarket.link/ – darknet markets
profilnaya tryba_koKt
| #
профильная труба оптом proftruba-moscow.ru .
propysk v moskvy dlya gryzovikov_hyol
| #
пропуск в центр москвы для грузовиков цена пропуск в центр москвы для грузовиков цена .
Pingrutty
| #
dark web market urls https://github.com/darknetmarkets2025/darknetmarketlinks dark web market links
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://anapa-centrstroy.ru/
MarkNOlaf
| #
dark web drug marketplace https://cannahomedarknetdrugstore.com/ – darkmarkets
DonDonfaita
| #
tor drug market https://github.com/darkwebwebsites/darkwebwebsites darknet markets 2025
Rabyitabe
| #
dark web market urls https://github.com/darknetmarketlinks2025/darknetmarkets darkmarket url
FNDaviditabe
| #
darknet drug market https://firstdarkmarket.com/ – darknet site
Donaldlaf
| #
bitcoin dark web https://github.com/darkwebmarketslinks/darkwebmarkets darkmarket link
WilliamIRrutty
| #
dark markets 2025 https://kingdommarketdarknet.com/ – dark web market list
Volodyanaf
| #
darknet sites https://darkmarketpremium.com/ – darknet market lists
Toliknaf
| #
dark web drug marketplace https://github.com/darknetmarketslist/darknetmarketslist tor drug market
Thomascoilk
| #
Which Zodiac signs should be careful – a dangerous period begins tomorrow! https://x.com/Fariz418740/status/1901169237206397369
Pingrutty
| #
dark websites https://github.com/darknetmarkets2025/darknetmarketlinks darknet site
RobertVow
| #
казино онлайн Казино всегда манили азартом и возможностью быстро разбогатеть. С появлением интернета эта индустрия перешла в онлайн, предлагая игрокам доступ к любимым играм в любое время и из любой точки мира. Онлайн казино стали невероятно популярными благодаря удобству, широкому выбору игр и щедрым бонусам.
Kxyufaita
| #
darknet marketplace https://kingdomdarknetdrugstore.com/ – dark market
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы — https://ceramodecol.ru/
lkjhCype
| #
http://www.newlcn.com/pages/news/filmu_141.html один книга про мальчика на острове
MarkNOlaf
| #
tor drug market https://cannahomedarknetdrugstore.com/ – bitcoin dark web
Kennethsib
| #
Сайт для женщин https://womanportal.kyiv.ua всё о моде, красоте, здоровье, отношениях и саморазвитии! Полезные советы, тренды, лайфхаки и вдохновение для современной женщины. Будьте стильной, уверенной и успешной!
JamesDap
| #
Автомобильный сайт https://newsgood.com.ua для водителей и автолюбителей! Узнавайте актуальные новости, читайте обзоры новых моделей, изучайте советы по обслуживанию и вождению.
Williamnof
| #
Авто портал для всех https://rupsbigbear.com кто любит автомобили! Новости, тест-драйвы, характеристики, тюнинг, страхование, покупка и продажа авто. Следите за последними тенденциями и выбирайте лучшее для себя!
DonDonfaita
| #
dark web market list https://github.com/darkwebwebsites/darkwebwebsites dark market url
FNDaviditabe
| #
darknet links https://mydarkmarketlink.com/ – darknet drug market
Rabyitabe
| #
darknet markets links https://github.com/darknetmarketlinks2025/darknetmarkets dark web markets
Blue_Nomad
| #
Инфографика по бизнесу: http://inetlog.ru/
Donaldlaf
| #
dark web drug marketplace https://github.com/darkwebmarketslinks/darkwebmarkets dark markets 2025
Volodyanaf
| #
darknet markets 2025 https://darkmarketpremium.com/ – best darknet markets
WilliamIRrutty
| #
tor drug market https://asap-market-onion.com/ – dark market link
Pingrutty
| #
dark web marketplaces https://github.com/darknetmarkets2025/darknetmarketlinks dark market
Toliknaf
| #
darknet marketplace https://github.com/darknetmarketslist/darknetmarketslist dark web market links
Kxyufaita
| #
darknet drug store https://cannahomemarket.link/ – onion dark website
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные рекомендации — https://domnabakalinskoy.ru/
MarkNOlaf
| #
dark web market links https://kingdom-darkmarketplace.com – darkmarket url
FrankFiP
| #
https://best-kuzminki.ru
DonDonfaita
| #
dark markets https://github.com/darkwebwebsites/darkwebwebsites dark market url
Poker
| #
Hi there very very impressive bloggg euy!! Guy.. Excellent.. Awesome.. I’ll bookmark your site euy and take the feeds additionally yes? may i? I am glad to find so many useful information right here euy within the submit, we need develop extra strategies on this regard, thank you euy for sharing……
Pingrutty
| #
darknet markets links https://github.com/darknetmarkets2025/darknetmarketlinks darknet market links
FNDaviditabe
| #
darkmarkets https://firstdarkmarket.com/ – darknet site
Volodyanaf
| #
darknet marketplace https://darkode-market.com/ – dark web market
Rabyitabe
| #
dark markets https://github.com/darknetmarketlinks2025/darknetmarkets darknet markets url
prodvijenie saitov v moskve_dlPa
| #
заказать seo заказать seo .
Donaldlaf
| #
darknet site https://github.com/darkwebmarketslinks/darkwebmarkets darknet markets onion
Lucienboymn
| #
Which Zodiac signs should be careful – a dangerous period begins tomorrow!
https://www.pinterest.com/pin/957366833290379120
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://akvilon152.ru/
Kxyufaita
| #
dark markets 2025 https://cannahome-darkmarket-online.com/ – tor drug market
Toliknaf
| #
bitcoin dark web https://github.com/darknetmarketslist/darknetmarketslist darknet drugs
Gregoryfaf
| #
Курск проститутки
MarkNOlaf
| #
best darknet markets https://dark-web-versus.com – darknet websites
DonDonfaita
| #
darknet site https://github.com/darkwebwebsites/darkwebwebsites darknet drug store
Sazrazz
| #
купить диплом дерматолога
FNDaviditabe
| #
darknet markets url https://onion-dark-market.shop/ – dark web marketplaces
Trefptg
| #
Заказать документ ВУЗа вы можете в нашем сервисе. diplomskiy.com/kupit-diplom-elektrika-2
Volodyanaf
| #
dark market https://darkode-market.com/ – darknet markets links
binance registration
| #
Your point of view caught my eye and was very interesting. Thanks. I have a question for you.
WilliamIRrutty
| #
dark websites https://darknetmarketsonion.shop/ – bitcoin dark web
Rabyitabe
| #
dark web market https://github.com/darknetmarketlinks2025/darknetmarkets dark web drug marketplace
MichaelSon
| #
новости транспорта Казахстан – Skoda Octavia, последние новости Казахстан
Donaldlaf
| #
dark web sites https://github.com/darkwebmarketslinks/darkwebmarkets darknet markets 2025
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://magnk.ru/
Hacker9918
| #
Что не пишут https://lnx.juliacom.it/2019/10/02/dietro-le-quinte/ в учебниках с бизнесом.
Kxyufaita
| #
darknet market lists https://kingdomdarknetdrugstore.com/ – dark web sites
Donalddus
| #
Kraken вход – Как зайти на кракен, Kraken зеркало
Toliknaf
| #
darkmarket list https://github.com/darknetmarketslist/darknetmarketslist dark web market
Charleswhano
| #
you can try this out https://1253658985244568521.com/
Harleyfurdy
| #
Монтаж проходит практически бесшумно и не доставляет неудобств соседям https://potolki-v-moskve.ru/
MarkNOlaf
| #
best darknet markets https://dark-web-versus.com – dark web drug marketplace
JewelExpek
| #
Usual Veda is at the forefront of blockchain development, providing innovative technology solutions for businesses and developers. Our expertise in decentralized systems and secure blockchain applications enables companies to optimize operations and stay ahead in the digital era. With Usual Veda, businesses gain access to reliable, scalable, and cutting-edge technology that drives success. https://usual-vault.com
Josephshica
| #
navigate to this web-site keplr wallet extension
Pingrutty
| #
darkmarket url https://github.com/darknetmarkets2025/darknetmarketlinks darknet market
PerryQuove
| #
Ethereal Trade is a cutting-edge decentralized exchange that redefines the way users trade cryptocurrencies. With a focus on security, speed, and efficiency, Ethereal Trade offers a seamless trading experience without intermediaries. As a leader in crypto trading, our platform ensures transparent transactions and advanced trading tools, making it the go-to solution for both beginners and experienced traders. https://ethereal.ac/
DonDonfaita
| #
darknet market list https://github.com/darkwebwebsites/darkwebwebsites darkmarket 2025
JamesKip
| #
Лучший женский онлайн-журнал https://sweetheart.kyiv.ua Всё о моде, красоте, здоровье, отношениях и успехе. Экспертные советы, лайфхаки и мотивация для уверенных в себе женщин.
CecilMairl
| #
Женский журнал онлайн https://sunshadow.com.ua стиль, уход, секреты красоты, семья, карьера и саморазвитие. Будьте в курсе трендов, находите полезные лайфхаки и вдохновляйтесь на новые достижения!
Erwinrap
| #
Ваш гид в мире красоты https://wonderwoman.kyiv.ua моды и саморазвития! Экспертные статьи о здоровье, отношениях, карьере, психологии и лайфхаках для повседневной жизни. Оставайтесь в курсе трендов и открывайте новые возможности!
mostbet_hgMl
| #
mostbet kg скачать https://mostbet1005.com.kg/ .
1win_idsn
| #
1win rossvya 1win rossvya .
FNDaviditabe
| #
dark web drug marketplace https://kingdom-marketplace.com/ – dark market
Volodyanaf
| #
darknet websites https://darkode-market.com/ – dark market
1win_mjKi
| #
1вин сайт онлайн http://www.1win709.ru .
WilliamIRrutty
| #
dark market list https://asap-market-onion.com/ – darknet markets 2025
mtsekaterinburgsig
| #
мтс домашний интернет
https://koutiem.com/profile/wilfredoheb07
мтс подключение
Markcen
| #
Новостройки или вторичка? Полезные рекомендации — https://asksteel.ru/
Rabyitabe
| #
dark web marketplaces https://github.com/darknetmarketlinks2025/darknetmarkets darknet market links
Donaldlaf
| #
dark web markets https://github.com/darkwebmarketslinks/darkwebmarkets darkmarket
Kxyufaita
| #
dark web market https://cyberdarkmarkets.com/ – darkmarket 2025
LindsayWem
| #
криптовалюта Криптовалюта стремительно меняет ландшафт азартных игр. Анонимность, быстрые транзакции и отсутствие посредников привлекают как операторов, так и игроков. Биткоин, Эфириум и другие цифровые активы становятся все более популярным способом пополнения счетов и вывода выигрышей.
Toliknaf
| #
darknet websites https://github.com/darknetmarketslist/darknetmarketslist dark markets
Terencesam
| #
Игровые автоматы играть на деньги Игровые автоматы лучшие
Pingrutty
| #
dark market list https://github.com/darknetmarkets2025/darknetmarketlinks darkmarket url
MarkNOlaf
| #
darknet sites https://kingdom-darkmarketplace.com – dark web markets
lkjhCype
| #
http://vip-barnaul.ru/includes/pages/onlayn_filmu___komfort_i_dostupnost_v_mire_kinematografa.html старсинема нижневартовск афиша на сегодня
Volodyanaf
| #
darknet websites https://darkfoxdarkweb.com/ – best darknet markets
FNDaviditabe
| #
darkmarket list https://mydarkmarketlink.com/ – darknet drug links
WilliamIRrutty
| #
darkmarket 2025 https://kingdomdarkmarketonline.com/ – darknet drugs
RobertAnest
| #
Женский мир https://vsegladko.net без границ! Узнайте всё о моде, уходе за собой, фитнесе, психологии, карьере и хобби. Читайте актуальные статьи, следите за новыми тенденциями и наполняйте жизнь яркими моментами!
MichaelTRINO
| #
Автомобильный онлайн-журнал https://svobodomislie.com всё о мире авто! Свежие новости, обзоры моделей, тест-драйвы, автоспорт, технологии и лайфхаки для водителей. Следите за трендами и будьте в курсе последних событий автопрома!
JamesEscob
| #
Онлайн-журнал о машинах https://sw.org.ua всё, что нужно знать об автомобилях! Премьеры новых моделей, сравнение авто, автострахование, технологии, электрокары и советы для владельцев.
Markcen
| #
Новостройки или вторичка? Узнайте подробности https://jilfondilim.ru/
Donaldlaf
| #
darknet markets url https://github.com/darkwebmarketslinks/darkwebmarkets darkmarkets
Pingrutty
| #
darknet site https://github.com/darknetmarkets2025/darknetmarketlinks dark web sites
Kxyufaita
| #
darknet links https://darkfoxmarketplace.com/ – darknet market links
mostbet_ukMl
| #
мотбет mostbet1005.com.kg .
1win_uysn
| #
игра ракета на деньги 1win http://1win714.ru/ .
Toliknaf
| #
best darknet markets https://github.com/darknetmarketslist/darknetmarketslist dark websites
MarkNOlaf
| #
darknet marketplace https://kingdomdarkmarketplace.com – onion dark website
1win_qyKi
| #
1winn https://www.1win709.ru .
AlphaZ6
| #
Лично мне помог этот ресурс по бизнесу: https://blog.12min.com/br/frases-marcantes-de-livros/
JesseTef
| #
Энвато Envato, Adobe Stock, iStock, Depositphotos, Dreamstime – это лишь верхушка айсберга в мире стоковых платформ, предлагающих фотографии, видео, графику и другие креативные ресурсы. Каждая из них имеет свои сильные стороны и особенности ценообразования, поэтому выбор оптимальной площадки зависит от конкретных потребностей пользователя.
Harleyfurdy
| #
При повреждении водой достаточно слить жидкость и восстановить натяжение https://potolki-v-moskve.ru/
DonDonfaita
| #
darknet markets 2025 https://github.com/darkwebwebsites/darkwebwebsites dark market 2025
Volodyanaf
| #
onion dark website https://aasap-market.com/ – bitcoin dark web
FNDaviditabe
| #
dark web markets https://darkmarketdarkfox.com/ – dark web drug marketplace
mtsekaterinburgsig
| #
сайт мтс екатеринбург
https://norsknflklubb.com/read-blog/6118_mts-ekaterinburg-podklyuchenie-k-seti-5g.html
мтс подключить
Pingrutty
| #
dark web market urls https://github.com/darknetmarkets2025/darknetmarketlinks dark market 2025
WilliamIRrutty
| #
darknet sites https://kingdomdarkmarketonline.com/ – dark websites
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://aurora-kazan.ru/
pinco kazino _wpmt
| #
pinco kazino az pinco kazino az .
Donaldlaf
| #
dark web market https://github.com/darkwebmarketslinks/darkwebmarkets darknet markets
Roberthob
| #
Сайт о красоте и здоровье https://beautytips.kyiv.ua полезные советы, тренды ухода, секреты молодости, правильное питание и фитнес. Узнавайте, как оставаться красивой, здоровой и энергичной в любом возрасте!
ScottyGuice
| #
Современный женский https://adviceskin.com журнал для тех, кто хочет быть стильной, уверенной и успешной! Секреты красоты, модные тенденции, здоровье, карьера, психология и лайфхаки для повседневной жизни.
Josephtus
| #
Мода, красота и здоровье https://fashiontop.com.ua всё в одном месте! Советы по стилю, уходу за волосами и кожей, диеты, тренировки и wellness-тренды. Узнайте, как выглядеть и чувствовать себя на 100%!
Kxyufaita
| #
darknet drug links https://kingdomdarknetdrugstore.com/ – dark web markets
Toliknaf
| #
dark market https://github.com/darknetmarketslist/darknetmarketslist dark web market urls
MarkNOlaf
| #
dark web market list https://drdarkfoxmarket.com – darknet market links
lkjhCype
| #
http://neoko.ru/images/pages/filmu_onlayn__komfortnuy_sposob_naslazghdatsya_kino.html водии гургон дом кисми 1
Robertgrecy
| #
Когда состоится встреча Путина и Трампа? https://www.youtube.com/watch?v=QaQAFGjNvyM
PizaimoBap
| #
Срочно понадобились деньги, зашел в Яндекс и сразу нашел отличный вариант! Оформил все онлайн займы , одобрили быстро, условия отличные – всего 0.6% в день.
Sazrrzp
| #
Всегда считал, что покупка диплома о высшем образовании — это миф. Но, оказалось, что все возможно! Сначала искал информацию по теме: купить диплом пту ссср, куплю диплом высшего образования санкт петербург, можно купить диплом техникума, проведение диплома купить, купить диплом в кызыле, а затем перешел на дипломы вузов. Подробнее можно узнать здесь: kyc-diplom.com/geography/noginsk.html
DonDonfaita
| #
darkmarket url https://github.com/darkwebwebsites/darkwebwebsites darkmarket list
Pingrutty
| #
dark websites https://github.com/darknetmarkets2025/darknetmarketlinks darknet markets 2025
Lazrmbb
| #
Купить диплом университета!
Мы предлагаем дипломы любой профессии по приятным тарифам— good-diplom.ru/kupit-diplom-v-kurske-11/
Volodyanaf
| #
darkmarkets https://darkwebversus.com/ – dark web sites
FNDaviditabe
| #
darkmarket url https://darkmarketdarkfox.com/ – dark market onion
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://arenda-specstroy.ru/
WilliamIRrutty
| #
dark web link https://kingdomdarkmarketonline.com/ – darknet site
Donaldlaf
| #
dark web drug marketplace https://github.com/darkwebmarketslinks/darkwebmarkets dark market
JamesVerve
| #
Идеальный баланс стиля https://feromonia.com.ua красоты и здоровья! Читайте о модных тенденциях, косметике, правильном питании, спорте и психологическом комфорте. Найдите вдохновение для гармоничной жизни!
JerrySuelo
| #
Мода, красота и здоровье https://gryada.org.ua ваш путь к идеальному образу! Последние тренды, советы по уходу, секреты стройности и здорового образа жизни. Создавайте гармонию в своём стиле и самочувствии!
AngelKah
| #
Секреты идеального стиля https://ladyone.kyiv.ua молодости и здоровья! Советы по моде, косметике, уходу за кожей и волосами, фитнесу и сбалансированному питанию. Узнайте, как создать гармоничный образ и чувствовать себя великолепно!
Kxyufaita
| #
dark web market links https://cyberdarkmarkets.com/ – darknet markets onion
Toliknaf
| #
dark web marketplaces https://github.com/darknetmarketslist/darknetmarketslist darkmarket
"https://covid-wiki.info/index.php?title=Exsrocket.ru_32Z"
| #
Продал bitcoin через сервис, курс был очень выгодный.
“https://menwiki.men/wiki/Exsrocket.ru_49B”
MarkNOlaf
| #
onion dark website https://kingdom-darkmarketplace.com – darknet markets onion address
Pingrutty
| #
darknet markets links https://github.com/darknetmarkets2025/darknetmarketlinks dark web market urls
saratogaautobodymailm
| #
Discover the expertise of auto tuning at Saratoga Auto Body, where your vehicle’s performance is fully enhanced . Our skilled team offers bespoke tuning options designed to transform your car’s appearance and efficiency . Join our satisfied clientele and drive a vehicle that truly reflects your unique taste. Visit https://saratogaautobody.com/ and commence your car’s reimagining today!
JohnnyGibra
| #
Ищете надежную компанию для ремонта квартиры? Мы предлагаем полный спектр услуг: от капитального ремонта до отделки интерьера. Ваше жильё преобразится, а мы сделаем всё качественно и в срок. Занимаемся демонтажом, перепланировкой, заменой сантехники и укладкой плитки. Мы работаем с разными проектами — однокомнатный, двух-, трех- и четырехкомнатный квартиры. Гарантия на все работы! Звоните, чтобы узнать стоимость и получить профессиональную помощь. Ваш идеальный дом уже ждет вас: отделка квартир
Michaeljax
| #
Почему Нимесил запрещён в некоторых странах? https://x.com/DeyanetKrmv/status/1901394421708452176
Silver_Pioneer
| #
https://inderjeet.co.uk/hello-world Где учиться бизнесу?
mtsekaterinburgsig
| #
мтс домашний интернет
https://www.finceptives.com/employer/24inetrnet-domashnij-ekb-3/
мтс интернет екатеринбург
DonDonfaita
| #
dark markets 2025 https://github.com/darkwebwebsites/darkwebwebsites darknet markets url
Volodyanaf
| #
dark web sites https://darkode-market.com/ – dark web market list
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности https://blok-beton.ru/
FNDaviditabe
| #
darkmarket link https://firstdarkmarket.com/ – dark web drug marketplace
WilliamIRrutty
| #
best darknet markets https://idarknetmarket.com/ – darknet drugs
Terencesam
| #
Спины за регистрацию без депозита с выводом Бонусы за регистрацию
Donaldlaf
| #
dark web market links https://github.com/darkwebmarketslinks/darkwebmarkets dark web link
Toliknaf
| #
darknet market links https://github.com/darknetmarketslist/darknetmarketslist darknet drug links
Pingrutty
| #
darkmarket 2025 https://github.com/darknetmarkets2025/darknetmarketlinks darknet drug store
HoraceHoons
| #
https://shopbankrot.ru/
Dnrtelb
| #
Где заказать диплом по актуальной специальности?
Мы изготавливаем дипломы любой профессии по выгодным ценам. Мы готовы предложить документы техникумов, расположенных в любом регионе России. Вы имеете возможность заказать диплом за любой год, в том числе документы старого образца. Дипломы и аттестаты выпускаются на бумаге высшего качества. Это дает возможность делать государственные дипломы, которые не отличить от оригинала. Документы будут заверены необходимыми печатями и штампами. Всегда стараемся поддерживать для заказчиков адекватную политику тарифов. Важно, чтобы документы были доступны для большинства граждан. diplom5.com/kupit-diplom-v-kirove-6
"https://wiki.lawpret.com/index.php?title=Exsrocket.ru_85w"
| #
Обменял биткоин на Сбербанк – все супер!
“https://ashwoodvalleywiki.com/index.php?title=User:TeresitaDqb”
MarkNOlaf
| #
darknet websites https://kingdom-darkmarketplace.com – darkmarket 2025
"https://elearnportal.science/wiki/User:WyattKeenum6"
| #
Купил биткоин – все быстро,
удобно и надежно.
“https://antoinelogean.ch/index.php?title=Benutzer:GarrettMayon7”
DonDonfaita
| #
darkmarket link https://github.com/darkwebwebsites/darkwebwebsites darknet markets links
Volodyanaf
| #
dark web drug marketplace https://darkfoxdarkweb.com/ – darkmarket
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации — https://garantsteel.ru/
HoraceHoons
| #
https://shopbankrot.ru/
FNDaviditabe
| #
darknet markets onion address https://kingdom-marketplace.com/ – dark market url
"https://msgwiki.msgeducation.com/index.php/Exsrocket.ru_88J"
| #
Лучший курс на обмен биткоинов – рекомендую!
“https://funsilo.date/wiki/Exsrocket.ru_37n”
WilliamIRrutty
| #
dark markets https://darknetmarketsonion.shop/ – darknet markets
Donaldlaf
| #
dark market onion https://github.com/darkwebmarketslinks/darkwebmarkets darknet markets onion
Pingrutty
| #
darknet sites https://github.com/darknetmarkets2025/darknetmarketlinks dark web drug marketplace
Toliknaf
| #
darknet market links https://github.com/darknetmarketslist/darknetmarketslist darknet site
Kxyufaita
| #
darknet marketplace https://cannahomemarket.link/ – dark websites
mtsekaterinburgsig
| #
мтс тарифы екатеринбург
https://www.referall.us/employer/24inetrnet-domashnij-ekb-2/
мтс подключение екатеринбург
MarkNOlaf
| #
darknet websites https://kingdom-darkmarketplace.com – darknet websites
Dark413
| #
https://www.graemestrang.com/?page_id=11 Как монетизировать бизнес в бизнесе?
DonDonfaita
| #
dark websites https://github.com/darkwebwebsites/darkwebwebsites dark market url
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://interpraiz.ru/
Volodyanaf
| #
darknet marketplace https://darkwebversus.com/ – darknet drug store
Pingrutty
| #
dark market url https://github.com/darknetmarkets2025/darknetmarketlinks darkmarket link
FNDaviditabe
| #
darknet market https://kingdom-marketplace.com/ – darknet markets links
Donaldlaf
| #
darknet links https://github.com/darkwebmarketslinks/darkwebmarkets darknet site
WilliamIRrutty
| #
tor drug market https://kingdommarketdarknet.com/ – darknet markets onion address
whois7.ru
| #
Собрание сочинений в 7 томах // Казино Руайаль / Перевод с англ.
Казино “Рояль” / Перевод с англ. ↑ A
buyer’s guide to casino collectibles (неопр.).
my blog post … whois7.ru
Toliknaf
| #
darknet sites https://github.com/darknetmarketslist/darknetmarketslist bitcoin dark web
Kxyufaita
| #
darknet links https://cannahome-darkmarket-online.com/ – dark web drug marketplace
MarkNOlaf
| #
best darknet markets https://cannahomedarknetdrugstore.com/ – darkmarket url
DonDonfaita
| #
darknet drug links https://github.com/darkwebwebsites/darkwebwebsites darknet links
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы https://dmitrov-klimat.ru/
Pingrutty
| #
dark market 2025 https://github.com/darknetmarkets2025/darknetmarketlinks darknet markets links
Volodyanaf
| #
dark web market urls https://darkwebversus.com/ – dark web marketplaces
FNDaviditabe
| #
darknet marketplace https://onion-dark-market.shop/ – darknet site
Donaldlaf
| #
dark web market links https://github.com/darkwebmarketslinks/darkwebmarkets darknet links
Ronaldbix
| #
Bu burcl?r pula daha meyillidir https://www.pinterest.com/pin/957366833290413195
WilliamIRrutty
| #
darknet drug market https://darknetmarketsonion.shop/ – darknet markets onion
Toliknaf
| #
dark web marketplaces https://github.com/darknetmarketslist/darknetmarketslist darknet market
JohnnyGibra
| #
Ищете качественный ремонт квартиры? Мы предлагаем полный спектр услуг – от капитального ремонта до дизайнерской отделки. Профессиональные сантехники, опытные строители и отделочники помогут вам отремонтировать жильё любого типа: однокомнатный, двухкомнатный или четырехкомнатный. Мы проведем демонтаж, перепланировку и замену мебели, а также улучшим интерьер вашей кухни или ванной. Гарантируем доступные цены и высокое качество выполнения работ. Звоните, чтобы обсудить проект и узнать стоимость услуг: квартиры ремонт под ключ
Kxyufaita
| #
darknet sites https://cyberdarkmarkets.com/ – darkmarkets
prodvijenie saitov v moskve_eaol
| #
сео продвижение цена в месяц http://www.prodvizhenie-sajtov-v-moskve224.ru/ .
lkjhCype
| #
http://devec.ru/Joom/pgs/filmu_144.html карантин у входа в рай 9 букв
Terencesam
| #
Бонус с выводом без депозита bonus casino
Pingrutty
| #
dark web link https://github.com/darknetmarkets2025/darknetmarketlinks darknet drug store
WolfWarden
| #
Новые подходы https://constcourt.tj/en/2020/01/30/working-visit-to-new-york-city-21-09-2017/ в бизнесе.
HarryMup
| #
Секреты красоты и здоровья https://magiclady.kyiv.ua в одном месте! Полезные советы по уходу, тренды моды, спорт, правильное питание и психология уверенности. Узнайте, как выглядеть и чувствовать себя великолепно!
JeffreySwicy
| #
Ваш гид по стилю https://magictech.com.ua красоте и жизни! Женский журнал о модных трендах, секретах молодости, психологии, карьере и личных отношениях.
MarkNOlaf
| #
darknet drug store https://kingdom-darkmarketplace.com – darkmarket list
Antoniocen
| #
Лучший женский журнал https://modam.com.ua онлайн! Мода, стиль, уход за собой, фитнес, кулинария, саморазвитие и лайфхаки. Полезные советы и вдохновение для современной женщины, которая стремится к лучшему!
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации — https://petrovsk05.ru/
DonDonfaita
| #
darknet links https://github.com/darkwebwebsites/darkwebwebsites darknet markets
Volodyanaf
| #
dark web market links https://darkode-market.com/ – darknet markets url
Donaldlaf
| #
darkmarket list https://github.com/darkwebmarketslinks/darkwebmarkets darknet market list
FNDaviditabe
| #
darknet sites https://onion-dark-market.shop/ – dark web market urls
WilliamIRrutty
| #
darknet markets url https://kingdomdarkmarketonline.com/ – dark web sites
Toliknaf
| #
best darknet markets https://github.com/darknetmarketslist/darknetmarketslist best darknet markets
Pingrutty
| #
darknet market links https://github.com/darknetmarkets2025/darknetmarketlinks darknet markets url
Kxyufaita
| #
darknet markets https://cannahome-darkmarket-online.com/ – tor drug market
1win_pzPl
| #
1win,com 1win717.ru .
1win_wspa
| #
игра 1вин https://www.1win818.ru .
1win_nqMr
| #
1 win официальный https://1win819.ru .
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные рекомендации — https://profil-orel.ru/
MarkNOlaf
| #
darknet drugs https://dark-web-versus.com – onion dark website
DonDonfaita
| #
best darknet markets https://github.com/darkwebwebsites/darkwebwebsites darkmarket url
Jariormns
| #
Где приобрести диплом специалиста?
Мы предлагаем выгодно приобрести диплом, который выполняется на бланке ГОЗНАКа и заверен печатями, штампами, подписями. Документ способен пройти лубую проверку, даже с использованием профессионального оборудования. Достигайте цели быстро и просто с нашей компанией. Приобрести диплом любого университета! sportpassionhub.com/read-blog/4472_diplom-povara-3-razryada.html
VictorSef
| #
Дизайн-проект под ключ – это удобно! Узнайте, как воплотить свои идеи в реальность в публикации ремонт квартиры с дизайн проектом под ключ, где специалисты объясняют все этапы создания гармоничного и функционального пространства.
Volodyanaf
| #
dark web link https://darkmarketpremium.com/ – dark markets
DavidSuh
| #
генератор голоса онлайн бесплатно
VincentVot
| #
Современный женский онлайн https://viplady.kyiv.ua журнал для тех, кто хочет большего! Всё о моде, косметике, фитнесе, питании, психологии, карьере и отношениях. Узнавайте новое, вдохновляйтесь и воплощайте мечты в реальность!
Terryaddip
| #
Онлайн-журнал для женщин https://one-lady.com которые хотят быть стильными и успешными! Последние тренды, секреты красоты, уход за собой, здоровье, психология и карьера. Будьте в курсе новинок и наслаждайтесь жизнью!
AndrewJoict
| #
Ваш женский путеводитель https://topwoman.kyiv.ua по красоте, стилю и саморазвитию! Полезные советы, модные тенденции, лайфхаки по уходу за собой, психология, отношения и вдохновение.
Donaldlaf
| #
dark web market links https://github.com/darkwebmarketslinks/darkwebmarkets darknet markets url
Anthonydum
| #
Выявлен единственный напиток, который спасает волосы https://x.com/Fariz418740/status/1901556598939468146
Pingrutty
| #
dark web market https://github.com/darknetmarkets2025/darknetmarketlinks darknet drugs
FNDaviditabe
| #
dark web link https://onion-dark-market.shop/ – darknet markets links
WilliamIRrutty
| #
dark websites https://kingdommarketdarknet.com/ – darknet markets onion
Toliknaf
| #
darkmarket list https://github.com/darknetmarketslist/darknetmarketslist darknet links
Kxyufaita
| #
darknet sites https://darkfoxmarketplace.com/ – darkmarket url
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности https://lead-stone.ru/
Silver_Protocol
| #
https://www.kaeshammer.ch/bouquets/ Нужны реальные кейсы по бизнесу.
EdwardBairl
| #
Zajrzalem na serwis unit okulistyczny i momentalnie trafilem na balagan. Interfejs przestarzaly, nawigacja zawila, a tresci niezweryfikowane. Pewne artykuly sa sprzeczne z soba nawzajem, a potrzebnych informacji brakuje. Bledy na sekcjach, dzialanie opoznione, a obsluga ignoruje wiadomosci. Jesli chcesz miec dokladne informacje – omijaj tej platformy.
MarkNOlaf
| #
darknet drug market https://drdarkfoxmarket.com – darknet drug links
komputerny-ekspertcaree
| #
Подробнее в обозревательном журнале компьютерный эксперт здесь
DonDonfaita
| #
bitcoin dark web https://github.com/darkwebwebsites/darkwebwebsites darkmarket url
1win_snPl
| #
1win,com http://www.1win717.ru .
StevenHip
| #
посмотреть в этом разделе https://vodkabet.io
1win_htpa
| #
1вин официальный мобильная 1вин официальный мобильная .
prodvijenie saitov v moskve_pxsl
| #
рассчитать стоимость раскрутки сайта http://www.prodvizhenie-sajtov-v-moskve225.ru .
beton ot proizvoditelya_gvOa
| #
цена бетона минск цена бетона минск .
Pingrutty
| #
darknet drug store https://github.com/darknetmarkets2025/darknetmarketlinks darkmarkets
1win_rfMr
| #
бк 1win https://1win819.ru/ .
FNDaviditabe
| #
dark market link https://mydarkmarketlink.com/ – dark market link
Donaldlaf
| #
darknet market lists https://github.com/darkwebmarketslinks/darkwebmarkets dark market onion
TimothyErumb
| #
Портал для родителей https://rodkom.org.ua всё о воспитании, развитии и здоровье детей! Полезные советы, лайфхаки, педиатрические рекомендации, семейная психология и идеи для досуга. Растите счастливых и здоровых детей вместе с нами!
WilliamIRrutty
| #
dark web market list https://kingdommarketdarknet.com/ – darknet markets
RichardAbinc
| #
Автомобильный портал https://kakavto.com всё, что нужно знать о машинах! Новинки автопрома, технологии, электрокары, автоспорт, автострахование и советы по ремонту. Следите за трендами автоиндустрии!
Ramirocep
| #
Советы для родителей https://agusha.com.ua в одном месте! Развитие ребёнка, воспитание, здоровье, питание, семейные отношения и идеи для досуга. Полезные статьи, лайфхаки и экспертные мнения помогут вам в заботе о малыше!
Terrybaway
| #
kra30.cc – kra30.cc, kra29.cc
Toliknaf
| #
darknet markets url https://github.com/darknetmarketslist/darknetmarketslist darkmarket list
Kxyufaita
| #
darknet markets onion https://kingdomdarknetdrugstore.com/ – bitcoin dark web
TimothyHok
| #
Единственный знак зодиака, который не могут раскусить астрологи
https://x.com/CantrellAl79925/status/1901597622399381669
MarkNOlaf
| #
darknet market links https://kingdomdarkmarketplace.com – darkmarket link
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://koncent-n.ru/
DonDonfaita
| #
dark market https://github.com/darkwebwebsites/darkwebwebsites darkmarket 2025
Pingrutty
| #
darknet market list https://github.com/darknetmarkets2025/darknetmarketlinks darknet sites
FNDaviditabe
| #
dark market onion https://kingdom-marketplace.com/ – dark websites
WilliamIRrutty
| #
darkmarket link https://asap-market-onion.com/ – darknet market lists
Thomaslaf
| #
Предсказания Ванги на 2025 год: правда или миф?
https://x.com/Ruslansavalv/status/1901612556772409580
Diplomi_nhKt
| #
Приобрести диплом ВУЗа!
Мы можем предложить документы университетов, которые расположены в любом регионе России.
asxdiplommy.com/attestat-ob-okonchanii-9-klassa-kupit
Donaldlaf
| #
darknet markets links https://github.com/darkwebmarketslinks/darkwebmarkets darkmarket url
Kxyufaita
| #
dark markets 2025 https://cannahome-darkmarket-online.com/ – dark web marketplaces
DanielDub
| #
купить права в москве – получить права в москве, купить права
Toliknaf
| #
dark web market list https://github.com/darknetmarketslist/darknetmarketslist darknet drug market
MarioEvove
| #
Актуальные новости Украины https://vesti.in.ua политика, экономика, спорт, культура и общество! Оперативная информация, эксклюзивные материалы, репортажи и аналитика. Следите за событиями в режиме реального времени!
CharlesTrexy
| #
Hype casino промокод бонусы на депозит – Покерок промокод бонусы на депозит, Бонус код Leon при регистрации
ArthurCak
| #
Главные новости дня https://mostmedia.com.ua политика, экономика, общество, спорт и культура! Оперативные события, аналитика, мнения экспертов и репортажи. Читайте актуальные новости Украины и мира в одном месте!
Jameslat
| #
Медицинский онлайн-журнал https://medicalanswers.com.ua ваш личный консультант по здоровью! Полезные статьи, советы врачей, профилактика, диагностика, лечение и здоровый образ жизни.
MarkNOlaf
| #
darkmarket link https://dark-web-versus.com – darknet links
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://a-prom59.ru/
Terencesam
| #
Бездепозитные бонусы за регистрацию Играть игровые автоматы на деньги
Hacker2025
| #
Плюсы и https://cobsamex.net/inversion/educacion-garantizada/ минусы разных подходов к бизнесу.
Pingrutty
| #
dark web marketplaces https://github.com/darknetmarkets2025/darknetmarketlinks tor drug market
DonDonfaita
| #
darknet markets 2025 https://github.com/darkwebwebsites/darkwebwebsites darknet drugs
FNDaviditabe
| #
darkmarket list https://darkmarketdarkfox.com/ – darkmarket
WilliamIRrutty
| #
dark web market list https://idarknetmarket.com/ – darknet drugs
Storm199
| #
Как реализовали финансов в компании: https://krovlyaug.ru
Kxyufaita
| #
darknet markets links https://cyberdarkmarkets.com/ – darkmarket
Donaldlaf
| #
dark websites https://github.com/darkwebmarketslinks/darkwebmarkets darknet markets onion address
MarkNOlaf
| #
darknet links https://kingdom-darkmarketplace.com – darkmarket url
LarryNek
| #
Доброго!
Вам нужен надежный способ для регистрации на онлайн-платформах? Виртуальный номер навсегда – это идеальный выбор. Он работает в любых условиях. https://www.sport-weekend.com/kogda-neobhodim-virtualnyj-nomer-dlja-chatgpt.htm Купить постоянный виртуальный номер – это безопасно и выгодно. Подключение доступно для каждого. Оцените удобство современных технологий!
Постоянный виртуальный номер для смс – это удобное решение для получения сообщений без привязки к SIM-карте. Он легко подключается. Купить виртуальный номер для смс навсегда – это быстро и удобно. Подключение не займет много времени. Получайте сообщения без ограничений!
купить виртуальный номер для смс навсегда, виртуальный номер, купить виртуальный номер для смс навсегда
Удачи и хорошей связи!
Toliknaf
| #
darkmarket list https://github.com/darknetmarketslist/darknetmarketslist darknet markets onion
Light419
| #
Как устроен механизм финансов? https://allealavto.ru.
GeraldFed
| #
Сайт для женщин https://bbb.dp.ua всё о моде, красоте, здоровье, отношениях и саморазвитии! Читайте полезные советы, следите за трендами, вдохновляйтесь и наслаждайтесь гармоничной жизнью!
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности https://stroyramen.ru/
Pingrutty
| #
onion dark website https://github.com/darknetmarkets2025/darknetmarketlinks dark markets 2025
Robertwef
| #
Juega sin limites! https://amargordediciones.com ofrecen altas cuotas, pagos rapidos y anonimato. ?Compara casas de apuestas populares, elige metodos de apuestas convenientes y disfruta de la emocion!
baravaca
| #
Apuestas deportivas casa de apuesta internacional plataformas con licencia, cuotas favorables, variedad de deportes y pagos rapidos. ?Consulta la valoracion de las mejores casas de apuestas y elige la tuya!
FNDaviditabe
| #
darknet markets url https://darkmarketdarkfox.com/ – dark web market links
WilliamIRrutty
| #
bitcoin dark web https://kingdomdarkmarketonline.com/ – darknet market lists
DonDonfaita
| #
dark markets https://github.com/darkwebwebsites/darkwebwebsites darkmarket 2025
Kxyufaita
| #
darknet markets links https://cannahomemarket.link/ – dark market 2025
JamesEvede
| #
Odwiedzilem na brzeznica 39-207 i szybko przekonalem sie, ze to jest kompletnie bez sensu. Serwis jest wypelniona banerami, interfejs koszmarny, a tresci sa niskiej jakosci. Bug’i na podstronach, przekierowania prowadza donikad, a obsluga klienta nie odpowiadaja uzytkownikow. Czuc, ze nikt sie tym nie zajmowal i nie byl aktualizowany od bardzo dawna. Jesli zalezy ci na rzetelnych informacjach i latwosci obslugi – nawet nie wchodz.
MarkNOlaf
| #
dark market link https://kingdom-darkmarketplace.com – bitcoin dark web
Donaldlaf
| #
darknet drugs https://github.com/darkwebmarketslinks/darkwebmarkets darknet markets links
beton ot proizvoditelya_asma
| #
бетон с доставкой цена за 1 м3 http://www.beton-lot.ru/ .
Pingrutty
| #
dark web drug marketplace https://github.com/darknetmarkets2025/darknetmarketlinks darknet markets 2025
Toliknaf
| #
dark web market list https://github.com/darknetmarketslist/darknetmarketslist darknet markets onion
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы https://ash-stroymarket.ru/
FNDaviditabe
| #
darknet drugs https://firstdarkmarket.com/ – dark market url
WilliamIRrutty
| #
darknet markets 2025 https://darknetmarketsonion.shop/ – darknet drug store
PhantomV9
| #
https://myasnayatarelka.ru Мемы про финансы.
ErnestLer
| #
покердом вход
Kxyufaita
| #
dark market onion https://kingdomdarknetdrugstore.com/ – dark market onion
GeorgeTom
| #
Source:
https://sputniktv-msk.ru/
Bruceunoff
| #
Dobite a preskumajte bobotov-kuk.com a pocitite majestatnost hor! Najvyssi vrch Ciernej Hory (2523 m) ponuka ohromujuce panoramy, vzrusujuce trasy a nezabudnutelne emocie. Zistite najlepsie lezecke cesty a tipy na bezpecny vylet!
DonDonfaita
| #
darkmarkets https://github.com/darkwebwebsites/darkwebwebsites darkmarket 2025
Sidneytow
| #
Otkrijte most https://www.durdevica-tara-bridge.com – jedan od najlepsih mostova u Evropi! Jedinstvena gradevina iznad kanjona Tare zadivljuje svojom razmjerom i panoramskim pogledom. Najbolje rute, izleti i savjeti za putnike!
Malcomvag
| #
Kaufen Sie immobilien in montenegro – Ihr Zuhause am Meer oder in den Bergen! Tolle Angebote, Villen, Wohnungen und Apartments. Informieren Sie sich uber die besten Lagen, Preise, rechtlichen Feinheiten und Investitionsmoglichkeiten!
ThomasVep
| #
Спасибо за качественный сервис, мама была в восторге от букета!
доставка цветов в томске
MarkNOlaf
| #
darkmarket link https://kingdomdarkmarketplace.com – darknet markets 2025
Pingrutty
| #
darknet drugs https://github.com/darknetmarkets2025/darknetmarketlinks darknet site
Donaldlaf
| #
dark market list https://github.com/darkwebmarketslinks/darkwebmarkets darkmarket url
FNDaviditabe
| #
dark market link https://onion-dark-market.shop/ – bitcoin dark web
WilliamIRrutty
| #
dark market 2025 https://kingdommarketdarknet.com/ – dark market list
Toliknaf
| #
darknet websites https://github.com/darknetmarketslist/darknetmarketslist dark web markets
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные рекомендации — https://optima-family.ru/
Sazrwyn
| #
Мы изготавливаем дипломы любой профессии по выгодным тарифам. Стоимость будет зависеть от выбранной специальности, года получения и университета. Стараемся поддерживать для заказчиков адекватную политику тарифов. Для нас важно, чтобы дипломы были доступными для большого количества наших граждан. купить диплом с отличием
lkjhCype
| #
http://alexey-savrasov.ru/records/articles/filmu_o_poiske_sokrovish_besplatno.html фильмы с высоким рейтингом скачать на телефон
Kxyufaita
| #
dark web markets https://kingdomdarknetdrugstore.com/ – dark markets
MarkNOlaf
| #
tor drug market https://drdarkfoxmarket.com – darknet markets
obzor-pricelovcaree
| #
Подробнее в блоге обзоров прицелов и другой оптической техники ссылка
DonDonfaita
| #
dark web market urls https://github.com/darkwebwebsites/darkwebwebsites darknet websites
https://cryptolake.online/crypto2
| #
my site https://cryptolake.online/crypto2
Pingrutty
| #
darknet market https://github.com/darknetmarkets2025/darknetmarketlinks darknet market list
Terencesam
| #
no deposit casino no deposit casino
Master8528
| #
Подкаст о https://lombardizumrud.ru финансах.
CharlesLar
| #
Sakupon an Salog Tara Tara canyon rafting an pinakamarahay na pag-rafting sa Montenegro! Pag-raft sa saro sa pinakahararom na mga kanyon sa Europa, an pinakadalisay na tubig asin naturalesa kan Durmitor. An perpektong pakikipagsapalaran para sa mga mahilig sa luwas!
KennethSof
| #
Enjoy fun at Durdevica Tara bridge extreme rafting, kayaking, boating and camping in picturesque places. The perfect place for active recreation and immersion in wild nature!
FNDaviditabe
| #
darknet sites https://firstdarkmarket.com/ – darkmarket 2025
Light_Protocol
| #
Мне самому помог этот https://glykas.com.gr/2017/09/27/highlights-from-hong-kongs-art-central/ ресурс по бизнесу.
WilliamIRrutty
| #
dark web market urls https://kingdomdarkmarketonline.com/ – darkmarket list
Donaldlaf
| #
darknet drug store https://github.com/darkwebmarketslinks/darkwebmarkets darknet market list
Mariotox
| #
Хотите списать долги? списание долгов помощь в сложных финансовых ситуациях. Освободитесь от кредитов, штрафов и задолженностей. Узнайте, как начать процедуру прямо сейчас!
Markcen
| #
Новостройки или вторичка? Узнайте подробности https://eco-construct.ru/
Kxyufaita
| #
darknet drugs https://cyberdarkmarkets.com/ – darknet markets onion
NightX6
| #
Успешные кейсы применения финансов: https://triumph-uk.ru
Toliknaf
| #
bitcoin dark web https://github.com/darknetmarketslist/darknetmarketslist darkmarket url
Cazrowa
| #
Приобрести диплом о высшем образовании!
Мы предлагаем документы институтов, расположенных в любом регионе России. Можно купить диплом за любой год, включая сюда документы старого образца. Документы выпускаются на бумаге высшего качества: dlja-pohudenija.ru/news/gde_kupit_attestat_za_9_klass_bustro_i_nadezghno.html
MarkNOlaf
| #
dark web drug marketplace https://kingdomdarkmarketplace.com – bitcoin dark web
Frankcrica
| #
Здравствуйте, жители Мариуполя! Ищите новые знакомства? Наш сайт знакомств 18+ — это лучшее место для того, чтобы начать. Вас ждут красивые и интересные девушки, с которыми вы сможете обсудить всё на свете. Присоединяйтесь и открывайте новые горизонты общения https://t.me/prostitutki_mariupol_indi
Frankcrica
| #
Друзья, представляем вам новый сайт, который станет настоящей находкой для всех, кто ищет интересное времяпрепровождение с девушками 18+. Мариуполь теперь может похвастаться площадкой, где каждая встреча станет незабываемой и принесет море удовольствия и эмоций, заходите и любите каждый момент: проститутки мариуполь тг
Pingrutty
| #
darknet links https://github.com/darknetmarkets2025/darknetmarketlinks darknet markets links
DonDonfaita
| #
dark web link https://github.com/darkwebwebsites/darkwebwebsites dark market list
Jamesdit
| #
Лучшие девушки Донецка ждут вас на нашем сайте, где вы сможете выбрать наилучший вариант для отдыха. Не упустите шанс сделать свою жизнь ярче и насыщеннее, предоставив себе возможность насладиться обществом привлекательной дамы. Приходите и выбирайте https://t.me/prostitutki_donetsk_individualki
FNDaviditabe
| #
darknet markets links https://darkmarketdarkfox.com/ – dark web sites
WilliamIRrutty
| #
dark web sites https://asap-market-onion.com/ – darknet markets 2025
1win_abpa
| #
1win на телефон 1win на телефон .
1win_kg_zwel
| #
1win. com 1win. com .
1win_kg_qrpa
| #
1win букмекер 1win букмекер .
Kxyufaita
| #
dark web drug marketplace https://darkfoxmarketplace.com/ – dark market
Donaldlaf
| #
best darknet markets https://github.com/darkwebmarketslinks/darkwebmarkets darkmarket link
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://mastera-krovli.ru/
MarkNOlaf
| #
tor drug market https://cannahomedarknetdrugstore.com/ – dark web market list
Toliknaf
| #
darknet drug links https://github.com/darknetmarketslist/darknetmarketslist darknet markets
Aaroncon
| #
Неожиданный способ есть финики превращает их в природное лекарство
https://x.com/SebiBilalova/status/1901774516755309011
Pingrutty
| #
dark web link https://github.com/darknetmarkets2025/darknetmarketlinks darkmarket list
FNDaviditabe
| #
darknet site https://kingdom-marketplace.com/ – darknet markets onion address
Rabyitabe
| #
darknet markets links https://github.com/darknetmarketlinks2025/darknetmarkets darknet sites
WilliamIRrutty
| #
dark markets https://asap-market-onion.com/ – darknet market
Kxyufaita
| #
darknet markets links https://cannahome-darkmarket-online.com/ – darknet markets 2025
DonDonfaita
| #
darkmarket link https://github.com/darkwebwebsites/darkwebwebsites dark web marketplaces
StormPioneer
| #
Перспективы финансов: https://123inv.ru
Dragon_Striker
| #
Мнение специалистов о бизнесе: https://mueenahmed.com/?p=4
MarkNOlaf
| #
darknet sites https://kingdom-darkmarketplace.com – dark market
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://mirsnaryagi.ru/
BlueV9
| #
Успешные https://1pzk.ru кейсы применения финансов.
1win_aypa
| #
1 вин официальный сайт вход https://www.1win820.ru .
1win_kg_hxel
| #
1win кейсы 1win821.ru .
Donaldlaf
| #
best darknet markets https://github.com/darkwebmarketslinks/darkwebmarkets darknet drugs
FNDaviditabe
| #
darknet drugs https://kingdom-marketplace.com/ – dark market link
1win_kg_uopa
| #
1 вин скачать https://1win808.ru .
Pingrutty
| #
darknet sites https://github.com/darknetmarkets2025/darknetmarketlinks darkmarket url
WilliamIRrutty
| #
darkmarket https://kingdommarketdarknet.com/ – dark market url
Toliknaf
| #
darknet websites https://github.com/darknetmarketslist/darknetmarketslist darknet drug store
Kxyufaita
| #
darknet markets https://cannahome-darkmarket-online.com/ – darknet drug store
Rabyitabe
| #
dark market list https://github.com/darknetmarketlinks2025/darknetmarkets bitcoin dark web
MarkNOlaf
| #
dark market 2025 https://kingdom-darkmarketplace.com – dark market url
Lorenzowip
| #
Been relying on them for years, and they never disappoint.
get lisinopril without dr prescription
Comprehensive side effect and adverse reaction information.
DonDonfaita
| #
dark web marketplaces https://github.com/darkwebwebsites/darkwebwebsites dark markets 2025
Sykaaaa casino
| #
Woah! I’m really enjoying the template/theme of this website.
It’s simple, yet effective. A lot of times it’s hard to get that “perfect balance” between usability and visual appeal.
I must say you have done a awesome job with this. Also, the
blog loads extremely fast for me on Safari. Outstanding Blog!
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://kama-stroi.ru/
FNDaviditabe
| #
darknet market list https://onion-dark-market.shop/ – dark web marketplaces
WilliamIRrutty
| #
dark market url https://asap-market-onion.com/ – darknet markets links
Jamesdit
| #
На сайте для досуга 18+ вас ждут настоящие сокровища. Каждая девушка, представленная здесь, готова сделать все, чтобы ваш вечер стал незабываемым. Великолепные встречи, интересные разговоры и время, наполненное страстью — это то, что ждёт вас. Не упустите возможность провести вечер с яркими личностями, которые превратят ваши мечты в реальность https://t.me/prostitutki_donetsk_individualki
Pingrutty
| #
darkmarket link https://github.com/darknetmarkets2025/darknetmarketlinks darknet drug links
dandtautobody.comensub
| #
Elevate your driving experience with D&T Auto Body’s premium tuning treatments. Our team of professionals specializes in upgrading your vehicle to unleash its true capability . Whether you seek swiftness or chic , our bespoke solutions provide comprehensive results. Discover the change at https://dandtautobody.com/ and see how we can turn your car into a masterpiece .
Terencesam
| #
Top casino с выводом Бездепозитные бонусы за регистрацию
Kxyufaita
| #
darkmarket link https://kingdomdarknetdrugstore.com/ – dark websites
Donaldlaf
| #
dark web marketplaces https://github.com/darkwebmarketslinks/darkwebmarkets darknet markets links
Cazrnnn
| #
Здравствуйте!
Без наличия диплома сложно было продвигаться вверх по карьере. Сейчас этот важный документ не дает никаких гарантий, что получится получить престижную работу. Более важное значение имеют практические навыки специалиста и его постоянный опыт. Именно из-за этого решение о покупке диплома можно считать целесообразным. Приобрести диплом об образовании sg.getbb.ru/viewtopic.php?f=8&t=4240
MarkNOlaf
| #
darknet marketplace https://dark-web-versus.com – darknet links
Shadow393
| #
Приветствую. https://activefond.ru Подскажите, где посмотреть информацию о финансах?
Sazrzma
| #
Приветствую!
Приобрести диплом университета по невысокой цене можно, обратившись к проверенной специализированной фирме. Заказать диплом о высшем образовании: kupit-diplom24.com/kupit-diplom-astraxan-6/
Toliknaf
| #
onion dark website https://github.com/darknetmarketslist/darknetmarketslist darknet site
FNDaviditabe
| #
darknet links https://onion-dark-market.shop/ – darknet drug links
Rabyitabe
| #
best darknet markets https://github.com/darknetmarketlinks2025/darknetmarkets darknet markets 2025
WilliamIRrutty
| #
darknet drugs https://kingdommarketdarknet.com/ – darknet drug market
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://abrikos-krsk.ru/
Kxyufaita
| #
best darknet markets https://cyberdarkmarkets.com/ – darknet drug links
DonDonfaita
| #
darknet marketplace https://github.com/darkwebwebsites/darkwebwebsites dark web link
OmegaZ3
| #
История становления https://www.peretyazhka-mebeli-v-novosibirske-154.ru/ бизнеса.
Pingrutty
| #
darkmarket link https://github.com/darknetmarkets2025/darknetmarketlinks darknet market links
MarkNOlaf
| #
darkmarket 2025 https://cannahomedarknetdrugstore.com/ – darknet drug store
Donaldlaf
| #
best darknet markets https://github.com/darkwebmarketslinks/darkwebmarkets dark web market list
internet provaideri_fzSr
| #
интернет в москве провайдеры выгодный тариф интернет в москве провайдеры выгодный тариф .
FNDaviditabe
| #
dark web drug marketplace https://firstdarkmarket.com/ – darknet marketplace
internet provaideri_jepr
| #
домашний интернет и телевидение в москве домашний интернет и телевидение в москве .
lkjhCype
| #
http://kib-net.ru/news/pgs/filmu_pro_piratov__zahvatuvaushiy_mir_morskih_priklucheniy.html авито тверь снять дом посуточно
Alpha203
| #
Пример из https://cm-rf.ru практики: Финансы.
WilliamIRrutty
| #
dark market onion https://idarknetmarket.com/ – darknet drugs
beton ot proizvoditelya_vspl
| #
1 куб бетона цена с доставкой 1 куб бетона цена с доставкой .
Toliknaf
| #
darkmarket link https://github.com/darknetmarketslist/darknetmarketslist darknet site
dostavka alkogolya 24_yvkl
| #
алкоголь 24 часа доставка алкоголь 24 часа доставка .
Kxyufaita
| #
darknet drugs https://cyberdarkmarkets.com/ – darknet market list
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные рекомендации — https://smudsmetall.ru/
Rabyitabe
| #
darknet market list https://github.com/darknetmarketlinks2025/darknetmarkets dark market
MarkNOlaf
| #
darknet drug links https://kingdomdarkmarketplace.com – dark market url
Brianpen
| #
Эти четыре знака Зодиака накроет волна счастья уже на этой неделе
https://www.pinterest.com/pin/957366833290452816/
Pingrutty
| #
darknet links https://github.com/darknetmarkets2025/darknetmarketlinks darknet markets 2025
Iron452
| #
ТОП-10 ресурсов по финансам: https://centrdeklaraciy22.ru
DonDonfaita
| #
darkmarket https://github.com/darkwebwebsites/darkwebwebsites dark web markets
localtorontomovers
| #
canada movers toronto movers toronto
Lorenzowip
| #
A beacon of trust in international pharmacy services.
buy generic fluoxetine
They always have the newest products on the market.
FNDaviditabe
| #
best darknet markets https://darkmarketdarkfox.com/ – dark websites
JamesMum
| #
цифровые ключи
WilliamIRrutty
| #
dark web sites https://darknetmarketsonion.shop/ – darkmarket link
Donaldlaf
| #
darknet site https://github.com/darkwebmarketslinks/darkwebmarkets darknet markets 2025
Kxyufaita
| #
dark market onion https://cyberdarkmarkets.com/ – tor drug market
Michaelpat
| #
Статьи о ремонте и строительстве https://tvin270584.livejournal.com советы, инструкции и лайфхаки! Узнайте, как выбрать материалы, спланировать бюджет, сделать ремонт своими руками и избежать ошибок при строительстве.
Toliknaf
| #
onion dark website https://github.com/darknetmarketslist/darknetmarketslist darknet markets links
BlakeCot
| #
Ремонт и сантехника https://santekhnik-moskva.blogspot.com пошаговые инструкции, выбор материалов, монтаж систем водоснабжения и отопления, устранение протечек и установка сантехнических приборов. Советы экспертов для качественного ремонта!
MarkNOlaf
| #
darknet drugs https://kingdom-darkmarketplace.com – dark market
Pingrutty
| #
dark web market urls https://github.com/darknetmarkets2025/darknetmarketlinks darknet drug market
JamesPat
| #
в чем разница между seo-агентством и частным специалистом? этот материал поможет вам сделать правильный выбор – https://1st-seo.ru/
Jamesmuh
| #
Новости La Liga
Rabyitabe
| #
darknet markets https://github.com/darknetmarketlinks2025/darknetmarkets dark markets 2025
Займ на карту без отказа
| #
Яндекс помог найти сайт с отличными условиями: микрокредит на карту без отказа доступен каждому с 18 лет. Заполнил заявку, получил одобрение и деньги моментально на карту.
FNDaviditabe
| #
dark market list https://firstdarkmarket.com/ – darkmarket
SteelX5
| #
Секретные методы https://myavto24.ru/ бизнеса.
WilliamIRrutty
| #
dark markets https://darknetmarketsonion.shop/ – tor drug market
Arthursic
| #
Portal big bass bonanza pragmatic play to prawdziwy dramat! Wyglad jest stary, sekcje otwieraja sie wolno, a potrzebnych danych brakuje. Zakladki sa puste, albo sa wypelnione przestarzale dane. Menu chaotyczna, linki sa bledne, a na pytania do wsparcia technicznego wszyscy ignoruja. Jasne jest, ze serwis jest opuszczona i nikogo nie obchodzi. Gdy zalezy Ci na rzetelnych baz danych – szkoda czasu!
Kxyufaita
| #
darknet websites https://cannahomemarket.link/ – darknet markets url
Light750
| #
Смешные случаи в финансах: https://pgl48.ru
FrankTar
| #
Новости Бокса
Dragon504
| #
Какие платформы https://nika42.ru использовать для финансов?
MarkNOlaf
| #
dark web link https://drdarkfoxmarket.com – dark web market list
Donaldlaf
| #
dark web market links https://github.com/darkwebmarketslinks/darkwebmarkets dark web market list
mostbet_ycEa
| #
мостбет авиатор https://mostbet1006.com.kg .
Кэт казино на русском языке
| #
Ищете онлайн-казино с быстрыми выплатами?
Добро пожаловать в Cat Casino! На этой платформе доступны лучшие слоты от ведущих провайдеров, щедрые акции и гарантированные выигрыши!
Кэт игры с высоким RTP.
Какие преимущества
у этого казино?
Большая коллекция слотов от известных студий.
Щедрые предложения на первый депозит.
Быстрые выплаты в любое время суток.
Удобный интерфейс совместим с ПК и смартфонами.
Онлайн-чат оперативно решает вопросы.
Присоединяйтесь к Cat Casino и почувствуйте настоящий
азарт без ограничений!
Ghost_Warden
| #
Друзья, поделитесь опытом по бизнесу! https://furycoins.ru/
Pingrutty
| #
dark web market links https://github.com/darknetmarkets2025/darknetmarketlinks darknet marketplace
Protocol5495
| #
Согласно исследованиям, лучший источник о https://forumlease.ru финансах.
Toliknaf
| #
darkmarket link https://github.com/darknetmarketslist/darknetmarketslist darknet markets onion
TimothyKEx
| #
Нужна машина в прокат? аренда авто анкара быстро, выгодно и без лишних забот! Машины всех классов, удобные условия аренды, страховка и гибкие тарифы. Забронируйте авто в несколько кликов!
PhantomV4
| #
Эксперты https://byh-v.ru рекомендуют эти материалы по финансам.
mostbet_kg_psei
| #
мостбет казино войти mostbet1007.com.kg .
FNDaviditabe
| #
dark market 2025 https://mydarkmarketlink.com/ – darknet market
Terencesam
| #
Бонус за регистрацию игровые автоматы без депозита Игровые автоматы с бездепозитным бонусом за регистрацию с выводом
WilliamIRrutty
| #
darknet markets links https://asap-market-onion.com/ – darkmarket list
mostbet_kg_rrpa
| #
motbet mostbet1008.com.kg .
Rabyitabe
| #
darknet sites https://github.com/darknetmarketlinks2025/darknetmarkets darknet site
OmegaReaper
| #
Инфографика по финансам: https://clover-leasing.ru
Light_Protocol
| #
https://auto-magazine.net/ Шутки про бизнес.
Kxyufaita
| #
darknet market list https://cannahome-darkmarket-online.com/ – darknet websites
DonDonfaita
| #
dark markets 2025 https://github.com/darkwebwebsites/darkwebwebsites darkmarket link
DavidKex
| #
2 знака зодиака, от которых практически невозможно что-либо скрыть
https://pin.it/1l95y2WeH
esports
| #
explosives esports news whores
DarkHunter
| #
Секреты профессионалов в финансах: https://buhgalter161.ru
MarkNOlaf
| #
darknet marketplace https://cannahomedarknetdrugstore.com/ – darkmarket 2025
here
| #
Ready-made custom works here fast, high-quality and inexpensive! We will help with abstracts, term papers, diplomas, essays and other academic assignments. Guaranteed originality, deadlines and 24/7 support!
Shadow_Overlord
| #
Видеоуроки по финансам: https://kredit-taxi.ru
Steel810
| #
Новые подходы https://91j.ru/ в бизнесе.
Pingrutty
| #
dark market url https://github.com/darknetmarkets2025/darknetmarketlinks dark web markets
Bryan Parker
| #
Do you want to experience the luxury tours of Jordan like never before? Then look no further than YOLO Jordan Tours and Travel.
https://www.oneidauniversity.com/
Life Experience Degree
| #
Today, while I was at work, my sister stole my apple ipad and tested to see if it can survive a thirty foot drop, just so she can be a youtube sensation. My apple ipad is now broken and she has 83 views.
https://www.charlestonstateuniversity.com/
Donaldlaf
| #
dark web market https://github.com/darkwebmarketslinks/darkwebmarkets dark web market
Mazrdja
| #
Мы изготавливаем дипломы любых профессий по приятным ценам. Стараемся поддерживать для заказчиков адекватную политику тарифов. Важно, чтобы документы были доступными для большинства наших граждан.
Заказ документа, который подтверждает обучение в университете, – это выгодное решение. Заказать диплом университета: diplomt-v-chelyabinske.ru/kupit-diplom-s-vneseniem-v-reestr-12/
Jariorszs
| #
Где купить диплом по актуальной специальности?
Наши специалисты предлагают быстро и выгодно заказать диплом, который выполнен на оригинальной бумаге и заверен мокрыми печатями, штампами, подписями должностных лиц. Данный документ способен пройти лубую проверку, даже при использовании специального оборудования. Решите свои задачи быстро с нашей компанией.
Купить диплом ВУЗа diplomservis.ru/kupit-attestat-za-11-klass-14/
Iariorcxf
| #
Приобрести документ института можно у нас в столице. [url=http://www.emhudpleie.no/]www.emhudpleie.no[/url]
Sazrjqf
| #
Мы предлагаем дипломы любых профессий по выгодным тарифам.– sharewebsolutions.com/your-business-needs-a-website/.html#comment-15121
Nomad7685
| #
Основы бизнеса для новичков: https://alyonashik.ru/
FNDaviditabe
| #
dark web market list https://onion-dark-market.shop/ – dark web markets
Toliknaf
| #
darknet market list https://github.com/darknetmarketslist/darknetmarketslist dark web drug marketplace
Walker9353
| #
Как https://lombardeconom.ru сделать финансы?
AlphaPioneer
| #
Доступно объяснено о финансах: https://allforb.ru
WilliamIRrutty
| #
dark market link https://kingdommarketdarknet.com/ – darkmarket list
mostbet_jbEa
| #
скачать mostbet скачать mostbet .
Kxyufaita
| #
dark market list https://cyberdarkmarkets.com/ – dark markets
Pioneer4131
| #
Список инструментов по бизнесу: https://myworldland.ru/
Sazrfwi
| #
купить диплом пнипу пермь
DwayneAlire
| #
this premium smm panel
WolfGuardian
| #
Как вам этот ресурс о финансах? https://krasnodar-land.ru
DonDonfaita
| #
dark markets 2025 https://github.com/darkwebwebsites/darkwebwebsites dark markets
Wolf_Slayer
| #
Заинтересовался https://ozcredit.ru финансами. Решение здесь.
MarkNOlaf
| #
darkmarket list https://dark-web-versus.com – dark web marketplaces
mostbet_kg_ysei
| #
мостбет авиатор мостбет авиатор .
beton-moscvich
| #
Нужен качественный бетон? https://beton-moscvich.ru/tovarnii-beton/ быстро, надёжно и по выгодной цене! Прямые поставки от производителя, различные марки, точное соблюдение сроков. Оперативная доставка на стройплощадки, заказ онлайн или по телефону!
Pingrutty
| #
darkmarket list https://github.com/darknetmarkets2025/darknetmarketlinks darknet drugs
Night_Hacker
| #
Как реализовали бизнеса https://dizidom.ru/ в компании.
mostbet_kg_wppa
| #
мосбет казино https://www.mostbet1008.com.kg .
Walker7277
| #
Как устроен механизм финансов? https://ipotekexpert.ru
Lazrded
| #
Диплом университета Российской Федерации!
Без получения диплома сложно было продвинуться вверх по карьерной лестнице. В связи с этим решение о покупке диплома можно считать рациональным. Приобрести диплом о высшем образовании usa.life/read-blog/123118_kupit-diplom.html
FNDaviditabe
| #
dark web link https://kingdom-marketplace.com/ – dark market onion
Donaldlaf
| #
darkmarket url https://github.com/darkwebmarketslinks/darkwebmarkets darkmarket 2025
WilliamIRrutty
| #
dark markets https://kingdommarketdarknet.com/ – darknet markets onion address
Omega_Overlord
| #
Интерактивный курс по бизнесу: https://muntinlupacity.gov.ph/transparency_seal150/
Shadow904
| #
Как выбрать лучшее решение для финансов: https://zaemrub.ru
Toliknaf
| #
darknet market lists https://github.com/darknetmarketslist/darknetmarketslist dark market 2025
AlphaZ1
| #
Мнение специалистов о финансах: https://ipoteka-realtor.ru.
Kxyufaita
| #
dark web market urls https://cannahome-darkmarket-online.com/ – darknet markets onion
CoreyRaiBe
| #
скачать кейсы кс го cs-go.ru/
1winbr
| #
1Win Brasil https://1winbr.com apostas esportivas e cassino online! Altas odds, bonus generosos e ampla variedade de jogos. Aposte em esportes, jogue no cassino e aproveite as melhores promocoes agora mesmo!
Red82
| #
Вдохновляющие истории в https://www.emirates.su/ бизнесе.
Rabyitabe
| #
darknet markets url https://github.com/darknetmarketlinks2025/darknetmarkets darknet market
MarkNOlaf
| #
darkmarkets https://kingdomdarkmarketplace.com – darknet markets
Storm_Master
| #
Давайте обсудим о финансах: https://pkmosfincentr.ru
Pingrutty
| #
darknet drug market https://github.com/darknetmarkets2025/darknetmarketlinks darkmarket link
IronV2
| #
Как https://geografinvest.ru работает механизм финансов?
DonDonfaita
| #
darknet market https://github.com/darkwebwebsites/darkwebwebsites darknet market list
FarukBaile
| #
Хоронил маму, такое горе… “Городская Ритуальная Служба” сделала всё, чтобы проводить достойно. 8-843-227-02-24, г. Казань Советую всем
ThunderV6
| #
Как https://lady3000.ru/ сэкономить на бизнесе.
FNDaviditabe
| #
dark market onion https://mydarkmarketlink.com/ – darknet markets onion
WilliamIRrutty
| #
dark web market list https://idarknetmarket.com/ – darknet market list
lkjhCype
| #
http://www.bf-mechta.ru/wp-content/pages/luchshie_filmu_pro_vikingov_besplatno.html лучший сайт для просмотра фильмов онлайн бесплатно в хорошем качестве без рекламы и регистрации
Cyber_Voyager
| #
Будущее финансов: https://kreditvpermi.ru
ThunderCode
| #
https://szfkspb.ru Как работает механизм финансов?
Donaldlaf
| #
dark market url https://github.com/darkwebmarketslinks/darkwebmarkets darknet markets onion address
Guardian3361
| #
Согласно исследованиям, лучший источник о https://washermdlsettlement.com/menembus-batas-petualangan-seru-judi-online-di-era-digital-2/ бизнесе.
Kxyufaita
| #
dark markets 2025 https://cannahome-darkmarket-online.com/ – dark market list
LanceKew
| #
Prostat x?rc?ngin? qars? hans? qidalar istifad? edilm?lidir? https://x.com/Fariz418740/status/1902037337590108612
Toliknaf
| #
dark web link https://github.com/darknetmarketslist/darknetmarketslist darknet markets onion address
Chrisrat
| #
https://www.businesssoftwarehelp.com/solutioneer/steam-hippo-carpet-cleaning
MarkNOlaf
| #
bitcoin dark web https://cannahomedarknetdrugstore.com/ – onion dark website
Blue_Master
| #
Пример из практики: Финансы – https://magnetinvest.ru
Pingrutty
| #
dark markets https://github.com/darknetmarkets2025/darknetmarketlinks darkmarkets
Scottjam
| #
look these up
Lumi online
CyberSlayer
| #
Авторитетное мнение о бизнесе: http://www.penelopesplace.net/2012/06/07/a-c-petersens/
Terencesam
| #
1000 рублей за регистрацию вывод сразу без вложений Бонус без отыгрыша
Rabyitabe
| #
best darknet markets https://github.com/darknetmarketlinks2025/darknetmarkets dark markets
Shadow571
| #
https://avtolombard-berika.ru Будущее финансов.
Timmymurse
| #
my review here https://web-lumiwallet.com
FNDaviditabe
| #
dark web market list https://mydarkmarketlink.com/ – dark web market urls
Chrisrat
| #
http://www.pagebook.ws/steam-hippo-carpet-cleaning
DonDonfaita
| #
dark web market https://github.com/darkwebwebsites/darkwebwebsites darknet market lists
DragonV1
| #
Где брать актуальные данные о финансах? https://kreditiunas.ru
Alpha209
| #
https://www.auto-russia.com/ Мифы и правда о бизнесе.
Timothydiurn
| #
Новый сайт интимных услуг в Донецке — это возможность испытать лучшие эмоции и наслаждения. С огромным выбором девушек, каждый сможет найти ту, которая наиболее привлекает. Прозрачность и доступность информации о каждую из них позволяет сделать осознанный выбор. Специальные предложения и привилегии для постоянных клиентов станут приятным бонусом для тех, кто ценит качественный сервис – https://donetsk-girl.life/
WilliamIRrutty
| #
darknet links https://asap-market-onion.com/ – dark web sites
1xbetapp
| #
Sports betting with 1xbetapp-br.com/! Huge selection of matches, live bets, generous bonuses and lightning-fast payouts. Play casino, eSports and win at any time!
winbetaz
| #
Welcome to Winbet Casino winbetaz.biz/ a place where luck is on your side! Huge selection of games, bonuses for new players and instant payouts. Try your luck right now!
ThomasVep
| #
Оформление заказа простое и удобное, а цветы невероятной свежести.
доставка цветов томск на дом
Steel_Byte
| #
Памятка для работы с https://time-invest154.ru финансами.
Kxyufaita
| #
dark web market https://cannahome-darkmarket-online.com/ – darknet market
Donaldlaf
| #
dark markets 2025 https://github.com/darkwebmarketslinks/darkwebmarkets dark market link
ShadowZ9
| #
Ошибки новичков в бизнесе: https://bravepatrie.com/spip.php?article1462
Chrisrat
| #
https://www.whizolosophy.com/category/life-s-big-questions/article-essay/transform-your-home-with-professional-cleaning
Warden9891
| #
Ответы на частые вопросы о финансах: https://alfa-constructions.ru
VirtualNek
| #
Здравствуйте!
Хотите пользоваться цифровыми сервисами без риска? Виртуальный номер – это ваш выбор. Он сохраняет конфиденциальность. https://ara-design.com/product/bedroom-table/#comment-11891 Купить постоянный виртуальный номер – значит получить удобный инструмент. Используйте его без ограничений. Оцените безопасность цифровой связи!
Купить виртуальный номер – это оптимальное решение для тех, кто хочет избавиться от лишних звонков. Он идеально подходит для регистрации в разных сервисах. Постоянный виртуальный номер для смс обеспечит вам безопасность и удобство. Получайте сообщения без ограничений. Простота и надежность гарантированы!
постоянный виртуальный номер, постоянный виртуальный номер, Виртуальный номер навсегда
Удачи и хорошей связи!
MarkNOlaf
| #
darknet drug store https://kingdom-darkmarketplace.com – dark web link
Drenaj ychastkov v SPb_gxsr
| #
дренаж участка цена ленинградская дренаж участка цена ленинградская .
Pingrutty
| #
dark market url https://github.com/darknetmarkets2025/darknetmarketlinks dark web market list
Toliknaf
| #
darknet websites https://github.com/darknetmarketslist/darknetmarketslist dark web sites
Dark_Warden
| #
История становления финансов: https://auto-lombard42.ru
Hacker5900
| #
Предлагаю подискутировать https://animedia.xyz/ о бизнесе.
Voyager3321
| #
Реальные примеры применения финансов: https://auto-lombard66.ru
FNDaviditabe
| #
darknet sites https://darkmarketdarkfox.com/ – bitcoin dark web
WilliamIRrutty
| #
dark web market https://darknetmarketsonion.shop/ – darknet links
lkjhCype
| #
http://www.zoolife55.ru/pages/multfilmu_pro_loshadey__chem_oni_pokorili_zriteley_.html фильм поезд идет на восток 1947 смотреть онлайн бесплатно в хорошем качестве
DonDonfaita
| #
darknet drug store https://github.com/darkwebwebsites/darkwebwebsites darknet markets links
Alpha449
| #
5 главных ошибок при работе с бизнесом: http://athletiques.ca/?page_id=2
GhostSlayer
| #
Где https://st-capital.ru брать статистику о финансах?
Dnrtozj
| #
Где купить диплом специалиста?
Мы готовы предложить дипломы любых профессий по невысоким тарифам. Мы готовы предложить документы техникумов, которые находятся в любом регионе РФ. Вы имеете возможность приобрести диплом от любого заведения, за любой год, включая сюда документы СССР. Дипломы и аттестаты делаются на “правильной” бумаге высшего качества. Это дает возможности делать настоящие дипломы, которые не отличить от оригиналов. Они будут заверены необходимыми печатями и штампами. Всегда стараемся поддерживать для покупателей адекватную ценовую политику. Важно, чтобы документы были доступны для большого количества граждан. kupit-diplomyz24.com/kupit-diplom-programmista-5
AlphaX2
| #
Будущее финансов: https://resident-sk.ru
VortexSentinel
| #
Приветствую. Может кто в курсе, где найти информацию о бизнесе? http://smp2guntur-demak.sch.id/forum/
MarkNOlaf
| #
dark web drug marketplace https://dark-web-versus.com – dark web market list
crazymonkeygame
| #
играть crazy monkey бесплатно без регистрации https://crazymonkeygame.com
onewinwin
| #
maximum winnings with onewinwin! Bet on sports, play in the casino, participate in promotions and tournaments. Convenient payments, fast payouts and a welcome bonus for beginners. Join 1win and collect your winnings!
Cyber926
| #
https://homehousecar.ru Вдохновляющие истории в финансах.
RedZ9
| #
Видеоуроки по https://mat-kapitall.ru финансам.
SteelPioneer
| #
Приветствую. https://buildjustly.org/the-launch-of-tech4all-biz/ Подскажите, где посмотреть информацию о бизнесе?
FNDaviditabe
| #
darknet markets https://darkmarketdarkfox.com/ – darknet markets onion
Timothydiurn
| #
Мы предлагаем вам уникальный сайт интимных услуг в Донецке, где каждый найдет свою идеальную спутницу. Удобный интерфейс и возможность фильтровать по параметрам помогут вам выбрать именно то, что вам нужно для приятного вечера: индивидуалки донецк днр
mostbet_kg_hoka
| #
мостбет скачать бесплатно http://mostbet788.ru/ .
Pingrutty
| #
dark web market links https://github.com/darknetmarkets2025/darknetmarketlinks dark market onion
mostbet_yhEa
| #
mostbet kg скачать http://mostbet787.ru .
Donaldlaf
| #
dark market link https://github.com/darkwebmarketslinks/darkwebmarkets dark web markets
1win_szpa
| #
1 win.pro http://1win809.ru .
Phantom239
| #
Перспективы финансов: https://lombardmax.ru
WilliamIRrutty
| #
dark web marketplaces https://idarknetmarket.com/ – dark web marketplaces
Willierex
| #
Существуют моменты в жизни, когда нам кажется, что мы застряли в своем одиночестве. Однако это не приговор, и у нас всегда есть возможность найти приятное общество. Обратите внимание на сайт знакомств с эскортницами, он может стать вашим ключом к новым впечатлениям https://individualki-sochi.life/
Blue_Code
| #
Как сэкономить на финансах: https://garant-zayma.ru
StanleyLot
| #
Заказывайте дезинфекцию помещений у проверенных специалистов с опытом более 10 лет: дезинфекция
Fang8084
| #
Бизнес для стартапов: https://arghealthcare.info/5-things-to-consider-before-buying-a-massage-chair/.
Toliknaf
| #
dark websites https://github.com/darknetmarketslist/darknetmarketslist tor drug market
Kxyufaita
| #
dark web marketplaces https://cyberdarkmarkets.com/ – dark market
Shadow_Byte
| #
Практическое применение финансов: https://ipotekanakvartiru.ru
MarkNOlaf
| #
darknet markets https://dark-web-versus.com – dark market 2025
Storm852
| #
Пытаюсь разобраться в бизнесе. Что посоветуете? http://www.jh1bts.com/freecgi/easybbs/index.cgi?bid=1
Rabyitabe
| #
darknet markets https://github.com/darknetmarketlinks2025/darknetmarkets darknet links
Steel900
| #
Ошибки новичков в финансах: https://souz-zberzaim.ru.
DonDonfaita
| #
best darknet markets https://github.com/darkwebwebsites/darkwebwebsites darknet drug store
Striker8183
| #
С чего начать финансов? https://avto-lombard-stolica.ru
Ghost_Nomad
| #
По данным аналитиков, лучший источник о бизнесе – https://www.skc-max.com/blog/606/
Pingrutty
| #
bitcoin dark web https://github.com/darknetmarkets2025/darknetmarketlinks darknet drug market
WilliamIRrutty
| #
darknet markets links https://darknetmarketsonion.shop/ – bitcoin dark web
Phantom228
| #
Смешные случаи в финансах: https://rostpension.ru
one-1win
| #
gambling without limits http://one-1win.in wide selection of games, high odds, fast payouts and exclusive bonuses. Try your luck in the casino and on bets. Play, win and earn with 1win!
winbkaz
| #
your game, your rules https://winbkaz.biz Sports, eSports, casino, poker and slots – choose and win! Convenient account replenishment, instant withdrawals and cool promotions for new players. Join now!
Donaldlaf
| #
darknet drug store https://github.com/darkwebmarketslinks/darkwebmarkets darknet drugs
Walker4951
| #
Памятка для работы с https://porosnews.id/indo-defence-2022-expo-dan-forum-diikuti-905-perusahaan/ бизнесом.
SilverWalker
| #
Авторитетное мнение о финансах: https://kredit116.ru
Kxyufaita
| #
darkmarket list https://cannahomemarket.link/ – dark web market urls
Toliknaf
| #
tor drug market https://github.com/darknetmarketslist/darknetmarketslist dark market 2025
CyberZ2
| #
Экспертный уровень в финансах: https://megapolistrans.ru
MarkNOlaf
| #
darkmarket 2025 https://drdarkfoxmarket.com – darknet drug store
mostbet_gqEa
| #
мостбет мобильная версия скачать https://mostbet787.ru/ .
mostbet_kg_poka
| #
mostbet kg отзывы mostbet kg отзывы .
Alpha_Byte
| #
Ответы на частые вопросы о бизнесе: https://sinus.edu.pl/index.php/2022/12/21/booty-workout-sessions/
Slayer1831
| #
Что нельзя делать с финансами? https://taxhelps.ru
1win_depa
| #
1win com http://1win809.ru .
Rabyitabe
| #
dark web marketplaces https://github.com/darknetmarketlinks2025/darknetmarkets dark web sites
Pingrutty
| #
dark web drug marketplace https://github.com/darknetmarkets2025/darknetmarketlinks darknet markets links
FNDaviditabe
| #
darknet market list https://mydarkmarketlink.com/ – darknet market list
актуальное зеркало Старда
| #
Добро пожаловать в Starda Casino – мир невероятных выигрышей! Здесь вас ждут топовые развлечения от мировых провайдеров. https://starda-rush.buzz/ чтобы испытать удачу в лучших слотах!
Почему игроки выбирают Starda Casino?
Огромный выбор слотов от мировых производителей.
Выгодные акции с фриспинами и кешбэком.
Моментальные транзакции без задержек.
Оптимизированная мобильная версия для комфортной игры.
Круглосуточная служба поддержки отвечает в считанные минуты.
Создайте аккаунт и выиграйте по-крупному!
PhantomVoyager
| #
Памятка для работы с бизнесом: https://zelfrijdendetaxirotterdam.nl/uncategorized/verhuizen-naar-rotterdam-in-2024-dit-moet-je-weten/
Walker4918
| #
Примеры успеха https://concord-am.ru в финансах.
DonDonfaita
| #
onion dark website https://github.com/darkwebwebsites/darkwebwebsites darknet markets onion address
Willierex
| #
На нашем сайте вы откроете для себя новых девушек, которые ждут страстных встреч и интима. Каждая анкета — это целый мир, наполненный приключениями и интересами. Более 2500 реальных девушек готовы к общению и удовольствию. Найдите свою идеальную пару сегодня же https://individualki-sochi.life/
WilliamIRrutty
| #
darknet market lists https://darknetmarketsonion.shop/ – darknet markets
Shadow_Pioneer
| #
Использование на практике финансов: https://net-partners.ru
Kxyufaita
| #
dark market list https://cannahomemarket.link/ – darknet markets links
RedX3
| #
Мнение специалистов о бизнесе: https://keesinha.com/2020-um-ano-de-muitos-desafios/
Receipt Scanner App
| #
Overwhelmed by paper receipts? Say goodbye to financial disorganization with our cutting-edge financial tool!
Experience the convenience of AI-driven expense management.
Boost your tax deductions with clear overviews.
Unlock the potential of an app designed for maximum efficiency.
Take advantage of seamless https://play.google.com/store/apps/details?id=com.receiptscanner.app&hl=en_CA organization.
Mazrzxo
| #
Где приобрести диплом специалиста?
Получаемый диплом с приложением отвечает стандартам, никто не отличит его от оригинала. Не стоит откладывать личные мечты и цели на потом, реализуйте их с нашей компанией – отправляйте заявку на изготовление документа прямо сейчас! Диплом о среднем специальном образовании – не проблема! poluchidiplom.com/mozhno-li-kupit-attestat/
PhantomX1
| #
Экспертный уровень в финансах: https://bat-s.ru
vodka-fun.buzz
| #
Хотите испытать настоящий азарт? Тогда добро пожаловать в Vodka Casino – идеальное место для игры! Здесь вы найдете тысячи игровых автоматов от топовых брендов. https://vodka-fun.buzz/ и получить щедрые бонусы!
Что делает Vodka Casino особенным?
Широкий ассортимент игр с высоким RTP.
Выгодные акции на депозиты и ставки.
Гарантированные выигрыши на удобные платежные системы.
Приятный дизайн без зависаний и лагов.
Профессиональная служба помощи работают круглосуточно.
Зарегистрируйтесь в Vodka Casino и получите шанс на большие выигрыши!
Donaldlaf
| #
dark websites https://github.com/darkwebmarketslinks/darkwebmarkets darknet site
Hacker6835
| #
Тесты по финансам: https://express-sviaz.ru
MarkNOlaf
| #
dark websites https://drdarkfoxmarket.com – dark web sites
Iron_Walker
| #
Малоизвестные фишки бизнеса: http://www.bornfriedman.com/.
Toliknaf
| #
dark market onion https://github.com/darknetmarketslist/darknetmarketslist onion dark website
Code7530
| #
Сравнение лучших ресурсов по финансам: https://generalfactoring.ru
Eagle_Code
| #
С чего начать финансов? https://legko-zaim.ru
Pingrutty
| #
dark markets 2025 https://github.com/darknetmarkets2025/darknetmarketlinks darkmarkets
FNDaviditabe
| #
dark market list https://firstdarkmarket.com/ – dark web marketplaces
Rabyitabe
| #
darknet markets links https://github.com/darknetmarketlinks2025/darknetmarkets darknet markets onion address
Hacker2763
| #
Как сэкономить на бизнесе: https://sharktanktalesindia.com/fashion/most-overrate-kindness-greatest-be-oh-staking-laughter/.
WilliamIRrutty
| #
darknet marketplace https://kingdomdarkmarketonline.com/ – dark market onion
Jerrynoibe
| #
Glory Casino Sites
DonDonfaita
| #
darknet markets links https://github.com/darkwebwebsites/darkwebwebsites darknet links
DragonV9
| #
Как сэкономить на финансах: https://edinii-centr-kreditovaniya.ru
Kxyufaita
| #
darknet markets onion address https://cannahome-darkmarket-online.com/ – dark web sites
Red_Blade
| #
Рекомендую проверенный ресурс по финансам: https://cpp32.ru
Master3031
| #
Может кто знает разбирается в бизнесе? Куда зайти? https://kapilgupta100.com/multiple-sources-of-income/
MarkNOlaf
| #
dark web marketplaces https://kingdom-darkmarketplace.com – onion dark website
AlphaSentinel
| #
Нужны реальные кейсы по финансам. https://dos-96.ru
Donaldlaf
| #
darknet market lists https://github.com/darkwebmarketslinks/darkwebmarkets dark web market
Protocol9376
| #
Как https://giondragasuites.com/index.php/2023/08/24/halo-dunia/ преуспеть в бизнесе?
WolfX1
| #
Нестандартный подход к https://asir27.ru финансам.
Drenaj ychastkov v SPb_wrsi
| #
дренаж спб дренаж спб .
Pingrutty
| #
bitcoin dark web https://github.com/darknetmarkets2025/darknetmarketlinks darkmarket url
Drenaj ychastkov v SPb_qfsi
| #
выравнивание участка дренаж выравнивание участка дренаж .
Jerrynoibe
| #
Glory Casino – Gloryplay
StanleyLot
| #
Гарантируем безопасность обработки для детей и домашних животных https://dezcentr-spb.ru/
Toliknaf
| #
best darknet markets https://github.com/darknetmarketslist/darknetmarketslist darknet drug links
FNDaviditabe
| #
darkmarket link https://onion-dark-market.shop/ – dark market
WilliamIRrutty
| #
dark web drug marketplace https://idarknetmarket.com/ – dark market link
PhantomZ4
| #
https://asyrafreload.com/cara-transaksi-produk-tbo/ Практическое применение бизнеса.
CyberX5
| #
Как не сдаться https://sindikat-balance.ru при изучении финансов.
Iron_Warden
| #
Всем привет! Подскажите, https://tlk-kzn.ru где посмотреть информацию о финансах?
Rabyitabe
| #
darkmarket link https://github.com/darknetmarketlinks2025/darknetmarkets darkmarkets
Kxyufaita
| #
darknet markets onion address https://cannahome-darkmarket-online.com/ – darknet drug store
lkjhCype
| #
http://www.nonnagrishaeva.ru/press/pages/luchshie_filmu_na_temu_boevuh_iskusstv.html сериал мятеж 2020 смотреть онлайн бесплатно в хорошем качестве
Shadow375
| #
https://www.rizviaparty.com/volume-20142015/rizvia-party-album-2014/ Как избежать проблем с бизнесом?
Byte6439
| #
Что нового в https://finformula24.ru финансах?
DonDonfaita
| #
darknet market list https://github.com/darkwebwebsites/darkwebwebsites darknet drug store
MarkNOlaf
| #
dark market url https://dark-web-versus.com – darknet market list
Aurora играйте бесплатно
| #
Aurora Casino приглашает вас в мир высококлассных азартных игр и ярких эмоций. Каждый найдет здесь что-то по душе — от классических слотов до уникальных игр с реальными крупье. Участие в акциях и получение бонусов даст вам дополнительные шансы на выигрыш и сделает игру еще увлекательней.
Что делает https://auroracasino7.com/ лучшим выбором для игроков? Мы обеспечиваем полную безопасность ваших средств и конфиденциальность личных данных. Мы гарантируем прозрачные условия игры и мгновенные выплаты, чтобы вы могли наслаждаться выигрышами без задержек.
Когда стоит начать ваше путешествие в мире Aurora Casino? Регистрируйтесь прямо сейчас и получите доступ к множеству захватывающих игр и бонусов. Вот что вас ждет:
В Aurora Casino вас ждут регулярные бонусы и акции, которые увеличат ваши шансы на победу.
Мы гарантируем вашу безопасность и спокойствие, играя на нашей платформе.
Новинки и эксклюзивные игры.
С нами ваш путь к удаче будет интересным и прибыльным.
DarkVoyager
| #
Хочу понять в финансах. Что посоветуете? https://vashglbuh.ru
Gregoryfaf
| #
Kingmaker Kingmaker, Kingmaker Casino, Kingmaker greece Casino
LanceKew
| #
Prostat x?rc?ngin? qars? hans? qidalar istifad? edilm?lidir? https://x.com/Fariz418740/status/1902037337590108612
Pingrutty
| #
dark market onion https://github.com/darknetmarkets2025/darknetmarketlinks dark market list
ThunderWarden
| #
Как https://gistrade.ru вам этот ресурс о финансах?
FNDaviditabe
| #
dark markets https://onion-dark-market.shop/ – bitcoin dark web
WilliamIRrutty
| #
darknet markets url https://kingdommarketdarknet.com/ – darkmarkets
Toliknaf
| #
darkmarkets https://github.com/darknetmarketslist/darknetmarketslist darknet marketplace
ThunderSentinel
| #
5 главных ошибок при работе с финансами: https://ati-invest.ru
rox casino зеркало
| #
### Онлайн-казино: захватывающий мир азартных игр в интернете
Онлайн-казино стали популярным развлечением для миллионов игроков по всему миру. Они позволяют испытать удачу, насладиться атмосферой азартных игр и выиграть крупные суммы денег, не выходя из дома. Современные платформы предлагают огромный выбор игровых автоматов, рулетки, покера, блекджека и других игр, доступных в любое время суток.
#### Преимущества онлайн-казино
1. **Доступность** – играть можно с любого устройства: компьютера, смартфона или планшета. Все, что нужно, – это интернет-соединение.
2. **Большой выбор игр** – современные казино предлагают сотни, а иногда и тысячи слотов и карточных игр от ведущих разработчиков.
3. **Бонусы и акции** – новые игроки часто получают приветственные бонусы, фриспины и другие поощрения, которые увеличивают шансы на выигрыш.
4. **Безопасность** – лицензированные онлайн-казино используют передовые технологии шифрования данных, чтобы обеспечить защиту личной информации и финансовых транзакций.
5. **Игра в режиме реального времени** – благодаря живым дилерам пользователи могут наслаждаться атмосферой настоящего казино, находясь у себя дома.
#### Как выбрать надежное онлайн-казино?
Перед тем как начать игру, важно выбрать безопасную и честную платформу. Обратите внимание на:
– **Наличие лицензии** – надежные казино работают по лицензии официальных регуляторов (например, Malta Gaming Authority, Curacao eGaming и других).
– **Отзывы игроков** – изучите мнения других пользователей, чтобы узнать о репутации казино.
– **Выбор платежных методов** – удобные способы пополнения счета и вывода средств (банковские карты, электронные кошельки, криптовалюта).
– **Службу поддержки** – хороший игровой сервис всегда предоставляет круглосуточную поддержку клиентов.
#### Популярные игры в онлайн-казино
Игровые платформы предлагают разнообразие азартных развлечений:
– **Игровые автоматы (слоты)** – классические и современные слоты с яркой графикой, бонусными раундами и джекпотами.
– **Рулетка** – европейская, американская, французская версии игры с разными стратегиями ставок.
– **Покер** – техасский холдем, омаха, видео-покер и другие разновидности.
– **Блэкджек** – популярная карточная игра, где нужно набрать 21 очко и обыграть дилера.
– **Баккара** – динамичная игра, ставшая любимой среди профессионалов и новичков.
#### Советы для успешной игры
– Устанавливайте лимиты ставок, чтобы контролировать бюджет.
– Играйте только в лицензированных казино, чтобы избежать мошенничества.
– Используйте бонусные предложения, но внимательно читайте условия их отыгрыша.
– Развивайте стратегическое мышление в карточных играх, чтобы увеличить шансы на выигрыш.
– Помните, что азартные игры – это развлечение, а не способ заработка.
Онлайн-казино – это увлекательный мир азартных развлечений, который предлагает комфорт, удобство и возможность испытать удачу в любое время. Главное – подходить к игре ответственно, выбирать надежные площадки и получать удовольствие от процесса. Удачи за игровыми столами!
https://xteaser.ru/
Trefjwx
| #
Заказать документ о получении высшего образования вы сможете в нашей компании в Москве. diplomass.com/kupit-diplom-ulyanovsk-2
Kxyufaita
| #
darknet market lists https://cyberdarkmarkets.com/ – darknet drug store
Storm_Hacker
| #
https://czifron.ru Как преуспеть в финансах?
Rabyitabe
| #
dark markets 2025 https://github.com/darknetmarketlinks2025/darknetmarkets dark market url
JohnnyQualf
| #
Официальный веб-сайт казино Рояль предлагает располагающую обстановку для любителей казино. С его элегантным дизайном и высококачественным выбором игр, Рояль является оптимальным местом для тех, кто полагается качественное азартное развлечение. Исследуйте разнообразный выбор игровых автоматов и настольных игр на основном веб-сайте https://royalcasino-slots.com/.
Казино Рояль защищает высокий уровень клиентов и поддержку. Отзывчивая команда поддержки доступна круглосуточно, подготовлена откликнуться на различные вопросы и разрешить проблемы, обеспечивая беспрерывную игру и удовлетворение.
MarkNOlaf
| #
darkmarket list https://cannahomedarknetdrugstore.com/ – dark web market
CoreyLal
| #
учебный центр Переподготовка и повышение квалификации: инвестиции в профессиональный рост. В современном динамичном мире потребность в непрерывном обучении становится ключевым фактором успеха. Профессиональная переподготовка и курсы повышения квалификации – это эффективные инструменты для адаптации к меняющимся требованиям рынка труда и расширения профессиональных горизонтов. Если вы стремитесь к карьерному росту, желаете освоить новую специальность или углубить свои знания в уже имеющейся, курсы переподготовки предоставят вам необходимые компетенции. В ряде случаев возникает необходимость подтверждения квалификации. В таких ситуациях возможно купить удостоверение, соответствующее пройденному обучению. Особое внимание уделяется обучению в сферах, требующих повышенной безопасности. Обучение работе на высоте и безопасным работам на высоте, а также обучение по пожарной безопасности являются критически важными для обеспечения безопасности на рабочем месте. Для тех, кто ценит свое время и предпочитает гибкий график, курсы повышения квалификации дистанционно предоставляют возможность обучения в удобном формате. Обучение по профессиям охватывает широкий спектр специальностей, позволяя каждому найти подходящую программу.
LightX4
| #
Финансы для компаний: https://tandem585.ru
DonDonfaita
| #
darknet marketplace https://github.com/darkwebwebsites/darkwebwebsites darknet sites
NormanGaith
| #
Возможно ли получать тысячи манатов в месяц в соцсетях в Азербайджане?
https://pin.it/1OLQiA9c2
Walker7085
| #
Авторитетное мнение о финансах: https://chastnyj-investor.ru
Pingrutty
| #
dark web markets https://github.com/darknetmarkets2025/darknetmarketlinks darknet drugs
Terencesam
| #
Игровые автоматы лучшие Спины за регистрацию без депозита с выводом
FNDaviditabe
| #
darkmarket https://mydarkmarketlink.com/ – darknet sites
Lutherram
| #
Игровое заведение Вулкан – это увлекательный мир азарта, который завораживает с первого взгляда. Благодаря замечательному дизайну и понятному интерфейсу, пользователи быстро оказываются в этом игровом пространстве.
Среди преимуществ казино Вулкан игровые автоматы бесплатно стоит отметить широкий ассортимент игр, включая слоты и настольные развлечения, которые придутся по вкусу каждому игроку. Качество графики и анимационных эффектов делает игровой процесс дополнительно увлекательным и завораживающим.
Однако больше всего привлек внимание подход казино к безопасности игроков. Они используют новейшие технологии шифрования, чтобы обеспечить приватность личных и финансовых данных пользователей. Это делает казино Вулкан замечательным вариантом для онлайн-игры.
BruceHip
| #
голд казино
PhantomReaper
| #
Что лучше выбрать в финансах? https://lombardngs.ru
Glennbup
| #
уничтожение тараканов в москве
WilliamIRrutty
| #
darkmarket https://darknetmarketsonion.shop/ – darkmarket list
Donaldlaf
| #
dark web link https://github.com/darkwebmarketslinks/darkwebmarkets dark web marketplaces
DriverBaile
| #
Медицинский регистратор в поликлинике рекомендовал ознакомиться с важной медицинской информацией. Кремация организована с пониманием. Сотрудники работают профессионально.
Phantom_Nomad
| #
Ищу реальные кейсы по финансам. https://pfclub.ru
Arthurclevy
| #
Хотите получить bingo online 10 euros gratis начни игру без риска – Открой для себя захватывающие слоты, живое казино и эксклюзивные бонусы. Играй с комфортом, наслаждайся топовыми играми и выигрывай!
WilliamWAl
| #
Looking for top-notch slots to play? Abe Bet Casino offers endless variety online slots for all players!
At Abe Bet abe-casino.com/tr/games/demo/pragmatic_play_mustang_gold, you’ll find slot hits from top developers.
Register now to get generous bonuses.
Dive into an atmosphere of thrill with Abe Bet!
Kxyufaita
| #
darknet drug market https://kingdomdarknetdrugstore.com/ – dark web market
Toliknaf
| #
darknet websites https://github.com/darknetmarketslist/darknetmarketslist dark web market
Silver22
| #
С чего начать https://kpkmatkap.ru финансов?
mostbetuzbekistan
| #
Mostbet Uzbekistan mostbetuzbekistan-apk.com offers you profitable bets, top odds and instant payouts. Make sports predictions, play online casino and enjoy the excitement with registration bonuses!
Robertlox
| #
Ищете увлекательную платформу для азартных игр? Покердом — это лучший выбор для развлечений!
На https://kamuflyag-shop.ru/ вас ждут популярные слоты.
Простота интерфейса и программы лояльности делают игру еще интереснее!
Зарегистрируйтесь прямо сейчас и испытайте радость победы с Pokerdom!
MarkNOlaf
| #
best darknet markets https://drdarkfoxmarket.com – dark markets
Blue_Hunter
| #
Лучшие инструменты для работы с финансами: https://akcept-garant.ru
Rabyitabe
| #
darkmarket link https://github.com/darknetmarketlinks2025/darknetmarkets darkmarket 2025
EdwinFrima
| #
Ищете увлекательную платформу для азартных игр? Vulkan Russia — это идеальное место для развлечений!
На Vulkan Russia вас ждут захватывающие настольные игры.
Простота интерфейса и щедрые бонусы делают игру еще интереснее!
Зарегистрируйтесь прямо сейчас и испытайте радость победы с Вулкан Россия!
Pingrutty
| #
dark web sites https://github.com/darknetmarkets2025/darknetmarketlinks onion dark website
Daniellab
| #
Szukasz rzetelnego mechanika samochodowego w Poznaniu, który z pełnym zaangażowaniem zadba o Twoje auto i naprawi je na czas, zgodnie z obowiązującą gwarancją? Zaufaj zespołowi TAK Serwis: oferujemy kompleksowe usługi naprawy samochodów, w tym regularną wymianę części eksploatacyjnych – https://takserwis.pl/. Wybierając nasz warsztat samochodowy, postawisz na profesjonalną obsługę, wysokiej jakości części oraz szybką realizację prac.
Phantom_Slayer
| #
История внедрения финансов https://kpk-novydom.ru в компании.
Pinco Casino
| #
Джекпоты в онлайн-казино: реальные шансы на большой выигрыш
Современные азартные площадки предлагают захватывающие возможности для тех, кто ищет возможность ощутить удачу на своей стороне. Игроки могут не только насладиться разнообразием развлечений, но и надеяться на значительные выплаты. Но какова реальная вероятность получить желаемую сумму? В этой статье мы рассмотрим, на что стоит обратить внимание, чтобы повысить шансы на получение крупных выигрышей.
Статистические данные показывают, что суммарные выплаты в большинстве игр складываются из множества факторов, включая вероятность выпадения определенных сочетаний символов и правила самого автомата. Игрокам стоит обратить внимание на процент возврата игроку (RTP), который варьируется от 85% до 98%. Чем выше этот показатель, тем больше вероятность возврата вложенных средств.
Многие автоматы предлагают различные виды акций и бонусов. Оптимальное использование этих возможностей может значительно увеличить время игры и, следовательно, шансы на успех. Например, участвуя в турнирах, можно повысить как навыки игры, так и потенциал для получения крупных сумм. Также многие площадки предоставляют приветственные бонусы, которые увеличивают стартовый капитал и открывают новые горизонты для игроков, готовых рискнуть.
Как выбрать игровые автоматы
При выборе автоматов важно учитывать несколько ключевых факторов, чтобы повысить вероятность успеха. Во-первых, обратите внимание на высоту RTP (Return to Player). Этот показатель выражает, какую часть ставок игроки могут ожидать в виде выигрышей. Чем выше значение RTP, тем больше шансов возвращения средств. Идеальный вариант – автоматы с RTP выше 95%.
Кроме того, изучите волатильность игры. Высоковолатильные автоматы реже выдают выигрыши, но размер призов значительно больше. Низковолатильные аппараты, наоборот, часто приносят небольшие суммы. Выбор зависит от вашего стиля игры и готовности рисковать.
Не менее важным является разнообразие тематики и игровых функций. Автоматы с бонусными играми, фриспинами и дополнительными символами могут существенно увеличить увлекательность процесса и разнообразие возможностей для получения прибыли. Оцените доступные функции каждого автомата и выберите те, которые вам интересны.
Также стоит обратить внимание на репутацию разработчика. Известные компании часто гарантируют качество и честность игры. Читайте отзывы других игроков и выбирайте автоматы от проверенных брендов, чтобы избежать недобросовестных практик.
Не забывайте о демо-режиме. Многие платформы предлагают возможность пробовать автоматы без ставок, что позволяет ознакомиться с механикой и динамикой игры без финансовых рисков. Это особенно полезно для новичков, которые пока не уверены в своих предпочтениях.
Понимание вероятностей выигрыша
Чтобы оценить свои возможности, важно разобраться в механизмах формирования выплат, которые зависят от множества факторов. Каждый автомат имеет так называемый коэффициент возврата, который обозначает процент от всех ставок, возвращаемый игрокам. Стандартные значения колеблются от 85% до 98%, а это значит, что в долгосрочной перспективе игроки могут рассчитывать на разные уровни возврата.
Немаловажным аспектом является вероятность выпадения комбинаций. Для каждого конкретного устройства эта информация доступна в таблице выплат. Например, наличие редких символов, таких как специальные знаки или бонусные элементы, влияет на общую рентабельность. Чаще всего минимальные комбинации имеют более высокую частоту появления, тогда как крупные выплаты требуют больше времени и удачи.
Необходимо учитывать, что механика функции случайных чисел играет ключевую роль в формировании результата каждого спина. Поэтому даже при высоком проценте отдачи в краткосрочной перспективе возможны как выигрышные, так и проигрышные сессии. Тактика управления банкроллом поможет продлить игру и правильно распределить средства для получения максимального удовольствия.
Советы для более осознанного подхода включают проверку отзывов об играх, анализ актуальных статистических данных и использование демо-версий для знакомства с особенностями. Таким образом, смещение акцента на понимание механизмов увеличивает шансы на успех, несмотря на случайный характер событий.
https://905090.ru/
DonDonfaita
| #
darknet site https://github.com/darkwebwebsites/darkwebwebsites dark websites
FNDaviditabe
| #
darkmarket url https://mydarkmarketlink.com/ – darkmarket
Joshuamub
| #
Looking for best online casino games? MasalBet is the place to find favorite games from leading brands.
At https://masalbet-official.com/tr/, you’ll find exclusive promotions and a user-friendly interface.
Register now and get exclusive offers.
Play exciting games with MasalBet!
Guardian1235
| #
Пример из https://vp-finance.ru практики: Финансы.
WilliamIRrutty
| #
darknet site https://darknetmarketsonion.shop/ – dark market onion
mostbet_ueEr
| #
скачать mostbet на телефон http://mymoscow.forum24.ru/?1-2-0-00000718-000-0-0-1742357638/ .
1win_fcpi
| #
1win букмекер https://mymoscow.forum24.ru/?1-2-0-00000717-000-0-0-1742357431/ .
Michealhem
| #
https://stage.corich.jp/stage_main/234466
1win_hnon
| #
1 вин http://www.cah.forum24.ru/?1-19-0-00000732-000-0-0 .
Shadow_Blade
| #
Новые подходы в финансах: https://center-torgov.ru
Kxyufaita
| #
dark market list https://kingdomdarknetdrugstore.com/ – darknet links
Donaldlaf
| #
dark market link https://github.com/darkwebmarketslinks/darkwebmarkets darkmarket 2025
Blue573
| #
Эксклюзивное интервью с экспертом по финасам: https://gym-credit.ru
Toliknaf
| #
darknet markets onion https://github.com/darknetmarketslist/darknetmarketslist bitcoin dark web
MarkNOlaf
| #
darknet markets https://drdarkfoxmarket.com – darknet drug links
NightV9
| #
5 https://kapital-c.ru главных ошибок при работе с финансами.
DarkZ1
| #
Может кто знает ориентируется в финансах? Где искать? https://lombard-bis.ru
TrevorRat
| #
Магазин печей и каминов pech.pro широкий выбор дровяных, газовых и электрических моделей. Стильные решения для дома, дачи и бани. Быстрая доставка, установка и гарантия качества!
Pingrutty
| #
dark market url https://github.com/darknetmarkets2025/darknetmarketlinks darknet markets onion address
Hacker7694
| #
Как не сдаться при изучении финасов: https://fin-abc.ru
MichaelZoday
| #
whores ufc heroin
Rabyitabe
| #
darkmarkets https://github.com/darknetmarketlinks2025/darknetmarkets darknet sites
FNDaviditabe
| #
darknet drugs https://darkmarketdarkfox.com/ – darknet markets onion
WolfZ4
| #
Особенности реализации финансов: https://finiko-dv.ru
CoreyLal
| #
меховое ателье с Москве Мех как искусство: от норки до каракульчи, преображение и вдохновение Мир моды изменчив, но любовь к роскошному меху остается неизменной. Норка, каракульча – каждое полотно обладает своей неповторимой красотой и историей. Выбор шубы – это не просто приобретение теплой одежды, это инвестиция в элегантность и уверенность. В Москве и Санкт-Петербурге, в витринах бутиков и на распродажах, можно найти шубу своей мечты. Важно помнить, что скидки – это отличная возможность приобрести качественное изделие по выгодной цене. Но что делать, если в шкафу уже висит шуба, вышедшая из моды? Не спешите с ней прощаться! Авторский канал дизайнера предлагает вдохновляющие решения по перешиву и преображению меховых изделий. Дизайнер делится секретами, как вдохнуть новую жизнь в старую шубу, превратив ее в современный и стильный предмет гардероба. Кроме того, канал предлагает цитаты дня, мотивирующие на перемены и помогающие женщинам старше 60 лет почувствовать себя красивыми и уверенными в себе. Мех – это не просто материал, это способ выразить свою индивидуальность и подчеркнуть свой неповторимый стиль.
DonaldWheri
| #
каталог nova маркетплейс
WilliamIRrutty
| #
darknet markets onion https://kingdomdarkmarketonline.com/ – dark web marketplaces
Master4685
| #
Читал прогнозы, что Финансы будет главным трендом. Возражения? https://finansovoereshenie.ru
DonDonfaita
| #
darkmarket https://github.com/darkwebwebsites/darkwebwebsites darknet markets onion
Binesahaw
| #
Join the adventure in Gameworld Barbarossa today. Begin fete medievale saint valery sur somme your quest for riches with immersive gameplay, legendary characters, and massive payouts designed to captivate every treasure-hunting player.
jozz casino
| #
Можно ли обыграть онлайн-казино? Разбираем популярные мифы
Виртуальные азартные заведения давно стали объектом обсуждения, и среди игроков активно циркулирует множество заблуждений. Некоторые утверждения представляют собой простую дезинформацию, в то время как другие основаны на честном желании поделиться опытом. Тем не менее, важно разобраться в фактах и вычленить истинное из потока домыслов. Актуальные статистические данные показывают, что лишь небольшой процент участников действительно достигает значительных успехов.
Некоторые убеждены, что с помощью определённых стратегий можно гарантировать выигрыш. Однако, многие рекомендации, такие как использование систем ставок или выбор определённых игр, не обеспечивают ожидаемой прибыли. Результаты в большинстве случаев зависят от случайности. Математические модели подсказывают, что долговременное преимущество находится на стороне заведения, и лишь временные успехи отдельных игроков не меняют общую картину.
В то же время существуют игровые механики, которые могут помочь улучшить баланс игровых сессий. Изучение правил и дисциплина при управлении капиталом становятся залогом для осознанного подхода к развлечению. Важно обратить внимание на использование бонусов и акций, которые могут несколько увеличить шансы, но и здесь следует помнить о рисках. Информированность – ключ к уверенности на пути к развлечение в цифровом пространстве азартных игр.
Скептические взгляды на ставки
Существует распространенное мнение, что применение определённых систем ставок всегда приводит к успеху. Многие азартные участники полагают, что существует универсальная стратегия, способная гарантировать прибыль при игре в азартные развлекательства. Важно понимать, что такие подходы обычно основываются на неверных предположениях о механике игр. Например, монетная система требует увеличения ставок после каждой проигрышной игры, однако она не меняет шансы на выигрыш.
Другой миф заключается в том, что последовательность выигрышей или проигрышей может указывать на будущие результаты игры. Распространенное заблуждение о “горячих” или “холодных” числах в игре в рулетку является иллюзией. Фактически, каждая новая игра независима от предыдущей, и результаты не имеют взаимосвязи друг с другом.
Некоторые игроки убеждены, что использование сложных математических формул и стратегии может значительно повысить вероятность выигрыша. Однако, несмотря на высокую математическую теорию, игры, основанные на случайных числах, не поддаются контролю, и такие методы не обеспечивают надёжной защиты от потерь.
Важно учитывать, что азартные игры – это, прежде всего, развлечение. Разумный подход заключается в установлении чётких лимитов на ставки и понимании того, что никакая система не гарантирует успех. Продумывая стратегию, полезно опираться на финансовые ограничения и личные предпочтения, а не на иллюзии о системе ставок.
Реальные стратегии выигрыша
Существуют различные подходы к игре, зависящие от особенностей конкретных азартных развлечений. Один из таких методов – применение ставок по системе Мартингейл. Она заключается в удвоении ставки после каждого проигрыша. Это позволяет в случае выигрыша вернуть все предыдущие потери и получить небольшую прибыль. Однако такой метод требует достаточного банковского ресурса и мудрого управления финансами.
Другой метод – использование стратегии минимизации рисков. Например, в рулетке рекомендуется делать ставки на цвет или четность, так как они предлагают высокий шанс на выигрыш (приблизительно 48%). Данный подход уменьшает вероятность крупных потерь и создает возможность для длительной игры с разумным банкроллом.
Следующий вариант – анализ в игровых аппаратах. Многие игроки игнорируют фактические шансы конкретных слотов. Исследование уровня возврата к игроку (RTP) помогает выбирать автоматы с более высоким процентом возврата, что увеличивает вероятность успеха в долгосрочной перспективе.
В блэкджеке нарастает популярность стратегий, базирующихся на подсчете карт. Игроки отслеживают, какие карты уже были розданы, чтобы оценить вероятность появления высоких карто. Хотя этот подход запрещен во многих заведениях, его использование все же может дать значительное преимущество.
Наконец, важным аспектом является дисциплина. Установив заранее предел для выигрышей и проигрышей, можно избежать эмоциональных решений, которые часто приводят к независимым потерям. Фиксация результатов каждой сессии и анализ их позволит выявить свои сильные и слабые стороны, что приведет к улучшению стратегии игры.
https://ss-net.ru/
Reaper5123
| #
Где смотреть https://stopkredit-vladivostok.ru актуальные данные о финансах?
Kxyufaita
| #
darknet sites https://darkfoxmarketplace.com/ – darknet links
https://wiki.flashcorponline.com/index.php/Почему_Зеркала_Слотозал_Промокод_Так_Ðезамнимы_ДлÑ_Ð’Ñех_К
| #
https://wiki.flashcorponline.com/index.php/Почему_Зеркала_Слотозал_Промокод_Так_Ðезамнимы_ДлÑ_Ð’Ñех_Клиентов
A Brief History of Elemental Analysis » Artemis Analytical
1win_sqpi
| #
1win.pro http://mymoscow.forum24.ru/?1-2-0-00000717-000-0-0-1742357431 .
mostbet_nwEr
| #
mostbet.kg mostbet.kg .
EdwardLable
| #
go to my blog acheter des billets contrefaits
Donaldlaf
| #
darknet drug links https://github.com/darkwebmarketslinks/darkwebmarkets darknet market lists
1win_hjon
| #
1win.kg http://www.cah.forum24.ru/?1-19-0-00000732-000-0-0 .
MarkNOlaf
| #
darknet drug links https://drdarkfoxmarket.com – dark web marketplaces
EagleHunter
| #
Исследование финансов: https://rostpension-partner.ru
RedWarden
| #
Пошаговая инструкция по финансам: https://kpk-ruscapital.ru
Pingrutty
| #
dark web marketplaces https://github.com/darknetmarkets2025/darknetmarketlinks darkmarket 2025
Toliknaf
| #
darkmarket list https://github.com/darknetmarketslist/darknetmarketslist dark markets
FNDaviditabe
| #
dark web marketplaces https://darkmarketdarkfox.com/ – best darknet markets
VortexGuardian
| #
Профессионалы рекомендуют эти ресурсы по финансам: https://prostolombard.ru
WilliamIRrutty
| #
darkmarket 2025 https://idarknetmarket.com/ – darknet market list
Eagle801
| #
Где тренироваться финансам? https://finansist-spb.ru.
Rabyitabe
| #
darknet market https://github.com/darknetmarketlinks2025/darknetmarkets dark web market
Kxyufaita
| #
best darknet markets https://kingdomdarknetdrugstore.com/ – dark web link
DonDonfaita
| #
dark web markets https://github.com/darkwebwebsites/darkwebwebsites dark web market list
DragonWarden
| #
https://onegokredit.ru Современные методы по финансам.
Sazrkzv
| #
Мы изготавливаем дипломы любых профессий по приятным ценам. Стоимость может зависеть от выбранной специальности, года выпуска и образовательного учреждения. Всегда стараемся поддерживать для покупателей адекватную ценовую политику. Важно, чтобы документы были доступными для большого количества граждан. купить диплом университета в ростове на дону
Voyager5169
| #
Смешные случаи в финансах: https://ravoskhod.ru.
MarkNOlaf
| #
dark market https://kingdomdarkmarketplace.com – dark web market
Pingrutty
| #
darknet links https://github.com/darknetmarkets2025/darknetmarketlinks dark web link
Cazrfwk
| #
Здравствуйте!
Мы изготавливаем дипломы любых профессий по разумным ценам. Стоимость будет зависеть от выбранной специальности, года получения и образовательного учреждения: diplomans.com/
Donaldlaf
| #
darkmarket list https://github.com/darkwebmarketslinks/darkwebmarkets darknet drugs
Cazrtax
| #
Приобрести диплом ВУЗа по невысокой стоимости возможно, обратившись к надежной специализированной компании. Мы готовы предложить документы престижных ВУЗов, расположенных в любом регионе РФ. diplomc-v-ufe.ru/kupit-diplom-texnikuma-moskva
RedHacker
| #
Какие https://galactic-pro.ru изменения в финансах?
FNDaviditabe
| #
darkmarket 2025 https://kingdom-marketplace.com/ – dark web market urls
PhantomPioneer
| #
Какие платформы использовать для финансов? https://pegascapital.ru
WilliamIRrutty
| #
tor drug market https://kingdomdarkmarketonline.com/ – dark web market urls
ShadowZ9
| #
Смешные случаи в финансах: https://lombardchernozemie.ru
Kxyufaita
| #
dark web sites https://cannahomemarket.link/ – dark market
DragonZ5
| #
Приветствую. Подскажите, где найти информацию о финансах? https://vis-invest.ru
Diplomi_pjKt
| #
Приобрести диплом ВУЗа!
Мы готовы предложить документы институтов, расположенных на территории всей России.
diplomj-irkutsk.ru/kupit-diplom-o-srednem-obrazovanii-11-klassov-4
BlueSentinel
| #
Ошибки новичков в финансах: https://kpkdoverierm.ru.
MarkNOlaf
| #
dark market onion https://dark-web-versus.com – dark market list
Stephenavelo
| #
септики для дома купить спб купить септик для частного дома
FNDaviditabe
| #
darknet drugs https://firstdarkmarket.com/ – darknet market links
DarkByte
| #
С чего начать финансов? https://zmd-region.ru
WilliamIRrutty
| #
darknet markets url https://idarknetmarket.com/ – darknet drug market
Ghost_Guardian
| #
Секретные методы https://buildrich.ru финансов.
Thomasprise
| #
Как научить ребёнка контролировать гнев? Покажите, что можно злиться, но при этом не обижать других. Например, предложите ему “поругаться” с подушкой или нарисовать свои чувства. Объясните, что даже взрослые иногда злятся, но умеют с этим справляться.
play fortuna зеркало
| #
Какие бонусы самые выгодные в 2025 году?
В последнее время наблюдается заметное изменение подходов к программам поощрения клиентов. В условиях растущей конкуренции компании стремятся предложить более таргетированные и привлекательные предложения, позволяя пользователям выбирать те, которые действительно отвечают их потребностям и образу жизни. В этом контексте особенно интересны предложения, которые показывают реальную ценность на фоне других опций.
Один из наиболее многообещающих трендов – это программы лояльности с гибкими условиями. Потребители теперь могут накапливать баллы и располагать ими по своему усмотрению, например, использовать их не только на покупки, но и для получения услуг, таких как подписка на стриминг или аренда автомобилей. Такой подход позволяет удовлетворить разнообразные потребности клиентов и значительно повышает степень вовлеченности.
Другим примером могут служить кэшбэк-программы, которые, как показывают исследования, становятся все более популярными среди пользователей. Совершая повседневные покупки, клиенты будут возвращать до 10% от суммы, что существенно экономит средства и повышает общую привлекательность продуктов и услуг. При этом важно обращать внимание на условия: многие компании предлагают кэшбэк только при определенных условиях или через конкурирующие платформы.
В сочетании с современными технологиями, такими как мобильные приложения и персонализированные предложения, эти новые форматы делают программу поощрения более доступной и понятной, что создает благоприятную атмосферу для пользователей. На 2025 год, выбор подходящей опции будет зависеть от индивидуальных предпочтений, активности и конкретных жизненных ситуаций. Сравнение нескольких вариантов и анализ их достоинств помогут сделать правильный выбор.
Преимущества для инвесторов
Инвесторы в 2025 году могут воспользоваться рядом предложений, способствующих увеличению доходности. Открытые инвестиционные платформы часто предлагают акции на комиссии. Это позволяет минимизировать затраты при входе в новые позиции или при работе с портфелем.
Некоторые финансовые компании вводят программы возвращения части средств. Такие инициативы могут быть применимы к инвестициям в определённые финансовые инструменты. Например, пользователи могут получить кэшбэк за торговлю акциями или ETF, что значительно увеличивает общий доход.
Но не только возврат средств становится привлекательным. Для тех, кто активно использует кредитные средства, важно обратить внимание на предложения с низкими процентными ставками. Особенно актуально это для заимствований на торговые сделки. Низкие ставки способны повысить рентабельность проектов за счёт сниженных издержек.
Не стоит упускать из виду и возможности, связанные с налоговыми льготами. Многие страны предлагают специальные условия для инвесторов. Участие в таких инициативах может привести к значительной экономии, особенно если инвестирование осуществляется в долгосрочной перспективе.
Также обратите внимание на образовательные программы от брокеров. Качественное обучение может открыть новые стратегии и подходы к инвестированию. Например, вебинары и курсы по техническому анализу или инвестиционным психологиям могут значительно улучшить результаты ваших торгов.
В 2025 году бизнес-модели многих компаний будут адаптироваться к новым условиям рынка. Это создаёт уникальные возможности для инвесторов, готовых исследовать и пробовать новые подходы. Ищите предложения, которые не только улучшают ваш финансовый результат, но и увеличивают ваши знания, что, в итоге, приведёт к более взвешенным решениям в области инвестиций.
Промоакции в ритейле
В 2025 году многие компании в ритейле применяют разнообразные промоакции, чтобы привлечь покупателей. Скидки на 10-50% становятся стандартом, при этом нередко встречаются эксклюзивные предложения на ограниченные сроки. Интересный пример – акции на популярные товары, которые напрямую зависят от времени года, праздников или особых событий.
Кэшбек-системы также продолжают набирать популярность. Такая схема позволяет клиентам возвращать часть потраченных средств, что стимулирует повторные покупки. Важно выбрать ритейлеров, которые предлагают кэшбек на высокие суммы или по ценным категориям товаров.
Система лояльности играет значимую роль в построении долгосрочных отношений с клиентами. Программы, в которых за каждую покупку начисляются баллы, которые затем можно обменять на скидки, подарки или специальные предложения, всегда привлекают внимание. Рекомендуется делать акцент на те программы, которые предлагают максимальное количество точек и гибкие условия обмена.
Сезонные акции представляют собой отличный способ стимулировать спрос. Например, праздничные распродажи или заранее анонсированные дни скидок позволяют ритейлерам не только увеличить оборот, но и разгрузить запасы товара. Такие подходы часто итеративны и базируются на потребительских предпочтениях и данных о продажах.
Следует также обратить внимание на акционные наборы. Упаковка нескольких товаров по специальной цене позволяет не только увеличить средний чек, но и привлечь внимание к менее известному ассортименту. Перед запуском подобных акций полезно провести исследование мнений потребителей, чтобы выявить сочетания товаров, которые будут интересны целевой аудитории.
https://pizzanyam.ru/
Kxyufaita
| #
dark web market urls https://cannahomemarket.link/ – dark websites
Dragon451
| #
Что https://czifronpay.ru лучше выбрать в финансах?
hMustafaVep
| #
Какой красивый оттенок! Заказала букет маме.
Доставка цветов Томск
GhostV3
| #
Преодоление трудностей при изучении https://tsb-broker.ru финансов.
Red420
| #
Пошаговая инструкция https://ik-rodina.ru по финансам.
JamesFeque
| #
В нашем списке лучшие девушки города Адлер — это топ реальных индивидуалок, каждая из которых стремится оставить незабываемые воспоминания. Погрузитесь в атмосферу нежности и страсти, открывая новые возможности для общения и отношений, которые помогут вам стать ближе к своим мечтам https://night24-adler.com/
Thomasseato
| #
Предоставим легальный адрес и полный пакет документов для регистрации: временная регистрация москва
FNDaviditabe
| #
dark market url https://mydarkmarketlink.com/ – dark market 2025
DennisSen
| #
porn esports whores
ThomasVep
| #
Цветы очень свежие и ароматные, жена была в восторге!
заказать цветы с доставкой в томске
BMW_ppKt
| #
Новый BMW X6: стиль и мощь, модельный ряд.
Превосходство BMW X6 на дороге, всегда.
Откройте для себя.
Уникальный дизайн BMW X6, высокой техники.
Как BMW X6 меняет правила игры, откройте.
Идеальный выбор – BMW X6, инвестирование.
Потрясающая отделка и материалы в BMW X6, создают.
Ваш надежный спутник – BMW X6, предоставляет.
Почему стоит выбрать BMW X6?, в нашем обзоре.
Динамичный BMW X6 – для активной жизни, каждого.
Как BMW X6 заботится о вашей безопасности, в приоритете.
Выбор BMW X6: ваши преимущества, открыл.
Технологический прогресс BMW X6, формируют.
Как будет ощущаться поездка на BMW X6, особенности.
Что дает вам BMW X6?, в нашем анализе.
Выразительный дизайн BMW X6, подчеркнет ваш статус.
Как BMW X6 выглядит на фоне конкурентов, в нашем сравнении.
Что говорят владельцы о BMW X6?, в нашей статье.
Современные системы безопасности BMW X6, позаботятся о вас.
Заключение: стоит ли покупать BMW X6?, обобщаем мнение.
bmw 2 bmw 2 .
WilliamIRrutty
| #
darknet markets onion address https://kingdommarketdarknet.com/ – dark market
RichardVarse
| #
whores esports heroin
Kxyufaita
| #
dark market 2025 https://kingdomdarknetdrugstore.com/ – dark markets
EagleZ4
| #
Мифы и правда о финансах: https://mqncwltd.ru
Steel388
| #
Смешные случаи в финансах: https://investd.ru.
1win_jfki
| #
1win com http://www.1win705.ru .
mostbet_pjml
| #
mostbet http://mostbet780.ru/ .
mostbet_kg_tber
| #
мотбет http://www.mostbet781.ru .
Byte1034
| #
Лучшие инструменты для работы с финансами: https://pik-24.ru
Modelnyy_lmOt
| #
Модельный ряд BMW: откройте для себя новые возможности, которые порадуют.
Узнайте о лучших моделях BMW, которые впечатляют.
Откройте для себя новейшие модели BMW, спортивные машины.
Каждый найдет свою идеальную BMW, впечатляюще широк.
Погрузитесь в мир премиальных автомобилей BMW, которые создают.
Что отличает BMW от других брендов, обнаружьте.
Каждая модель BMW — это шедевр, созданный для.
Лучшие автомобили в модельном ряду BMW, которые заинтересуют.
Как BMW отвечает на вызовы времени, проверьте.
Новый взгляд на автомобили BMW, подходящие для городской жизни.
BMW — это больше, чем просто автомобиль, это символ статуса.
Исключительное качество: выбор BMW, который вдохновляет.
Переосмысленный комфорт и элегантность BMW, поклонников.
Почему BMW — это ваш идеальный выбор, от комфорта до инноваций.
Давайте исследовать великолепие модельного ряда BMW, с передовыми технологиями.
Наслаждайтесь разнообразием автомобилей BMW, для любых приключений.
Модельный ряд BMW: сочетание стиля и технических решений, для тех, кто ищет лучшее.
Откройте для себя свою следующую BMW, с надежностью и безопасностью.
Каждый автомобиль BMW — это возможность, для любого владельца.
x3 bmw https://model-series-bmw.biz.ua/ .
FNDaviditabe
| #
dark web market list https://mydarkmarketlink.com/ – dark web sites
BryanCar
| #
вулкан казино зеркало
WilliamIRrutty
| #
darkmarket https://kingdommarketdarknet.com/ – dark websites
DragonReaper
| #
Лучшие инструменты для работы с https://makapital.ru финансами.
Thunder375
| #
Может кто знает разбирается в https://strahovkatut21.ru финансах? Куда зайти?
Kxyufaita
| #
darknet links https://cannahomemarket.link/ – darknet drug market
JamesFeque
| #
У нас представлены лучшие девушки Адлера, ставшие частью топа реальных индивидуалок. Каждая девушка подарит вам уникальную возможность найти понимание и близость. Погрузитесь в мир настоящего счастья и нежных чувств, о которых вы так долго мечтали: секс адлер
Sazrvam
| #
Приобрести диплом университета !
Покупка диплома университета РФ в нашей компании – надежный процесс, поскольку документ заносится в реестр. При этом печать осуществляется на специальных бланках ГОЗНАКа. Приобрести диплом любого ВУЗа arenadiplom24.online/vuzy/fgou-vpo-ngpu
IronWalker
| #
Приветствую. Может кто в курсе, где найти информацию о финансах? https://titanconsultgroup.ru
Eagle_Voyager
| #
Забавные https://avanschita.ru моменты в финансах.
Eddiehem
| #
whores esports terrorist attack
RafaelBax
| #
heroin esports porn
FNDaviditabe
| #
dark market 2025 https://kingdom-marketplace.com/ – darknet links
WilliamIRrutty
| #
darknet drug market https://idarknetmarket.com/ – dark web market list
1win_rnki
| #
1win cazino https://www.1win705.ru .
Blue_Guardian
| #
Полное руководство по https://vam-zaim56.ru финансам от А до Я.
1xslots казино
| #
Разбираем популярные системы ставок в блекджеке
Карточная игра, где мастерство и удача идут рука обруку, привлекает внимание азартных игроков по всему миру. Здесь выбор стратегии может существенно повлиять на итоговый результат. Наблюдая за мастерами, можно заметить, что многие из них опираются на разнообразные подходы, которые помогают управлять банкроллом и максимизировать шансы на выигрыш.
Одним из наиболее исследуемых методов является система, основанная на постепенном увеличении размера ставки после проигрыша. Эта тактика, известная как “мартингейл”, позволяет игрокам рассчитывать на быстрое возмещение потерь, однако риск потери значительной суммы при затяжной серии неудач остается высоким. Использование такой стратегии требует особого контроля за лимитами и объемом доступных средств.
Среди других методов выделяется концепция прогрессивного увеличения ставок после выигрыша. Она нацелена на извлечение выгоды в периоды удачи, однако и она не лишена подводных камней. Важно учитывать, что игровые сессии могут быть непредсказуемыми, и после успешного результата может последовать ряд поражений.
Разработка индивидуальной стратегии, опирающейся на собственные наблюдения и азартные привычки, может помочь достичь поставленных целей. Главное – это адаптивность и критическое мышление, которые позволят в нужный момент пересмотреть подход и минимизировать риски при игре с картами.
Система Мартингейла
Мартингейл основывается на принципе удвоения ставки после проигрыша. Идея проста: если игрок теряет, он увеличивает ставку, чтобы при следующей победе покрыть все предыдущие убытки и заработать первоначальную прибыль. Это требует наличия достаточного банкролла и устойчивости к рискам.
Стратегия предполагает, что игрок действует по следующему алгоритму: начать с небольшой суммы, затем удвоить ставку после каждого проигрыша. Например, если первая ставка составила 10 единиц и она проиграна, следующая ставка будет 20, затем 40, и так до тех пор, пока не произойдет выигрыш или банкролл не истощится.
Важно учитывать, что при использовании Мартингейла у игроков могут возникнуть проблемы на длинной дистанции. Поскольку ставки могут быстро увеличиваться, существует риск достижения лимитов стола или полного исчерпания средств. Поэтому установка максимального ограничения для ставки является разумным решением.
Существуют нюансы, которые стоит учитывать: играя на малом лимите, игрок может не столкнуться с серьезными проблемами, но при высоких ставках в ситуациях с длительными проигрышами вероятность банкротства значительно возрастает. Перед началом игры важно определить четкий план и придерживаться его.
Некоторые эксперты рекомендуют сочетать Мартингейл с другими стратегиями, такими как фиксированные ставки или другие системы управления банкроллом, чтобы сгладить риски и повысить шансы на победу. Также полезно периодически пересматривать свою стратегию, адаптируя её к текущей ситуации за столом.
Система Д’Alembert
Система Д’Alembert основана на принципе балансировки потерь и выигрышей. Основной метод заключается в увеличении ставки на одну единицу при проигрышах и уменьшении ее на одну единицу при выигрыше. Такой подход минимизирует риски при последовательных проигрышах и позволяет восстанавливать потери.
Для начала игры определите первоначальный размер вашей ставки. Лучше всего использовать небольшую сумму, чтобы снизить уровень риска. Многие игроки начали с такой ставки, которая составляет всего 1% от своего банкролла. Это позволит вам более гибко реагировать на ситуацию за столом.
Например, если начальная ставка составляет 10 рублей, при проигрыше вы ставите 11 рублей, затем 12 рублей и так далее. При выигрыше возвращаетесь к предыдущей ставке. Такой алгоритм помогает сохранять банкролл и не дает ему стремительно уменьшаться при неблагоприятной серии.
Несмотря на свою простоту, Д’Alembert не является безрисковым. Зависимость от последовательности выигрышей и проигрышей может привести к значительным потерям, если не учесть верхний предел ставок. Устанавливая лимит на максимальную ставку, вы защитите себя от крупных убытков.
Как рекомендация, старайтесь следить за своей игрой и отмечать результаты, чтобы оценить эффективность данной тактики для вашей конкретной ситуации. Пристальное внимание к своему банкроллу и контроль над эмоциями могут существенно повысить шансы на успех в долгосрочной перспективе.
https://elainexpress.ru/
mostbet_ptml
| #
мосбет казино http://www.mostbet780.ru .
Kxyufaita
| #
darknet markets https://darkfoxmarketplace.com/ – darknet market list
mostbet_kg_eser
| #
mosbet https://www.mostbet781.ru .
binance register
| #
Thanks for sharing. I read many of your blog posts, cool, your blog is very good.
Blade2343
| #
Видел прогнозы, что Финансы https://at-agency.ru будет главным трендом. Возражения?
OmegaStriker
| #
Вебинар о финансах: https://prostzaim.ru
Howardrat
| #
Kraken darknet market – Кракен вход через тор, Даркнет вход
FNDaviditabe
| #
dark market 2025 https://firstdarkmarket.com/ – dark web drug marketplace
SteelZ1
| #
Технические нюансы https://kmv-lombard.ru финансов.
BryanCar
| #
казино онлайн играть
WilliamIRrutty
| #
dark web market list https://idarknetmarket.com/ – darknet markets links
Guardian9690
| #
Лучшие материалы по финансам: https://metiz-krovly.ru
Thomasseato
| #
Мы поможем оформить регистрацию по месту пребывания без очередей и бюрократии: сколько стоит временная регистрация в спб на 1 год для граждан рф
Kxyufaita
| #
darknet market list https://darkfoxmarketplace.com/ – darknet markets links
SteelHunter
| #
Топовый ресурс по теме финансов: https://kreditchelny.ru
GhostX1
| #
Предлагаю подискутировать о финансах: https://ber-kar.ru.
Warden5606
| #
https://vp-ipoteka.ru Рекомендую качественный ресурс по финансам.
FNDaviditabe
| #
dark web drug marketplace https://firstdarkmarket.com/ – bitcoin dark web
WilliamIRrutty
| #
dark web market list https://kingdommarketdarknet.com/ – darknet markets 2025
Iron_Overlord
| #
Особенности реализации финансов: https://buhgalterskaya-pomosh.ru
Kxyufaita
| #
tor drug market https://cannahomemarket.link/ – dark websites
lasuzFus
| #
Все о новом фильме говорят, но если вы его не хотите смотреть, тогда на сайте fankino.ru прочтите краткое описание, чтобы в курсе быть. На сайте найдете много нового и интересного для себя. Мы собрали для вас смс поздравления, красивые картинки, народные приметы. Ищете адвокатъ ардашевъ тайна персидского обоза сериал с 2019 г? Fankino.ru – портал с понятным и простым интерфейсом. Тут размещаем актуальные ответы и вопросы. Вы узнаете, где можно покупаться на Крещение. Найдете на умение и способность любить тест. Также вас ждут увлекательные головоломки. Уверены, что вам понравится у нас!
Cyber_Reaper
| #
Профессионалы рекомендуют эти ресурсы по финансам: https://metiz-saiding.ru
CyberSpark
| #
Преодоление трудностей при изучении https://finanskusa.ru финансов.
FNDaviditabe
| #
darknet drug links https://firstdarkmarket.com/ – dark web link
WilliamIRrutty
| #
darknet market https://darknetmarketsonion.shop/ – dark market 2025
Byte9796
| #
Полезные сервисы для работы с финансами: https://kpk-ugra.ru
Storm639
| #
Как работает механизм финансов? https://ygroup-sochi.ru
Kxyufaita
| #
darknet marketplace https://cannahomemarket.link/ – darknet market
Sazrnay
| #
Мы готовы предложить дипломы любой профессии по доступным тарифам. Основные преимущества заказа документов в нашем сервисе
Вы покупаете диплом в надежной и проверенной временем компании. Такое решение позволит вам сэкономить не только много средств, но и время.
На этом плюсы не заканчиваются, их куда больше:
• Документы печатаются на настоящих бланках с мокрыми печатями и подписями;
• Дипломы всех ВУЗов и ССУЗов России;
• Цена значительно ниже той, которую пришлось бы заплатить за обучение в университете;
• Доставка в любые регионы Российской Федерации.
Заказать диплом о высшем образовании– http://infinityaccess.org/read-blog/385_kupit-attestat-8-klass.html/ – infinityaccess.org/read-blog/385_kupit-attestat-8-klass.html
Nomad9891
| #
Всем привет! Может кто в курсе, где посмотреть информацию о финансах? https://pbmotsenka.ru
Eanrxao
| #
Для быстрого продвижения по карьере требуется наличие официального диплома института. Выгодно приобрести диплом ВУЗа у надежной организации: diplomh-40.ru/kupit-diplom-v-ekaterinburge-15/
Byte8651
| #
Видеоуроки по финансам: https://lombard-goldenstone.ru.
FNDaviditabe
| #
dark web market https://firstdarkmarket.com/ – dark web markets
Voyager6277
| #
Как реализовали финансов в компании: https://winfinsochi.ru
WilliamIRrutty
| #
dark web market urls https://darknetmarketsonion.shop/ – darknet markets url
Kxyufaita
| #
darkmarket 2025 https://cannahomemarket.link/ – darknet market
SilverCode
| #
Подкаст о финансах: https://financegroup24.ru
Red160
| #
Авторитетное мнение о финансах: https://lombard2x2.ru
Davidcal
| #
https://ruspress24.ru/
BluePioneer
| #
Примеры успеха https://vsekredity-tmn.ru в финансах.
Bryanonefs
| #
Some are medicines that help people when doctors prescribe.
can you get lisinopril
A touchstone of international pharmacy standards.
FNDaviditabe
| #
dark web market https://darkmarketdarkfox.com/ – darkmarket list
Silver_Overlord
| #
Предлагаю подискутировать о финансах: https://yard-capital.ru
WilliamIRrutty
| #
dark market 2025 https://asap-market-onion.com/ – darkmarket
StormCode
| #
Как выбрать лучшее решение для финансов: https://samtrader.ru
Kxyufaita
| #
dark web markets https://cannahome-darkmarket-online.com/ – tor drug market
Davidcal
| #
Северо-Запад РФ
StormV4
| #
Как сэкономить на финансах: https://kredit-ivanovo.ru
JeffreyVet
| #
Bukmacher oraz kasyno internetowe Mostbet to jeden z najdłużej działających serwisów hazardowych w sieci z ofertą dla Polaków, gdyż został założony jeszcze w 2009 roku. Platforma może pochwalić się posiadaniem aktywnej licencji od Curacao, która gwarantuje bezpieczeństwo. Gracze będą mogli spotkać tutaj ponad 3000 zróżnicowanych gier hazardowych, które zostały podzielone na automaty, gry stołowe oraz gry na żywo. Nie zabraknie też interesującego dodatku w formie różnorodnych gier natychmiastowych, z czego najbardziej wyróżniają się gry natychmiastowe. Platforma Mostbet Casino udostępnia jednocześnie wiele zróżnicowanych tytułów od najbardziej popularnych dostawców oprogramowania, wśród których znajdują się takie uznane marki jak na przykład Play’n GO, Pragmatic Play, Evolution, Microgaming czy NetEnt. Do tego dochodzą różnorodne bonusy, promocje czy turnieje.
Light_Slayer
| #
По данным аналитиков, лучший источник о финансах – https://antey-anapa.ru
Miguelbok
| #
казино
FNDaviditabe
| #
darknet drugs https://firstdarkmarket.com/ – darknet markets onion
WilliamIRrutty
| #
darkmarket list https://darknetmarketsonion.shop/ – tor drug market
Spark7634
| #
Новые подходы в финансах: https://balykovgroup.ru
Uazroer
| #
Привет!
Для многих людей, купить диплом университета – это острая потребность, удачный шанс получить хорошую работу. Но для кого-то – это понятное желание не терять множество времени на учебу в университете. Что бы ни толкнуло вас на это решение, наша фирма готова помочь. Оперативно, профессионально и недорого сделаем диплом любого ВУЗа и любого года выпуска на подлинных бланках с реальными подписями и печатями.
Основная причина, почему люди покупают документ, – желание занять хорошую работу. Предположим, знания позволяют человеку устроиться на работу, однако документального подтверждения квалификации нет. В том случае если работодателю важно наличие “корочки”, риск потерять место работы достаточно высокий.
Приобрести документ университета можно в нашей компании в столице. Мы оказываем услуги по производству и продаже документов об окончании любых университетов России. Вы получите необходимый диплом по любой специальности, любого года выпуска, включая документы образца СССР. Гарантируем, что в случае проверки документа работодателями, никаких подозрений не возникнет.
Ситуаций, которые вынуждают приобрести диплом очень много. Кому-то срочно нужна работа, а значит, нужно произвести хорошее впечатление на руководителя при собеседовании. Некоторые планируют попасть в престижную компанию, чтобы повысить собственный статус и в будущем начать свое дело. Чтобы не терять время, а сразу начать удачную карьеру, применяя врожденные таланты и приобретенные навыки, можно приобрести диплом прямо в онлайне. Вы сможете быть полезным в социуме, обретете денежную стабильность в кратчайший срок- купить аттестат
Kxyufaita
| #
darkmarket url https://cyberdarkmarkets.com/ – dark web market urls
Sazrswd
| #
Купить документ ВУЗа можно в нашей компании. Мы оказываем услуги по продаже документов об окончании любых университетов России. Вы получите необходимый диплом по любым специальностям, любого года выпуска, в том числе документы образца СССР. Гарантируем, что при проверке документов работодателями, никаких подозрений не возникнет. click-boutique.ru/forum/?PAGE_NAME=profile_view&UID=48595
LightMaster
| #
Простой https://ufalex.ru язык о финансах.
TimothyTop
| #
Казино 7k — это современная платформа для азартных развлечений с разнообразным выбором игр. Здесь можно найти популяризированные видеослоты, настольные игры и игры в реальном времени с мастерскими дилерами. Интерфейс интуитивно понятный и легкий в использовании, что делает игру удобной и неповторимой. На 7k740.casino предусмотрены классные бонусы для гемблеров. В целом, казино предоставляет отличный опыт для каждого игрока.
Striker7398
| #
Новые https://bisstrahovanie.ru подходы в финансах.
Matthewciz
| #
Онлайн-Казино Вулкан Платинум — это новейшая платформа для онлайн-гемблинга с большим выбором игр. Здесь можно найти лучшие игровые автоматы, настольные игры и игры с живыми дилерами с квалифицированными дилерами. Дизайн пространный и простой в навигации, что делает игру комфортной и неповторимой. На shint4.ru предусмотрены бонусные бонусы для гемблеров. В целом, казино предоставляет неповторимый опыт для каждого игрока.
Jerrymop
| #
cocaine esports gay
TimothyCob
| #
cocaine esports heroin
PhantomFang
| #
Малоизвестные фишки https://flagman-kpk.ru финасов.
Sazrsxi
| #
купить диплом электрогазосварщика
FNDaviditabe
| #
darkmarket https://onion-dark-market.shop/ – bitcoin dark web
OmegaV4
| #
Заинтересовался финансами. Решение здесь: https://credit-pushkino.ru
mostbet_edOr
| #
mostber https://www.agility.forum24.ru/?1-0-0-00000756-000-0-0-1742360323 .
1win_fyPt
| #
официальный сайт 1win https://agility.forum24.ru/?1-0-0-00000755-000-0-0-1742359870/ .
WilliamIRrutty
| #
darknet websites https://asap-market-onion.com/ – darknet site
SilverOverlord
| #
Где смотреть актуальные данные о финансах? https://uksojuz.ru
1win_pbEn
| #
descărca 1win https://www.1win5002.ru .
Ralphtriny
| #
Odwiedzilem strone ??? liczac na, ze przetestuje dobra strone, ale to po prostu porazka! Interfejs starodawny, menu niewygodne, a rozgrywka ciagle sie zawieszaja. Zbyt duzo banerow, czas ladowania jest zbyt dlugi, a pomoc nie reaguje na prosby. Informacje na stronie sa chaotyczne i bezuzyteczne. Wydaje sie, ze strona zostala porzucona. Polecam przeznaczyc czas na rzeczywiscie wiarygodne strony!
Kxyufaita
| #
darkmarkets https://cyberdarkmarkets.com/ – darknet drug links
LightCode
| #
https://lombard-garant-ug.ru Чек-лист для работы с финасами.
Byte5909
| #
Давайте обсудим о финансах: https://car-leazing.ru
JefferyEvilm
| #
rolex for women
https://systemcheck-wiki.de/index.php?title=Benutzer:MonteCanales07
JeffreyVet
| #
Bukmacher oraz kasyno internetowe Mostbet to jeden z najdłużej działających serwisów hazardowych w sieci z ofertą dla Polaków, gdyż został założony jeszcze w 2009 roku. Platforma może pochwalić się posiadaniem aktywnej licencji od Curacao, która gwarantuje bezpieczeństwo. Gracze będą mogli spotkać tutaj ponad 3000 zróżnicowanych gier hazardowych, które zostały podzielone na automaty, gry stołowe oraz gry na żywo. Nie zabraknie też interesującego dodatku w formie różnorodnych gier natychmiastowych, z czego najbardziej wyróżniają się gry natychmiastowe. Platforma Mostbet Casino udostępnia jednocześnie wiele zróżnicowanych tytułów od najbardziej popularnych dostawców oprogramowania, wśród których znajdują się takie uznane marki jak na przykład Play’n GO, Pragmatic Play, Evolution, Microgaming czy NetEnt. Do tego dochodzą różnorodne bonusy, promocje czy turnieje.
Bryanonefs
| #
They consistently exceed global healthcare expectations.
cytotec over the
A true champion for patients around the world.
Dragon_Master
| #
Нужны https://avtozalogkazan.ru реальные кейсы по финансам.
FNDaviditabe
| #
darknet markets onion https://firstdarkmarket.com/ – darkmarket
Light_Overlord
| #
Что нового в финансах? https://rieltorovich.ru.
WilliamIRrutty
| #
dark web drug marketplace https://darknetmarketsonion.shop/ – darknet drugs
Kxyufaita
| #
darknet drugs https://cannahomemarket.link/ – darkmarket
WillieViemy
| #
https://xnudes.app/
Phantom593
| #
https://dh068.ru Как сэкономить на финансах.
Red142
| #
Коллеги, поделитесь опытом по финансам! https://dengicapital.ru
1win_eaPt
| #
1вин вход с компьютера http://www.agility.forum24.ru/?1-0-0-00000755-000-0-0-1742359870 .
mostbet_otOr
| #
mostbet kg mostbet kg .
Marlinzef
| #
whores esports whores
ThomasFek
| #
heroin esports porn
Jamesjenda
| #
юрист по уголовным делам
1win_jfEn
| #
1win молдова 1win молдова .
FNDaviditabe
| #
darknet links https://mydarkmarketlink.com/ – best darknet markets
WolfV7
| #
Рекомендую проверенный ресурс по финансам: https://antikvar63.ru
WilliamIRrutty
| #
dark market 2025 https://kingdomdarkmarketonline.com/ – darkmarket 2025
StephenEpige
| #
перейдите на этот сайт
оборудование для переработки пластика
Silver647
| #
Как сэкономить на финансах: https://stolichniy-coop.ru
Kxyufaita
| #
dark web market links https://kingdomdarknetdrugstore.com/ – darknet market links
BarryNip
| #
Миру могут угрожать вспышки неизвестных ранее болезней https://x.com/Fariz418740/status/1902696858473910276
SilverX9
| #
Подробный разбор финасов: https://dohodpk.ru
JefferyEvilm
| #
duplicate rolex
https://telegra.ph/Rolex-herrenuhr-gold-03-20
RickyNax
| #
купить права категории б
FNDaviditabe
| #
dark markets https://kingdom-marketplace.com/ – darkmarket url
LeroyRaign
| #
https://midler.ru/portfolio/
Blue_Hacker
| #
Оптимизация процессов через финансы: https://rbgarant.ru.
Miguelbok
| #
pin up
WilliamIRrutty
| #
darknet drug store https://idarknetmarket.com/ – dark web market urls
CoreyLal
| #
https://t.me/top_mobile_casino_2025/3 Остановитесь на нашем рейтинге мобильных казино онлайн — здесь только проверенные и надежные онлайн-платформы, которые уже завоевали доверие тысяч пользователей. Мы отобрали для вас ТОП-6 казино, предлагающих оптимальные условия для игры, щедрые бонусные программы и моментальные выплаты. Каждая платформа дарит бесплатные вращения за регистрацию, чтобы вы могли начать играть без риска и лишних вложений!
Thunder86
| #
Где смотреть https://saint-expert.ru статистику о финасах?
JefferyEvilm
| #
replicas rolex
https://telegra.ph/Rolex-sea-dweller-03-20
Richardaluby
| #
gay esports terrorist attack
CesarDam
| #
Free steam accounts http://t.me/s/freesteamaccountc
DriverBaile
| #
Сотрудник коммунальной службы подсказал медицинскую информацию. Организация похорон прошла без проблем. Спасибо за профессионализм.
AlphaX7
| #
Оптимизация процессов через финансы: https://vf-partner.ru
Reaper6618
| #
Исследование https://financgarant.ru финасов.
Bryanonefs
| #
Their private consultation rooms are a great addition.
how long do gabapentin take to work
A true asset to our neighborhood.
JefferyEvilm
| #
rolex kopie
https://e-webwiki.co.uk/index.php/Rolex_Yacht_Master_40_Rose_Gold
Dragon833
| #
Как https://quickzaim24.ru преуспеть в финансах?
NightZ5
| #
Преодоление трудностей https://finmoli.ru при изучении финасов.
RobertFal
| #
rolex oyster perpetual date blue
https://cannabis-cultivation.wiki/index.php?title=Rolex_69I
RobertFal
| #
rolex two tone women’s watch
https://orphelins-apocalypse.synology.me/mediawiki/index.php?title=Rolex_9X
BlueV1
| #
Финасы для компаний: https://aviscar.ru.
Steel_Nomad
| #
Где смотреть актуальные данные о финансах? https://skpknarod.ru
NewRetroCasino_ppel
| #
Зеркало Ретро Казино http://www.newretromirror.ru .
Voyager9386
| #
Мифы и правда о финасах: https://magazin583.ru
Fang6234
| #
https://primfermer25.ru Какие платформы использовать для финансов?
Robbiepew
| #
Easy piano sheet music free piano
RobertFal
| #
rolex tiffany blue 36mm price
https://telegra.ph/Datejust-white-03-20
DennisZen
| #
Klavier lernen noten kostenlos noten fur klavier zum ausdrucken
JefferyEvilm
| #
rolex daytona ice blue
https://morphomics.science/wiki/Rolex_Submariner_Date_126610lv
RobertFal
| #
how.much are rolex watches
https://championsleage.review/wiki/Rolex_66E
ThomasSlupt
| #
Noten von klavier noten vom klavier
nvblfurn
| #
https://rossahar.ru/rynok/pgs/igru_na_dvoih__ogon_i_voda.html играть в карты косынка c компьютером бесплатно и без регистрации
RobertFal
| #
rolex watch silver and blue
https://openbouffalo.org/index.php/User:Aretha53T2017
WolfVoyager
| #
https://cnbd-kredit-pod-zalog.ru Сравнительный анализ финасов.
RobertFal
| #
replica rolex watches for sale
https://reparatur.it/index.php?title=Benutzer:Kellye8981
Fang4987
| #
https://kpkd.ru Интерактивный курс по финансам.
maslyanie transformatori kypit_svKt
| #
масляные трансформаторы купить масляные трансформаторы купить .
VortexX1
| #
Конкретные шаги для работы с финасами: https://avtolombard02.ru
RobertFal
| #
https://indigenouspedia.com/index.php?title=Rolex_19Z
soglasovanie pereplanirovki kvartiri_xdMn
| #
перепланировка комнаты http://www.soglasovanie-pereplanirovki-kvartiry14.ru .
programmi 1s _gzKi
| #
программы 1с программы 1с .
Brandonsof
| #
накрутка лайков в Тик Ток бесплатно онлайн
Herbertdow
| #
find Kreativstorm
Harolddax
| #
Learn More Here
Kreativstorm Reviews
StormSpark
| #
Новые подходы в финансах: https://ums-rostov.ru
private jet charter flights
| #
Kudos, I value it!
https://niadd.com/article/1280000.html
1win_iyKi
| #
1вин бет официальный сайт 1вин бет официальный сайт .
Herbertdow
| #
here
Kreativstorm Training Program
1win_pdMi
| #
1 вин 1 вин .
Kxyufaita
| #
darkmarket url https://cannahomemarket.link/ – darknet site
RobertFal
| #
womens rolex sizes
https://www.ventura.wiki/index.php/Rolex_38H
Sazrrsz
| #
Приобретение подходящего диплома через надежную фирму дарит массу достоинств для покупателя. Данное решение дает возможность сберечь как продолжительное время, так и серьезные финансовые средства. Впрочем, только на этом выгоды не ограничиваются, преимуществ значительно больше.Мы можем предложить дипломы любой профессии. Дипломы производят на оригинальных бланках государственного образца. Доступная стоимость в сравнении с серьезными тратами на обучение и проживание. Заказ диплома об образовании из российского института станет выгодным шагом.
Быстро заказать диплом о высшем образовании: diploml-174.ru/diplom-med-kolledzha-kupit-2/
NightMaster
| #
С чего начать финасов? https://kpk-farvater.ru
mostbet_omei
| #
скачать mostbet http://www.ongame.forum24.ru/?1-18-0-00001219-000-0-0-1742360461 .
Robbiepew
| #
Piano sheet music free piano
MarkNOlaf
| #
onion dark website https://dark-web-versus.com – darknet site
Edwardwed
| #
Хотите списать долги? центр банкротство в казани законное освобождение от кредитных обязательств. Работаем с физлицами и бизнесом. Бесплатная консультация!
Thunder_Code
| #
https://antikvariatekb.ru Реальные примеры применения финансов.
StephenPhema
| #
Site-ul https://betsysrealfood.com este o resursa cu diverse articole in limba romana, acoperind subiecte precum sanatatea, nutri?ia, moda ?i stilul de via?a. Titlurile articolelor includ teme precum „?tiri de sanatate”, „?tiri pentru bebelu?i”, „?tiri de Medicina Alternativa” ?i „?tiri de via?a”. Site-ul este destinat publicului vorbitor de romana, interesat de un stil de via?a sanatos ?i re?ete de casa.
PhilipDycle
| #
https://football-today.uk/
NightX2
| #
Как сэкономить на финасах: https://lombardkazna.ru
FNDaviditabe
| #
darknet market list https://firstdarkmarket.com/ – darknet marketplace
RobertFal
| #
rolex for sale near me
https://telegra.ph/Womens-green-rolex-03-20
WilliamIRrutty
| #
darknet markets 2025 https://darknetmarketsonion.shop/ – dark web market links
Spark3723
| #
Креативное решение к финансам: https://urfirma-biko.ru
Kxyufaita
| #
dark web markets https://cannahomemarket.link/ – darknet markets 2025
profis
| #
Быстрая продажа и покупка https://profis.com.ua/ размещайте объявления о продаже товаров, поиске услуг, аренде недвижимости и работе. Простой интерфейс, удобный поиск, бесплатные публикации!
Night_Warden
| #
https://matkap35.ru Свежие тренды в финасах.
eurshallsautobodyensub
| #
Transform your car into a spectacular creation with Eurshall’s Auto Body’s outstanding tuning programs. Our experienced team excels at upgrading each vehicle to fulfill its full potential . Whether you’re aiming for agility or chic, our customized solutions ensure holistic results. Visit https://eurshallsautobody.com/ today to witness the enhancement and drive a vehicle that truly embodies your character .
RobertFal
| #
cheapest womens rolex
https://wiki.lanvollon.info/index.php/Rolex_78u
MarkNOlaf
| #
dark web market urls https://cannahomedarknetdrugstore.com/ – darkmarkets
1win_wrKi
| #
1win букмекер http://belbeer.borda.ru/?1-6-0-00001555-000-0-0-1742473542/ .
RedX2
| #
Давайте обсудим о финансах: https://sport-pay.ru
1win_yjMi
| #
1win com https://taksafonchik.borda.ru/?1-14-0-00002041-000-0-0/ .
nedodjorse
| #
Koch Market гарантирует индивидуальный подход к каждому клиенту. Специализируемся на услугах дооснащение, полировка, тюнинг, детейлинг, оклейка. Обеспечиваем высокое качество работы. Ответим на вопросы и расскажем детальнее о действующих акциях. Ищете установка задней камеры? Koch-market.ru – здесь публикуем полезные статьи для автолюбителей. Цель Koch Market – увеличить доходность и обороты вашего бизнеса, а также сократить издержки. Оставьте на портале заявку, и мы в ближайшее время вам перезвоним. Задачи обсудим, запланируем работы и найдем лучшее решение.
mostbet_flei
| #
мостбет войти http://www.ongame.forum24.ru/?1-18-0-00001219-000-0-0-1742360461 .
FNDaviditabe
| #
darknet market https://mydarkmarketlink.com/ – darknet drug market
Sentinel7135
| #
Будущее финасов: https://premium196.ru
RobertFal
| #
rolex clone watches for men
https://flynonrev.com/airlines/index.php/Rolex_25N
WilliamIRrutty
| #
darknet site https://kingdommarketdarknet.com/ – bitcoin dark web
Night_Spark
| #
Какие изменения в финансах? https://mai-vc.ru.
Kxyufaita
| #
best darknet markets https://cannahome-darkmarket-online.com/ – dark web drug marketplace
RobertFal
| #
https://chrisophia.wiki/index.php/Rolex_77g
huxojeMaype
| #
Мечтаете встретить свою любовь? Тогда вам непременно стоит посетить Лилибум! Сайт предлагает простую регистрацию, открывает двери к новым знакомствам и дружбе. Стараемся безопасность вам и анонимность обеспечить. Вас ждет доброжелательная атмосфера. Ищете замужние желают познакомиться? Lilibum.ru – сайт, где можете прекрасно провести время. Порадует красивый и удобный интерфейс. Модераторы профили пользователей и фотографии внимательно проверяют. С Лилибум вы обязательно найдете свою половинку. Знакомьтесь, общайтесь и создавайте новые отношения!
PhilipDycle
| #
football
RobertFal
| #
rolex super clones for sale
https://semantische-richtlijnen.wiki/wiki/Rolex_62w
Steel167
| #
Как монетизировать https://kpk-proekt.ru финасов в бизнесе?
MarkNOlaf
| #
dark web market links https://kingdom-darkmarketplace.com – dark web marketplaces
DarkZ4
| #
Подкаст о финансах: https://atmosfera-fin.ru
FNDaviditabe
| #
dark market list https://mydarkmarketlink.com/ – darknet markets
WilliamIRrutty
| #
dark market onion https://asap-market-onion.com/ – darknet drugs
RobertFal
| #
https://telegra.ph/Gifts-for-roller-coaster-enthusiasts-03-20
Blue532
| #
Изучите https://roks22alt.ru этот источник о финасах.
AlphaWalker
| #
Основы финансов для новичков: https://msk-dt.ru
Kxyufaita
| #
dark web marketplaces https://kingdomdarknetdrugstore.com/ – darknet markets links
RobertFal
| #
rolex day date 41mm price
https://msgwiki.msgeducation.com/index.php/Rolex_92D
MarkNOlaf
| #
darkmarket link https://dark-web-versus.com – darkmarket url
Protocol7822
| #
Экспертный уровень в https://almaz-yar.ru финасах.
yinemgut
| #
Stakecasino.tech предоставляет полезную информацию. Stake Casino вас честными условиями и большим разнообразием азартных игр порадует. Сайт в применении довольно прост. Регистрация меньше минуты занимает. Проблем с выводом средств нет. Ищете stake casino промо? Stakecasino.tech – тут вы узнаете, как в Стейк казино начать играть. Платформа интуитивно понятный интерфейс предлагает, это делает для опытных игроков и новичков ее доступной. Стейк предоставляет специальные бонусы и акции. Консультанты службы поддержки доброжелательные и оперативно отвечают.
RobertFal
| #
rolex fake
https://wikistars5.com/index.php?title=Rolex_26D
Omega_Sentinel
| #
Где тренироваться финансам? https://furgongrad.ru
FNDaviditabe
| #
dark market list https://darkmarketdarkfox.com/ – dark market onion
RobertFal
| #
2023 prestige mega box
https://mediawiki.uedemersv.de/index.php?title=Benutzer:GloriaPeeples
WilliamIRrutty
| #
dark web market https://darknetmarketsonion.shop/ – darknet websites
DarkZ4
| #
Давайте обсудим о финасах: https://avtozaim1.ru
Sazrgpb
| #
Задумался а действительно можно купить диплом государственного образца в Москве, и был удивлен, все реально и главное официально!
Сначала серфил в сети и искал такие темы как: купить диплом мгюа, купить диплом о среднем образовании в абакане, купить диплом парикмахера цена, купить диплом перманентный макияж, купить диплом поступить в вуз, получил базовую информацию.
Остановился в итоге на материале proffdiplomik.com/sochi
Kxyufaita
| #
dark market list https://cannahome-darkmarket-online.com/ – dark web market
ShadowV3
| #
Подробный разбор финансов: https://buhgalter-vladikavkaz.ru
RobertFal
| #
datejust price
https://telegra.ph/Replica-vendors1-03-20
RobertFal
| #
https://disgaeawiki.info/index.php/User:Tangela0373
RobertFal
| #
aaa rolex
http://projectingpower.org:80/w/index.php/Rolex_78k
MarkNOlaf
| #
darknet market lists https://drdarkfoxmarket.com – dark web link
Blue771
| #
Что https://lombard-masterok.ru нового в финасах?
RedSlayer
| #
Вебинар о финансах: https://biznes-plani.ru.
FNDaviditabe
| #
darknet market list https://darkmarketdarkfox.com/ – dark market
WilliamIRrutty
| #
darkmarket link https://idarknetmarket.com/ – darkmarket url
RobertFal
| #
rolex datejust green dial price
http://wiki.wild-sau.com/index.php?title=Rolex_69e
Kxyufaita
| #
best darknet markets https://kingdomdarknetdrugstore.com/ – darknet markets onion
Byte6317
| #
Столкнулся с проблемой финасами. https://senexa.ru Решение здесь.
IronV2
| #
Мемы про финансы: https://autostrh.ru
RobertFal
| #
ss clone for sale
https://flynonrev.com/airlines/index.php/User:LavondaGilbreath
RobertFal
| #
nice rolex
https://www.openlongevityproject.org/index.php?title=User:UtaStapley9690
MarkNOlaf
| #
darknet market list https://drdarkfoxmarket.com – dark market
Jefferysmeap
| #
В Красной Поляне с радостью представляем вам сайт для знакомств с девушками 18+. Здесь вы сможете найти множество интересных предложений для общения и веселья, открывая для себя мир новых знакомств и ярких эмоций вместе с привлекательными дамами https://night24-krasnaya-polyana.com/
Iron_Hunter
| #
Читал прогнозы, что Финансы будет главным трендом. Соглашусь? https://finanexpert.ru
CyberWarden
| #
Нестандартный подход https://domoteka-omsk.ru к финасам.
FNDaviditabe
| #
darkmarket 2025 https://onion-dark-market.shop/ – dark web market links
RobertFal
| #
why is rolex watches so expensive
https://telegra.ph/Cheapest-place-to-buy-rolex-watches-03-20
RobertFal
| #
datejust rolex ebay
https://menuaipedia.menuai.id/index.php/Rolex_62z
fajoridox
| #
Copy General предоставляет широкий спектр качественных печатных услуг. Используем современное оборудование и технологии. Готовы ваши требования обсудить. К нам проявляют доверие множество частных, а также корпоративных клиентов по всему миру. Свяжитесь с нами прямо сегодня и убедитесь в качестве нашей работы. Ищете фотопечать? Copygeneral.ru – здесь представлены отзывы заказчиков, ознакомьтесь с ними. Типография работает без выходных и дает гарантию на отменное качество печати. Оплату можно произвести удобным способом. Стараемся ваши ожидания превзойти.
WilliamIRrutty
| #
dark market https://idarknetmarket.com/ – darkmarket list
RobertFal
| #
moissanite dealer near me
https://waselplatform.org/blog/index.php?entryid=208
nvblfurn
| #
https://veimmuseum.ru/news/pgs/ogon_i_voda_na_dvoih_onlayn_besplatno__uvlekatelnaya_igra_dlya_vseh.html какие игры можно поиграть дома на четверых
Kxyufaita
| #
onion dark website https://cannahomemarket.link/ – dark web market links
RobertFal
| #
1 1 watches
https://wiki.excito.org/index.php?title=User:AlfieRoldan56
ShadowZ7
| #
Коллеги, поделитесь опытом по финансам! https://abfns.ru
Light106
| #
https://finkorp.ru Коллеги, поделитесь опытом по финасам!
RobertFal
| #
milgauss for sale
https://telegra.ph/41mm-oyster-perpetual-03-20
Jefferysmeap
| #
Друзья, у нас для вас отличная новость! В Красной Поляне открылся сайт для досуга с девушками 18+. Теперь у вас есть возможность пообщаться с ними и провести время так, как вы мечтали. Откройте новый мир и наслаждайтесь уникальными моментами встреч https://night24-krasnaya-polyana.com/
RobertFal
| #
rolex deep sea-dweller 44mm
https://telegra.ph/Look-a-like-rolex1-03-20
MarkNOlaf
| #
darkmarket link https://cannahomedarknetdrugstore.com/ – darknet drug links
RobertFal
| #
superclone rolex
https://cannapedia.icu/index.php/Usuario:BryantFollansbee
RobertFal
| #
datejust 36mm vs 41mm
https://reparatur.it/index.php?title=Rolex_10S
Eagle_Sentinel
| #
Продвинутые техники финансов: https://mrparks.ru
RobertFal
| #
oyster pearl worth
https://telegra.ph/Ebay-oyster-perpetual-rolex-03-20
FNDaviditabe
| #
dark web market links https://firstdarkmarket.com/ – darknet drugs
GhostV6
| #
Оптимизация процессов через финасов: https://ffr24.ru.
RobertFal
| #
rolex submariner white face
https://mediawiki.novastega.me/index.php/Rolex_16b
WilliamIRrutty
| #
darknet market links https://darknetmarketsonion.shop/ – dark web sites
RobertFal
| #
best place to buy rolex in miami
https://shikhadabas.com/2025/03/18/rolex-77z/
RobertFal
| #
rolex blue daytona
https://Ragnafrost.wiki/index.php/Rolex_16v
Kxyufaita
| #
bitcoin dark web https://cannahomemarket.link/ – onion dark website
Hunter5739
| #
Особенности реализации https://ctroidom74.ru финансов.
Cyber_Fang
| #
Лично мне помог этот ресурс по финасам: https://mts-rus.ru
MarkNOlaf
| #
darkmarket https://kingdomdarkmarketplace.com – dark web sites
Bobbyinhex
| #
размещение рекламных конструкций светящиеся буквы
RobertFal
| #
submariner rolex band
https://reparatur.it/index.php?title=Rolex_46c
DonaldPap
| #
оформление вывесок изготовление рекламных конструкций спб
FNDaviditabe
| #
darknet drugs https://mydarkmarketlink.com/ – darknet websites
Ghost_Slayer
| #
Что не https://prof25.ru пишут в учебниках с финансами.
Matthewvet
| #
медицинские справки санкт петербург https://kupit-spravku-spb.ru
WilliamIRrutty
| #
dark web market links https://asap-market-onion.com/ – dark market onion
CyberZ1
| #
Свежие тренды в финансах: https://crowdautoinvest.ru
RobertFal
| #
https://Ragnafrost.wiki/index.php/Utilizador:CorinneDespeissi
WolfV6
| #
Ошибки новичков в финасах: https://family-stav.ru
RobertFal
| #
best online rolex dealer
https://andyfreund.de/wiki/index.php?title=Rolex_49z
Cazrnku
| #
Привет!
Без присутствия диплома очень нелегко было продвинуться вверх по карьерной лестнице. В настоящее время документ не дает гарантий, что удастся получить привлекательную работу. Намного более важное значение имеют навыки и знания специалиста и его опыт. Именно из-за этого решение о заказе диплома можно считать мудрым и рациональным. Приобрести диплом о высшем образовании tricityfriends.com/read-blog/30370
Charlesviank
| #
https://promocode.game/cs2/csfail/
Dnrtfwj
| #
Где заказать диплом по нужной специальности?
Мы изготавливаем дипломы любых профессий по приятным тарифам. Мы готовы предложить документы техникумов, которые находятся на территории всей РФ. Можно приобрести диплом от любого заведения, за любой год, в том числе документы старого образца. Дипломы и аттестаты печатаются на “правильной” бумаге самого высокого качества. Это позволяет делать государственные дипломы, которые не отличить от оригинала. Документы будут заверены всеми обязательными печатями и штампами. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Важно, чтобы документы были доступными для большого количества граждан. diplomdoc.ru/kupit-attestat-shkoli-7
RobertFal
| #
rolex 1992
https://telegra.ph/Gold-mens-rolex-datejust1-03-20
RobertFal
| #
superclone watch
https://telegra.ph/How-much-are-rolex-watches-worth-03-20
Kxyufaita
| #
darknet market list https://kingdomdarknetdrugstore.com/ – darkmarket 2025
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы https://17metrov.ru/
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://1domovoy.ru/
RobertFal
| #
https://haloleagues.com/rolex-38h/
1win_eeei
| #
wan win wan win .
MarkNOlaf
| #
dark market url https://cannahomedarknetdrugstore.com/ – darknet links
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://1eve1.ru/
Thunder_Nomad
| #
Что лучше выбрать в финасах? https://prostozaimi.ru
PhantomWarden
| #
Как https://alfaexpert32.ru сделать финансы?
Dark326
| #
Столкнулся с проблемой https://kredtaxi.ru финансами. Решение здесь.
finuvTus
| #
Сомово – компания, которая большой ассортимент мебельной продукции предоставляет. Мы рады общению с нашими дорогими клиентами. Стараемся вам исключительно лучшее дать. Одна из главных целей – формирование длительных отношений. Somovo-Ищете купить прихожие в Москве? mebel.ru/catalog/prikhozhii – здесь можно купить прихожие недорого с оперативной доставкой. При возникновении вопросов, позвоните нам по телефону, который на ресурсе указан. С радостью поможем с выбором и детали заказа подскажем. В интернет-магазине «Сомово» найдется товар, который необходим именно вам!
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы https://2052285.ru/
ForrestVag
| #
кракен маркет – кракен вход, kraken ссылка
Michaelmiz
| #
Автоматизация ПДС также включает поддержку реестра НПФ, что позволяет фондам оперативно обновлять данные об участниках и их накоплениях. Система веб-доступа к счетам НПФ обеспечивает участникам возможность отслеживать состояние своих счетов в режиме реального времени, что повышает доверие к фонду. Бэк офис НПФ
RobertFal
| #
blue rolex submariner
https://syria-wiki.org/index.php?title=Rolex_61S
elektrokarnizi dlya shtor_hxot
| #
электрокарнизы цена электрокарнизы цена .
hMustafaVep
| #
Заказала пионы подруге, она сказала, что это лучший подарок!
Пионы томск
FNDaviditabe
| #
best darknet markets https://firstdarkmarket.com/ – dark market 2025
RobertFal
| #
https://telegra.ph/Rolex-lady-datejust-on-wrist-03-20
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://2204000.ru/
WilliamIRrutty
| #
darknet websites https://asap-market-onion.com/ – darknet drug links
mostbet_vsmn
| #
служба поддержки мостбет номер телефона http://www.mostbet782.ru .
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы https://22058.ru/
nvblfurn
| #
https://suvredut.ru/img/pgs/igru_bez_interneta_besplatno__luchshie_variantu_dlya_vashego_ustroystva.html цветные линии шарики 98 играть онлайн
EagleX7
| #
Преимущества разных подходов к финасам: https://algcons.ru
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://2231109.ru/
RobertFal
| #
mens knock off watches
https://kreosite.com/index.php/User:MillardQ22
Kxyufaita
| #
darknet market list https://darkfoxmarketplace.com/ – bitcoin dark web
Overlord4586
| #
Что лучше выбрать в финансах? https://afr54-ipoteka.ru
Thunder595
| #
Нестандартный подход к финансам: https://myasnaya-zastava.ru
ThomasVep
| #
Букет подарил море эмоций, спасибо за красоту!
букет цветов томск
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://2k-metalwork.ru/
1win_gtei
| #
1вин вход 1вин вход .
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://337700.ru/
Sazrbzd
| #
Добрый день!
Приобрести диплом университета по выгодной цене можно, обращаясь к проверенной специализированной компании. Приобрести диплом о высшем образовании: diplom-top.ru/kupit-diplom-farmatsevta-10/
MarkNOlaf
| #
darknet sites https://kingdomdarkmarketplace.com – dark markets 2025
Trefjmg
| #
Приобрести документ о получении высшего образования вы имеете возможность в нашем сервисе. diplomd-magazinp.ru/kupit-diplom-moskva-4-2
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://33panorama.ru/
Pioneer4647
| #
Продвинутые техники финасов: https://troika-zaim.ru
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://3855154.ru/
RobertFal
| #
https://wiki.radiomurf.com/wiki/User:Arden15M27731851
GhostX7
| #
Конкретные шаги для работы с финансами: https://finconstrall.ru
FNDaviditabe
| #
darknet markets onion address https://onion-dark-market.shop/ – dark websites
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы https://3909102.ru/
EagleCode
| #
Экспертный уровень в финансах: https://kredit19.ru
WilliamIRrutty
| #
dark web sites https://idarknetmarket.com/ – darknet marketplace
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы https://417-017.ru/
mostbet_symn
| #
mostbet игры https://mostbet782.ru/ .
Kxyufaita
| #
darknet market list https://cannahome-darkmarket-online.com/ – dark websites
1win_kg_xhKn
| #
win 1 https://1win823.ru/ .
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные статьи и советы — https://4masterss.ru/
Spark3371
| #
Эксклюзивное интервью с экспертом https://arifmetika-mkk.ru по финасам.
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://565606.ru/
1win_nfei
| #
1wln http://1win822.ru/ .
Russellovank
| #
Casino Nomini http://nomini-casino.ru
RedV4
| #
Как войти https://fif124.ru в тему финансов?
1win_kg_ltKn
| #
1win.kg 1win.kg .
Markcen
| #
Новостройки или вторичка? Полезные рекомендации — https://740-789.ru/
MarkNOlaf
| #
dark web market links https://dark-web-versus.com – dark market
Red480
| #
Как сэкономить на финансах: https://aksay-rich.ru
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://74evakuator.ru/
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы https://7832206.ru/
Blade2195
| #
Будущее финасов: https://el-1.ru
nvblfurn
| #
http://svidetel.su/jsibox/article/index.php?ogon_i_voda_onlayn___uvlekatelnoe_prikluchenie_dlya_vseh.html пасьянс гольф играть бесплатно и без регистрации
FNDaviditabe
| #
darknet markets onion https://darkmarketdarkfox.com/ – darknet markets url
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://7lestnic.ru/
WilliamIRrutty
| #
dark web sites https://darknetmarketsonion.shop/ – dark web drug marketplace
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://9002781.ru/
mostbet_ahmn
| #
мосбет https://www.mostbet782.ru .
1win_kg_jyKn
| #
1 цшт http://1win823.ru .
Blade5911
| #
Конкретные шаги для работы с финансами: https://carcash-vdk.ru
cevinuUnins
| #
МВПОЛ услуги по выполнению промышленных полов предоставляет. Наша команда состоит из грамотных сотрудников, которые применяют новейшее оборудование, а также инструменты. Все работы совершаются в сжатые сроки. Ищете полимерные полы цена м2? Mvpol.ru – тут есть возможность посмотреть условия сотрудничества и стоимость услуг. На портале наши объекты представлены. Мы тщательно контролируем то, чтобы любой наш проект был закончен в соответствии с высочайшими стандартами. Мы знаем, что надо заказчику. У вас останутся приятные впечатления от сотрудничества с компанией МВПОЛ.
Kxyufaita
| #
dark web market urls https://darkfoxmarketplace.com/ – dark web markets
Markcen
| #
Покупка квартиры без рисков? Полезные статьи и советы — https://999555.ru/
GhostBlade
| #
Мне самому помог этот ресурс по финансам: https://ifa-invest.ru
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://a1realtyspb.ru/
Blue_Overlord
| #
ТОП-10 ресурсов по финасам: https://pravoved59.ru.
MarkNOlaf
| #
dark web marketplaces https://cannahomedarknetdrugstore.com/ – darknet markets 2025
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы https://abmodul.ru/
Markcen
| #
Новостройки или вторичка? Узнайте подробности https://abrikos-krsk.ru/
Blue314
| #
Всем привет! Может кто в https://pawntrade.ru курсе, где найти информацию о финансах?
Jariorpfx
| #
Где купить диплом специалиста?
Мы предлагаем выгодно и быстро приобрести диплом, который выполняется на бланке ГОЗНАКа и заверен мокрыми печатями, штампами, подписями официальных лиц. Данный документ способен пройти лубую проверку, даже с применением профессионального оборудования. Достигайте цели быстро с нашим сервисом.
Купить диплом о высшем образовании diplomt-v-chelyabinske.ru/kupit-diplom-motorista/
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://azovcity-kuban.ru/
FNDaviditabe
| #
onion dark website https://firstdarkmarket.com/ – darknet markets onion address
Iariorbvr
| #
Купить документ университета вы имеете возможность у нас в Москве. usawc.org/submit-mailbag-item-for-magazine
Lazrfgb
| #
Приобрести диплом об образовании!
Мы можем предложить дипломы любой профессии по приятным тарифам— diplom-kaluga.ru/kupit-diplom-v-novosibirske-20/
StormVoyager
| #
Видеоуроки по финасам: https://spkk-kazna.ru.
WilliamIRrutty
| #
darknet market https://asap-market-onion.com/ – darkmarkets
Night777
| #
Как применять финансы в бизнесе? https://kpkveles.ru
Markcen
| #
Как выбрать надёжного застройщика? Читайте проверенные советы https://b-import.ru/
soyt moskva_xbPa
| #
провести соут москва провести соут москва .
soyt moskva_fpkn
| #
заказать соут в москве заказать соут в москве .
Michaelsat
| #
hop over to these guys
galaxyswapper
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы https://baget-mos.ru/
Kxyufaita
| #
dark market link https://cannahome-darkmarket-online.com/ – darknet markets onion
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://baltimkom.ru/
BarryNip
| #
Миру могут угрожать вспышки неизвестных ранее болезней https://x.com/Fariz418740/status/1902696858473910276
ShadowGuardian
| #
Интерактивный курс по финансам: https://b-act.ru
Alpha_Pioneer
| #
Технические нюансы финасов: https://lombard-altyn.ru
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы https://baltstirol39.ru/
MarkNOlaf
| #
dark web drug marketplace https://dark-web-versus.com – darknet markets
Jasonquinc
| #
Доставка в раннее время прошла без проблем.
букет невесты
lacohiflops
| #
СанОбработка – компания, которая для выполнения чистоты и безопасности вашего дома предоставляет комплексные решения. Гарантируем индивидуальный подход к каждому клиенту. С удовольствием вам поможем. Вы останетесь довольны приемлемыми ценами и качеством обслуживания. Ищете дезинфекция вентиляции Тюмень? Sanobrabotkarf.ru – здесь представлены отзывы наших клиентов. Персонал вежливый и компетентный. Специалисты детально объяснят все этапы работы и ответят на вопросы. Они используют сертифицированные препараты и современные технологии. Защитите собственное жилище уже сегодня!
Cazrsku
| #
Мы изготавливаем дипломы любых профессий по приятным тарифам. Купить диплом бурильщика — kyc-diplom.com/diplomy-po-professii/kupit-diplom-burilshhika.html
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы https://basban31.ru/
LightV5
| #
Вебинар о https://doveriecredit.ru финансах.
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://bastion-bezheck.ru/
FNDaviditabe
| #
darkmarkets https://darkmarketdarkfox.com/ – dark market url
WilliamIRrutty
| #
darknet drug market https://kingdomdarkmarketonline.com/ – darkmarket url
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://batstroimat24.ru/
DarkNomad
| #
https://berirubli.ru Мифы и правда о финасах.
Red955
| #
Чек-лист https://salon-novosel.ru для работы с финансами.
RobinIrono
| #
Хотите перекусить вкусно? купить Хлеб для сэндвичей питу, шаверму, донер, гирос и тортилью. Ароматная свежая выпечка, мясо на гриле и фирменные соусы. Отличный выбор для перекуса, обеда или вечеринки.
Kxyufaita
| #
dark market https://cannahome-darkmarket-online.com/ – dark web market links
Pioneer5691
| #
Что вы думаете https://oboi42.ru этот ресурс о финансах?
WolfOverlord
| #
Эволюция развития финасов: https://zaim-vkarmane.ru.
MarkNOlaf
| #
best darknet markets https://cannahomedarknetdrugstore.com/ – dark markets 2025
NightV2
| #
Шутки про финансы: https://sidimdoma35.ru
FNDaviditabe
| #
dark market 2025 https://darkmarketdarkfox.com/ – dark web market
WilliamIRrutty
| #
dark web market urls https://darknetmarketsonion.shop/ – dark web market list
Hunter2785
| #
Согласно исследованиям, лучший https://ciframix.ru источник о финасах.
SteelWarden
| #
Смешные случаи в финансах: https://investassistant.ru
nvblfurn
| #
http://tankfront.ru/axis/pages/?igru_bez_ustanovki_besplatno__naslazghdaytes_razvlecheniyami_onlayn.html занятие с детьми 5 6 лет
Kxyufaita
| #
darkmarket url https://kingdomdarknetdrugstore.com/ – dark market link
DragonNomad
| #
Новые https://lombard-club.ru подходы в финансах.
Fang5293
| #
5 главных ошибок при https://sakhtools.ru работе с финасами.
MarkNOlaf
| #
darknet site https://kingdomdarkmarketplace.com – dark market 2025
tipografiya
| #
заказать журналы в типографии заказать книгу в типографии
IronWalker
| #
Полезные сервисы https://mkk-pfk.ru для работы с финансами.
FNDaviditabe
| #
darknet links https://mydarkmarketlink.com/ – dark market
WilliamIRrutty
| #
dark market 2025 https://asap-market-onion.com/ – darknet markets
Shadow373
| #
Что не пишут в учебниках с финасами: https://partner31.ru
ShadowSpark
| #
Ответы на частые вопросы о финансах: https://prliz.ru
Wallacesoolo
| #
типография печать табличка пвх
Lonniezem
| #
Эта клиника предлагает комфортное и безопасное лечение зубов детям, подробности тут https://500px.com/p/tschelkunchik?view=photos
Kxyufaita
| #
dark market https://cannahomemarket.link/ – darknet drugs
Lazrlad
| #
Где заказать диплом специалиста?
Приобрести диплом университета по доступной стоимости вы сможете, обращаясь к проверенной специализированной компании.: peoplediplom.ru
AlphaZ4
| #
https://kpksssr.ru Список инструментов по финасам.
Eagle_Warden
| #
Успешные кейсы применения https://kpk-lider.ru финансов.
WilliamDix
| #
Мы изготавливаем дипломы любой профессии по доступным ценам. Дипломы изготавливаются на настоящих бланках Заказать диплом института diplomidlarf.ru
MarkNOlaf
| #
darknet markets links https://kingdomdarkmarketplace.com – dark markets 2025
1win_pwPa
| #
1win казино https://www.1win810.ru .
Sazrymn
| #
Заказать документ ВУЗа вы сможете в нашем сервисе. Мы оказываем услуги по продаже документов об окончании любых университетов России. Вы сможете получить диплом по любой специальности, любого года выпуска, в том числе документы Советского Союза. Даем гарантию, что при проверке документов работодателем, никаких подозрений не появится. bookkarocallgirls.copiny.com/question/details/id/1069080
mostbet_agpr
| #
служба поддержки мостбет номер телефона http://www.mostbet783.ru .
Jamesflums
| #
Розыгрыш сертификатов OZON и WB – участвуй бесплатно!
Хотите получить подарочный сертификат Ozon или подарочный сертификат Wildberries совершенно бесплатно? Тогда подписывайтесь на наш Telegram-канал и участвуйте в розыгрышах!
В нашем канале регулярно разыгрываются сертификаты Ozon и Wildberries номиналом от 500 до 5000 рублей. Получить их можно легко:
1.Подписывайтесь на канал
2.Участвуйте в розыгрышах, выбирая билеты
3. Ожидайте результаты – победители определяются случайным образом!
Подарочные карты Ozon и Wildberries – это удобный способ оплатить любые покупки на популярных маркетплейсах. Участвуйте в розыгрышах и выигрывайте!
Присоединяйтесь прямо сейчас: Розыгрыш сертификатов OZON WB
Ghost139
| #
Как сэкономить на финансах: https://carzaym24.ru
1win_edPa
| #
игра 1вин игра 1вин .
mostbet_irpr
| #
mostbet casino http://mostbet783.ru .
nvblfurn
| #
https://www.colonoscopy.ru/PHPMailer/?igroutka__besplatnue_onlayn_strelyalki_dlya_malchikov___zahvatuvaushie_igrovue_priklucheniya_zghdut_vas.html огонь и вода 4 в хрустальном храме
mostbet_amSt
| #
мосбет mostbet784.ru .
FNDaviditabe
| #
tor drug market https://onion-dark-market.shop/ – dark web drug marketplace
Code6243
| #
Профессионалы рекомендуют эти ресурсы по финасам: https://protos39.ru
WilliamIRrutty
| #
dark market url https://asap-market-onion.com/ – darkmarket list
mostbet_bfSt
| #
mostbet kg скачать https://mostbet784.ru .
Fang5453
| #
Мифы и правда о финансах: https://kristelin.ru
1win_hsPa
| #
1win kg 1win kg .
mostbet_jupr
| #
мос бет https://mostbet783.ru/ .
Kxyufaita
| #
darknet markets 2025 https://darkfoxmarketplace.com/ – tor drug market
Dianna
| #
I always spent mmy half an hour to read this
blog’s content everyday along with a mug of coffee. https://Distribuidorafp.com.co/bdmbet-casino-evaluations-engage-in-new-games-through-your-account-and-secure-up-to-hours-of-fun-terms-and-more/
Voyager4790
| #
Что вы думаете этот https://alpari-cabinet.ru ресурс о финансах?
DarkX4
| #
Как думаете, где https://finanspg.ru лучше всего понять финасов?
DavidViedy
| #
B-52-ואדים הנהן בשלווה, מסכים להצעה “לטפל” – וגם פולינקה-הוא קרץ לנסטה – אני ואדיה! – הוא הציג את עצמו, לחץ ידיים עם החבר ‘ ה תזיין את התוכנית כמובן, הנה חמישה כוכבים, אתה לא יכול להגיד כלום. ומה אנחנו עושים במצבים האלה? נכון-רווהם. מתלונן, מתחנן. – לא, read this article
mostbet_trSt
| #
поддержка мостбет https://www.mostbet784.ru .
MarkNOlaf
| #
darknet markets url https://kingdomdarkmarketplace.com – darknet markets 2025
Liwuhssoomo
| #
На сайте http://ammely.ru представлены интересные, увлекательные статьи, которые точно понравятся женской аудитории. Опубликован контент о моде, красоте, здоровом образе жизни. Также имеются и полезные рецепты, которые идеально подойдут на каждый день либо на праздник, если внезапно нагрянули гости. Есть увлекательные рекомендации на тему дома, дизайна. Вам будет интересно узнать и о звездах, о том, как лучше и веселей провести праздник.
LesterZog
| #
https://plusgestio.com/pag/1xbet_today_promo_code_bonus_up_to_130.html
Dragon_Overlord
| #
Как https://elecland.ru реализовать финансы?
Lonniezem
| #
Для бизнеса: ускоренное получение лицензии за 14 дней с полной гарантией, подробнее по ссылке http://prsync.com/ilya-filippov/
FNDaviditabe
| #
darknet market list https://firstdarkmarket.com/ – dark market 2025
Dark874
| #
Оптимизация процессов через финасов: https://max-lombard.ru
WilliamIRrutty
| #
dark web marketplaces https://asap-market-onion.com/ – darknet markets links
WanuxnelalI
| #
На сайте https://xn—-8sbkf3cqel4a6c.xn--p1ai/ рассчитайте стоимость такой услуги, как ДТФ-печать на сувенирной продукции, текстиле. Все изображения отличаются безупречным качеством и цветопередачей, высокой детализацией. При необходимости вы сможете получить профессиональную консультацию. Главное направление деятельности компании – это DTF-печать на повседневной, корпоративной одежде, включая худи, футболки, толстовки. Все заказы выполняются с применением инновационных материалов, качественного оборудования.
Byte1878
| #
Какие изменения в финансах? https://pic-capital.ru
wplay
| #
whoah this weblog is wonderful i really like studying your posts.
Keep up the great work! You already know, lots of persons are searching around for this information, you can aid them greatly.
Nord-West
| #
Сияние вашего авто начинается здесь! Nord-West детейлинг работает с автомобилями премиум-класса уже более 10 лет. Оклейка кузова, полировка и керамика высшего качества доступны в Великом Новгороде. Доверьте свой автомобиль профессионалам.
Kxyufaita
| #
darknet markets onion https://cannahomemarket.link/ – darknet websites
Silver992
| #
Практическое применение https://mihbat.ru финансов.
Protocol2961
| #
Мой опыт показывает, https://zolotaya-pora.ru что по финасам лучше всего читать здесь.
wplay
| #
I’d like to thank you for the efforts you have put in penning this site.
I am hoping to view the same high-grade blog posts
from you in the future as well. In fact, your creative writing
abilities has encouraged me to get my own blog now 😉
MarkNOlaf
| #
darknet markets url https://kingdomdarkmarketplace.com – darknet links
jewejDyeds
| #
Calypso – комфортный для аренды яхт сервис. Наш подход ориентирован на удовлетворение персональных потребностей каждого клиента. Мы готовы обеспечить высочайший уровень сервиса и безопасность. Ваше путешествие на яхте станет невероятным! Ищете аренда катера Сочи? Sochi.calypso.ooo – тут можете с условиями заказа ознакомиться. На сайте вы найдете самые выгодные предложения. Поможем вам яхту, выбрать, которая вашим пожеланиям и требованиям соответствует. Присоединяйтесь к Calypso. Позвольте нам ваши мечты о безупречном отдыхе в реальность превратить!
NightX1
| #
С чего начать финансов? https://sopranocapital.ru
FNDaviditabe
| #
darknet marketplace https://darkmarketdarkfox.com/ – onion dark website
Look At This
| #
Hello there! Do you use Twitter? I’d like to follow you if that would be ok.
I’m absolutely enjoying your blog and look forward to new posts.
OmegaSentinel
| #
5 главных https://dengi-ontime.ru ошибок при работе с финасами.
WilliamIRrutty
| #
onion dark website https://asap-market-onion.com/ – darkmarket link
Vortex158
| #
Как сделать финансы? https://zaymlombard-top.ru
Kxyufaita
| #
dark web markets https://darkfoxmarketplace.com/ – darknet marketplace
Thunder601
| #
ТОП-10 ресурсов по финансам: https://777uspeh.ru
Alpha681
| #
Обнаружил, что по финасам лучше всего смотреть здесь: https://lombard-chronograph.ru
امنیت سایت
| #
Howdy! I could have sworn I’ve been to this web site before
but after going through many of the articles
I realized it’s new to me. Nonetheless, I’m definitely
delighted I found it and I’ll be bookmarking it and checking back regularly!
CyberProtocol
| #
Видел прогнозы, что Финансы будет главным трендом. Возражения? https://reteilbank.ru
MarkNOlaf
| #
dark web link https://cannahomedarknetdrugstore.com/ – darknet markets url
FNDaviditabe
| #
dark markets https://onion-dark-market.shop/ – dark web market list
WilliamIRrutty
| #
darknet marketplace https://kingdommarketdarknet.com/ – darknet websites
DarkOverlord
| #
https://a-leksmebel.ru Что нового в финасах?
аркада официальный сайт
| #
Nice post. I learn something new and challenging on blogs I stumbleupon everyday.
It’s always useful to read articles from other authors and practice
something from their web sites.
Omega_Master
| #
Как не сдаться при изучении https://insidersgroup.ru финансов.
DragonV1
| #
Эксперты https://rtl56.ru рекомендуют эти ресурсы по финансам.
Kxyufaita
| #
darkmarket list https://cannahome-darkmarket-online.com/ – darknet markets onion
DukeselSlorb
| #
На сайте http://kinomanhdc.online представлены фильмы в огромном многообразии. Перед вами как популярное кино, которое понравится всем без исключения, так и ожидаемые новинки, которые точно никого не оставят равнодушным. Есть возможность подобрать решение по определенной тематике: про вампиров, космос, подростков, девушек, любовь и многое другое. Здесь вы найдете и сериалы, киноленты за прошедшие годы. На сайте также представлены и мультфильмы для детей разного возраста, семейного просмотра. Все это вы сможете смотреть прямо сейчас и в режиме реального времени.
BlueX7
| #
Где смотреть https://vap-invest.ru статистику о финасах?
MarkNOlaf
| #
darkmarket link https://kingdom-darkmarketplace.com – darknet sites
constructive-m.ru
| #
Jetton Casino – это ваш билет в мир настоящего азарта и захватывающих побед. В нашем казино представлены только лучшие игровые автоматы, покер, рулетка и многое другое. Регистрируйтесь и начинайте выигрывать уже сегодня.
https://constructive-m.ru/ – это не просто казино, а целая вселенная для любителей азартных игр. Получайте кэшбэк, участвуйте в акциях и выигрывайте эксклюзивные призы. Гарантируем безопасные транзакции, а также удобные способы пополнения счета и вывода выигрышей.
Огромный ассортимент развлечений для всех любителей азартных игр.
Ежедневные подарки, кэшбэк и выгодные предложения для новых и постоянных игроков.
Быстрое пополнение счета и мгновенные выплаты без скрытых комиссий.
Эксклюзивные турниры с высокими призовыми фондами и шанс сорвать джекпот.
Начните игру в Jetton Casino и сделайте первый шаг к крупным победам.
игры с высоким RTP
| #
Добро пожаловать в Водка Казино — место,
где азарт встречается с невероятными бонусами и возможностями для выигрыша.
В нашем казино каждый игрок
найдет большой выбор слотов, карточных игр и рулетки, независимо от того, начинающий вы или опытный игрок.
Здесь вас ждет не только удовольствие от игры, но и множество выгодных предложений.
Все игры в Водка Казино
имеют высокий RTP, что увеличивает шансы на успешную игру.
Для вас мы подготовили интерактивные слоты, а также классические настольные игры, которые делают вашу игру более увлекательной и прибыльной.
Почему не начать прямо сейчас?
Водка Казино предоставляет игрокам мгновенную регистрацию и удобные способы пополнения счета, чтобы вы могли сосредоточиться на главном
— выигрыше!
У нас всегда есть для вас интересные бонусы, которые позволят вам начать играть с дополнительным капиталом.
Присоединяйтесь к Водка Казино, чтобы наслаждаться азартными играми с лучшими шансами на победу!
Зарегистрироваться можно
всего за несколько секунд.
Наслаждайтесь щедрыми бонусами сразу после регистрации.
Регулярные турниры и акции для тех, кто хочет увеличить
свои шансы на выигрыш.
Поддержка 24/7 для решения любых вопросов.
Играйте в любимые игры в любое время и в любом месте.
Начните выигрывать прямо сейчас и испытайте
удачу! https://vodka-777-spinwin.autos/
Byte8092
| #
Как сэкономить на финансах: https://credit-help32.ru
Red717
| #
Друзья, поделитесь опытом по финансам! https://hankukwan.ru
deneme bonusu veren siteler
| #
Kudos. Awesome stuff!
FNDaviditabe
| #
dark web market list https://darkmarketdarkfox.com/ – darknet markets links
WilliamIRrutty
| #
darknet markets url https://kingdommarketdarknet.com/ – darknet market
Hunter4783
| #
Повышение эффективности через финасов: https://an-biznespartner.ru
Kxyufaita
| #
dark market list https://cannahome-darkmarket-online.com/ – darkmarket link
DragonX6
| #
https://pts812.ru Вебинар о финансах.
StormOverlord
| #
Столкнулся с https://feniks-invest.ru проблемой финасами. Решение здесь.
VortexZ6
| #
Нюансы работы с https://maxlombard.ru финансами.
LesterZog
| #
https://nytalkradio.net/pag/1xbet_new_registration_promo_code_4.html
MarkNOlaf
| #
darknet site https://kingdom-darkmarketplace.com – dark market link
Aaronfek
| #
Срочная установка пластиковых окон за 3 дня с момента заказа — оставить заявку на замер можно здесь https://gitlab.com/AlexStone725
nvblfurn
| #
https://mytaganrog.com/media/madzghong_igrat_besplatno__zahvatuvaushee_prikluchenie_v_mir_drevney_igru.html игра красный шар 4 часть 1
siracajslusa
| #
К-ЖБИ гарантирует высочайшее качество продукции и строгое соблюдение сроков. Завод обладает гибкими производственными мощностями для выполнения заказов по чертежам заказчика. С удовольствием на все вопросы ответим, позвоните нам по номеру телефона. Ищете лестничные марши и бетонные площадки серии л лм лз и лп? Gbisp.ru – тут есть возможность заявку оставить. Укажите в форме ваш e-mail, имя и контактный номер. Далее нажмите кнопку «Отправить». Выполняем быструю доставку продукции. Обеспечиваем новейший уровень облуживания любому заказчику. К взаимовыгодным партнерским отношениям стремимся!
Aaronfek
| #
Средства индивидуальной защиты для работы в лаборатории – узнайте больше информации https://blacksocially.com/Janescience
FNDaviditabe
| #
darknet drugs https://mydarkmarketlink.com/ – darknet markets onion
Ghost873
| #
Топовый https://vipautosale.ru ресурс по теме финасов.
GeorgeWak
| #
Названы доказательства пребывания на Земле более высоких цивилизаций https://x.com/Fariz418740/status/1903334968513745235
WilliamIRrutty
| #
dark web link https://darknetmarketsonion.shop/ – darknet market lists
Storm586
| #
Что нельзя делать с финансами? https://kitleasing.ru
NightVoyager
| #
Как преуспеть https://rosa-rai.ru в финансах?
Jamesflums
| #
Церковь Валенсии Церковь в Валенсии – это не просто место отправления религиозных обрядов, это духовный центр, объединяющий верующих разных конфессий. Среди многообразия храмов и общин особое место занимают протестантские церкви, предлагающие альтернативный взгляд на христианство.
Kxyufaita
| #
dark web markets https://cannahome-darkmarket-online.com/ – best darknet markets
AlphaZ3
| #
Подкаст о финасах: https://inberi.ru.
Aurora casino онлайн
| #
Aurora Casino приглашает вас в мир высококлассных азартных игр и ярких эмоций. Каждый найдет здесь что-то по душе — от классических слотов до уникальных игр с реальными крупье. Мы также предлагаем различные бонусы и акции, которые сделают каждый момент игры еще более захватывающим.
Что делает https://auroracasino.live/ лучшим выбором для игроков? В Aurora Casino ваша безопасность и защита личных данных — наша первоочередная задача. Мы гарантируем прозрачные условия игры и мгновенные выплаты, чтобы вы могли наслаждаться выигрышами без задержек.
Когда вам лучше начать играть в Aurora Casino? Не откладывайте! Присоединяйтесь к нам и начните выигрывать прямо сейчас. Вот что вас ждет:
Большие бонусы и постоянные акции.
Мы гарантируем вашу безопасность и спокойствие, играя на нашей платформе.
Новинки и эксклюзивные игры.
В Aurora Casino вы всегда найдете яркие эмоции и шансы на большие выигрыши.
Sazrvrx
| #
Мы изготавливаем дипломы любой профессии по разумным ценам. Цена зависит от определенной специальности, года получения и образовательного учреждения. Всегда стараемся поддерживать для покупателей адекватную политику тарифов. Важно, чтобы дипломы были доступными для большого количества наших граждан. купить диплом на фармацевта
StormZ8
| #
Тесты по финансам: https://sokol-dv.ru
PhantomReaper
| #
Новые подходы https://aliansauto29.ru в финасах.
SteelX4
| #
https://arealdecor.ru Будущее финансов.
FNDaviditabe
| #
dark websites https://onion-dark-market.shop/ – dark web marketplaces
WilliamIRrutty
| #
darknet markets links https://kingdommarketdarknet.com/ – darknet market list
Jamesflums
| #
путешествие В мире, где горизонты становятся ближе, а мечты о приключениях — осязаемыми, наше агентство открывает двери в мир безграничных возможностей. Мы – ваш надежный компас в океане туризма, предлагая не просто путешествия, а мастер-туры, сотканные из ваших желаний и нашей экспертизы. Солнечная Турция, роскошные ОАЭ, загадочный Египет – мы подберем идеальное направление, соответствующее вашему вкусу. Оформление виз, организация трансфера, бронирование отелей и авиабилетов – все это мы берем на себя, чтобы вы могли полностью расслабиться и наслаждаться каждым моментом. Мечтаете о морском круизе или отдыхе в живописной Черногории? Хотите исследовать бескрайние просторы России? Мы поможем воплотить в жизнь самые смелые планы. Наша цель – сделать ваше путешествие незабываемым, комфортным и безопасным. Доверьтесь профессионалам, и ваш отпуск станет настоящим шедевром!
Kxyufaita
| #
darknet markets links https://cyberdarkmarkets.com/ – darknet markets onion address
Diplomi_sfKt
| #
Купить диплом любого ВУЗа!
Мы готовы предложить документы институтов, расположенных в любом регионе России.
diplomg-kurerom.ru/gde-kupit-diplom-texnikuma-5
mostbet_poMi
| #
mostbet mostbet .
AlphaX6
| #
Повышение эффективности https://fkarussia.ru через финасов.
EugeneKex
| #
печать листовок буклетов буклет офсетная печать
Henryged
| #
печать 3д наклеек https://pechat-nakleek-spb.ru
mostbet_eyEt
| #
mostbet casino https://taksafonchik.borda.ru/?1-14-0-00002042-000-0-0-1742473173 .
Vortex754
| #
Как вам этот ресурс https://kr-ankb.ru о финансах?
DerekRah
| #
печать логотипа на сувенирной продукции сувениры с печатью на заказ
mostbet_lrMi
| #
mostbest mostbest .
1win_vqoi
| #
wan win fanfiction.borda.ru/?1-3-0-00000125-000-0-0-1742475251 .
MarkNOlaf
| #
onion dark website https://cannahomedarknetdrugstore.com/ – darknet drug store
Mazrcdc
| #
Мы предлагаем дипломы любой профессии по приятным ценам. Стараемся поддерживать для покупателей адекватную ценовую политику. Важно, чтобы документы были доступными для большого количества граждан.
Заказ документа, подтверждающего обучение в университете, – это грамотное решение. Купить диплом любого ВУЗа: diplomt-v-chelyabinske.ru/kuplyu-diplom-o-visshem-obrazovanii-8/
mostbet_hfEt
| #
поддержка мостбет http://taksafonchik.borda.ru/?1-14-0-00002042-000-0-0-1742473173 .
ThunderWarden
| #
Профессионалы рекомендуют эти ресурсы по финансам: https://ivctt.ru
1win_ykoi
| #
1win.pro http://www.fanfiction.borda.ru/?1-3-0-00000125-000-0-0-1742475251 .
Cazraar
| #
Купить диплом о высшем образовании!
Мы можем предложить документы университетов, которые расположены на территории всей РФ. Дипломы и аттестаты выпускаются на бумаге самого высокого качества: opengroup.net/include/pgs/kupit_diplom_o_srednem_obrazovanii_29.html
DarkSlayer
| #
Изучите этот источник о финасах: https://kpkdelo.ru
wawembib
| #
Карамельная Фабрика мармелад, леденцы на палочке и другое предлагает. Каждый день наши карамелье шедевры создают. Купить вкусные и красивые сладости можно в нашем интернет-магазине. При возникновении вопросов, обращайтесь к нам, мы с удовольствием на них ответим. Ищете мастер класс по изготовлению карамели? Karamelfabrika.ru – здесь узнаете, как мы делаем карамель. Не останавливаемся на достигнутом и постоянно расширяем ассортимент. Проводим интересные мастер-классы, которые для детей станут источником радости и дадут возможность взрослым в детство ненадолго вернуться. Создаем для вас карамельное настроение!
mostbet_oiMi
| #
mostbet kg отзывы http://www.eisberg.forum24.ru/?1-0-0-00000327-000-0-0-1742579529 .
FNDaviditabe
| #
darknet markets onion address https://kingdom-marketplace.com/ – dark market
WilliamIRrutty
| #
darknet drug market https://kingdomdarkmarketonline.com/ – darknet drugs
mostbet_fvEt
| #
скачать мостбет официальный сайт http://www.taksafonchik.borda.ru/?1-14-0-00002042-000-0-0-1742473173 .
1win_vuoi
| #
1вин официальный http://fanfiction.borda.ru/?1-3-0-00000125-000-0-0-1742475251 .
Vortex407
| #
Приветствую. Может кто в курсе, где посмотреть информацию о финансах? https://sakhprokat.ru
Mazrftw
| #
Где заказать диплом специалиста?
Полученный диплом с нужными печатями и подписями целиком и полностью отвечает требованиям и стандартам, никто не отличит его от оригинала. Не следует откладывать собственные мечты на потом, реализуйте их с нашей помощью – отправьте быструю заявку на диплом уже сегодня! Диплом о среднем специальном образовании – легко! diplomus-spb.ru/nadezhnij-sposob-kupit-gotovij-attestat-bistro/
LightV1
| #
Практическое применение финасов: https://kpksodrug.ru
Davidzic
| #
Заказать пиццу Жаждете сочного итальянского наслаждения, не покидая Кемерово? Добро пожаловать в мир аппетитной пиццы, где каждый кусочек – это маленький праздник. Наша пицца на тонком, хрустящем итальянском тесте – это симфония вкусов, созданная из свежайших ингредиентов. Живете в Теплом ключе? Мы доставим ваш заказ прямо к вашей двери, быстро и аккуратно. А для тех, кто предпочитает забрать заказ самостоятельно, предусмотрена удобная опция самовывоза. Но это еще не все! Наша пиццерия предлагает не только восхитительную пиццу, но и сытные осетинские пироги, приготовленные по традиционным рецептам. Это идеальный выбор для тех, кто хочет попробовать что-то новое и аутентичное. Ищете идеальное место для празднования дня рождения? Мы дарим подарки именинникам, делая ваш особенный день еще более радостным и запоминающимся. Закажите пиццу прямо сейчас на нашем сайте и насладитесь непревзойденным вкусом!
Kxyufaita
| #
dark web markets https://kingdomdarknetdrugstore.com/ – darknet site
Cazreyf
| #
Заказать диплом любого ВУЗа мы поможем. Купить аттестат в Барнауле – diplomybox.com/kupit-attestat-v-barnaule
Alpha964
| #
Сравнение лучших ресурсов по финансам: https://lomkorona.ru
Volodyanaf
| #
dark market https://darkmarketpremium.com/ – dark market
MarkNOlaf
| #
dark market list https://kingdomdarkmarketplace.com – dark websites
Storm_Pioneer
| #
Как избежать проблем с финасами? https://real-credit24.ru
CyberZ5
| #
Как монетизировать финансы в бизнесе? https://cppnvrsk.ru
уборка после ремонта
| #
Безупречная чистота и комфорт в Вашем Доме:
Экспертные Услуги Клининговой Компании в Санкт-Петербурге городе на Неве
Измучились от нескончаемых забот по
уборке? Позвольте нам попечься о
чистоте вашего родного очага или офиса!
Наша клининговая компания в Санкт-Петербурге предлагает комплексные услуги, которые удовлетворят самые высокие
требования к качеству и сервису.
Доступные цены и прозрачные условия;
Высокое качество обслуживания и педантичное деталям;
Персональный подход к каждому клиенту;
Точное соблюдение сроки выполнения работ.
Не ждите, пока грязь и беспорядок станут катастрофой.
Подарите нам шанс возродить вашему пространству чистоту и порядок!
Узнайте всё о наших услугах и оставьте заявку на сайте
: уборка после ремонта
Toliknaf
| #
dark web market urls https://github.com/abacuslink4jjku/abacuslink – dark web link
FNDaviditabe
| #
dark market onion https://mydarkmarketlink.com/ – dark websites
Vortex709
| #
Полезные сервисы для работы с https://caratlombard.ru финансами.
клининговые компании спб
| #
Безупречная чистота и комфорт в Вашем Доме: Экспертные Услуги Клининговой Компании в Санкт-Петербурге СПб
Измучились от нескончаемых забот по уборке? Дайте возможность нам позаботиться о чистоте вашего дома или офиса! Наша клининговая компания в Санкт-Петербурге предлагает широкий спектр услуги, которые придутся по вкусу самые взыскательные требования к качеству и сервису.
Доступные цены и прозрачные условия;
Высокое качество обслуживания и внимание к деталям;
Персональный подход к каждому клиенту;
Чёткие сроки выполнения работ.
Не ждите, пока грязь и беспорядок станут катастрофой. Дайте нам шанс вернуть вашему пространству чистоту и порядок! Узнайте всё о наших услугах и оставьте заявку на сайте : https://cadpower.iitcsolution.com/bbs/board.php?bo_table=free&wr_id=269341
WilliamIRrutty
| #
dark web link https://kingdommarketdarknet.com/ – darknet websites
Silver_Guardian
| #
Изучите https://time27.ru этот источник о финасах.
Rabyitabe
| #
darknet websites https://github.com/darkwebsitesyhshv/darkwebsites – bitcoin dark web
Kxyufaita
| #
dark web market urls https://cannahome-darkmarket-online.com/ – darknet websites
Jariordme
| #
Где заказать диплом специалиста?
Наша компания предлагает быстро и выгодно купить диплом, который выполняется на оригинальном бланке и заверен мокрыми печатями, водяными знаками, подписями. Данный документ пройдет лубую проверку, даже при помощи профессионального оборудования. Достигайте свои цели быстро и просто с нашими дипломами. Приобрести диплом о высшем образовании! cosmeticsworld.org/read-blog/18084_svidetelstvo-o-brake.html
Xazrndi
| #
Купить диплом ВУЗа!
Мы предлагаем дипломы психологов, юристов, экономистов и других профессий по доступным тарифам. Вы приобретаете диплом через надежную и проверенную временем компанию. : diplom-kaluga.ru/kupit-diplom-mbi-o-visshem-obrazovanii-na-blanke-goznak-3/
GregoryTah
| #
Приобретите качественные купели для бани, или спа-зоны в нашем магазине! Мы предлагаем широкий ассортимент: традиционные модели из лиственницы и кедра, современные чаши с гидромассажем, варианты для небольших помещений https://kupeli-ural.ru/
Vortex125
| #
Мне самому помог этот ресурс по финансам: https://lombardsterling.ru
DonDonfaita
| #
darknet market https://github.com/nexusdarkrtv1u/nexusdark – darknet site
Volodyanaf
| #
darknet sites https://darkode-market.com/ – dark markets
Master9502
| #
https://spkk-optimist.ru Как реализовали финасов в компании.
Pingrutty
| #
dark web link https://github.com/aresonioncq0a7/aresonion – dark web market links
Thunder_Spark
| #
Доступно объяснено о финансах: https://avtozaem-moskva.ru
MarkNOlaf
| #
darkmarket link https://cannahomedarknetdrugstore.com/ – darknet sites
Vortex_Byte
| #
Может кто знает ориентируется в финасах? https://sebito.ru Где искать?
Donaldlaf
| #
dark web market links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet market
FNDaviditabe
| #
dark websites https://kingdom-marketplace.com/ – dark web link
WilliamIRrutty
| #
darkmarket link https://kingdommarketdarknet.com/ – darknet markets
nvblfurn
| #
https://ma.by/docs/pages/?igru_na_73.html мультики про трансформеров для мальчиков 5 лет
Cyber_Voyager
| #
Предлагаю подискутировать о финансах: https://doer-group.ru
هایک ویژن
| #
سلام مقاله خوبی بود خرید هایک ویژن https://sites.google.com/view/hikvisiontehran/
Toliknaf
| #
dark market list https://github.com/abacuslink4jjku/abacuslink – darkmarkets
ThunderV1
| #
Глубокий анализ финансов: https://centrzaymov19.ru
Lonniezem
| #
Эксклюзивные ювелирные изделия с зубами динозавров – подробная информация здесь https://www.anobii.com/en/011a8d0efaca3880ef/profile/activity
Kxyufaita
| #
darknet markets onion address https://kingdomdarknetdrugstore.com/ – best darknet markets
Shadow_Hacker
| #
Секретные методы финасов: https://nfinanse.ru
Mazrvtn
| #
Где купить диплом по нужной специальности?
Мы оказываем услуги по продаже документов об окончании любых университетов России. Документы производят на подлинных бланках. ctivefanconnect.com/read-blog/14517_oficialnyj-attestat.html
Volodyanaf
| #
darknet markets url https://darkode-market.com/ – darknet sites
Rabyitabe
| #
darknet market lists https://github.com/darkwebsitesyhshv/darkwebsites – dark web sites
Pingrutty
| #
darknet market https://github.com/aresonioncq0a7/aresonion – darknet websites
MarkNOlaf
| #
dark web market urls https://cannahomedarknetdrugstore.com/ – darknet markets onion
ShadowNomad
| #
Продвинутые техники финасов: https://lombardych27.ru
dovezabanny
| #
Служба Эвакуации 911 предлагает качественные услуги. Мы оперативно и безопасно любой транспорт перевозим. Владеем новейшим автопарком эвакуаторов. Можете не сомневаться в ответственном подходе к вашей проблеме решению. Ищете Эвакуатор Пенза? Penza-evakuator.ru – сайт, где вы узнаете, какие виды техники мы готовы эвакуировать. Можем предложить наши услуги дешевле, чем у конкурентов. Работаем 24/7 без выходных. Помогаем клиентам порой в самых непростых ситуациях. Доверьте службе спасения 911 вашего авто транспортировку.
DonDonfaita
| #
darknet markets links https://github.com/nexusdarkrtv1u/nexusdark – darknet sites
Shadow_Guardian
| #
Коллеги, поделитесь опытом по финансам! https://tevsretni.ru
Sazrnuq
| #
Привет!
Купить диплом института по выгодной цене вы сможете, обратившись к надежной специализированной фирме. Заказать диплом: good-diplom.ru/kupit-diplom-tula-3/
Lazrxab
| #
Приобрести диплом о высшем образовании!
Мы изготавливаем дипломы любой профессии по приятным ценам— diplom-ryssia.com/kupit-meditsinskij-diplom-bistro-i-bez-lishnix-problem/
Nomad4060
| #
Мемы про финансы: https://ah19.ru
Sazrmfx
| #
Покупка документа о высшем образовании через надежную компанию дарит ряд достоинств для покупателя. Это решение помогает сберечь время и значительные финансовые средства. Однако, преимуществ намного больше.Мы можем предложить дипломы любых профессий. Дипломы производят на настоящих бланках. Доступная цена по сравнению с большими издержками на обучение и проживание. Покупка диплома университета станет выгодным шагом.
Быстро купить диплом о высшем образовании: diplom-ryssia.com/kupit-v-moskve-attestat-za-11-klass-2/
Mazrtca
| #
Мы изготавливаем дипломы любой профессии по выгодным ценам. Стараемся поддерживать для заказчиков адекватную политику тарифов. Для нас очень важно, чтобы документы были доступными для большинства наших граждан.
Покупка документа, который подтверждает окончание университета, – это грамотное решение. Купить диплом университета: diplomius-docs.com/kupit-diplom-visshee-obrazovanie/
FNDaviditabe
| #
tor drug market https://kingdom-marketplace.com/ – darknet markets 2025
Scottlek
| #
https://zarcasinorank.online/
WilliamIRrutty
| #
dark market onion https://asap-market-onion.com/ – dark market 2025
Fang9200
| #
Коллеги, поделитесь опытом по финасам! https://shikfin.com
Donaldlaf
| #
darknet markets url https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – tor drug market
Cazreum
| #
Заказать диплом о высшем образовании поможем. Ваше решение – купить диплом сварщика – diplomybox.com/diplom-svarshchika
Kxyufaita
| #
darknet markets onion address https://kingdomdarknetdrugstore.com/ – darkmarket 2025
Toliknaf
| #
darknet markets onion https://github.com/abacuslink4jjku/abacuslink – darknet site
DarkWarden
| #
Где тренироваться https://vsekredity-rostov.ru финансам?
Volodyanaf
| #
darknet market lists https://aasap-market.com/ – dark market url
RedV8
| #
https://kpkmska.ru Сравнительный анализ финасов.
SilverX5
| #
Мифы и правда о финансах: https://fartzaim.ru
Pingrutty
| #
darknet site https://github.com/aresonioncq0a7/aresonion – darknet markets onion
WesleyZerly
| #
печать на упаковке коробках печать упаковки типография
MarkNOlaf
| #
dark market onion https://drdarkfoxmarket.com – darknet markets 2025
Kennethartes
| #
печать фото на холсте недорого https://holst-pechat-spb.ru
Dustinmon
| #
уф печать на блокноте изготовление и печать блокнотов
Rabyitabe
| #
darkmarket https://github.com/darkwebsitesyhshv/darkwebsites – dark market onion
подскажите хороший хостинг
| #
подскажите хороший хостинг
Red_Code
| #
https://lombard-54.ru Видеоуроки по финасам.
DonDonfaita
| #
best darknet markets https://github.com/nexusdarkrtv1u/nexusdark – darknet market
FNDaviditabe
| #
darknet markets 2025 https://onion-dark-market.shop/ – bitcoin dark web
WilliamIRrutty
| #
dark markets 2025 https://idarknetmarket.com/ – dark web market links
Wolf317
| #
Список инструментов https://grant59.ru по финансам.
SteelStriker
| #
Где учиться финансам? https://aipru.ru
Kxyufaita
| #
darknet markets 2025 https://cannahomemarket.link/ – dark market 2025
SilverX7
| #
Креативное решение к финасам: https://centr-lombard.ru
Donaldlaf
| #
dark web market links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet drug store
Volodyanaf
| #
darknet market https://darkode-market.com/ – darkmarket
Pingrutty
| #
darkmarket 2025 https://github.com/aresonioncq0a7/aresonion – darknet markets onion address
Toliknaf
| #
dark markets https://github.com/abacuslink4jjku/abacuslink – darknet market list
Alpha_Overlord
| #
Кейс: https://centr-fbi.ru Финансы.
MarkNOlaf
| #
dark web market list https://cannahomedarknetdrugstore.com/ – dark web market list
Silver_Hacker
| #
Интерактивный курс по финансам: https://fond-investor.ru
ThunderWalker
| #
Примеры https://iphone78.ru успеха в финасах.
Rabyitabe
| #
darknet market links https://github.com/darkwebsitesyhshv/darkwebsites – dark market list
FNDaviditabe
| #
darknet links https://mydarkmarketlink.com/ – dark web market links
WilliamIRrutty
| #
darknet market links https://idarknetmarket.com/ – tor drug market
StephenRow
| #
До конца марта эти 4 знака Зодиака обретут личное счастье https://x.com/Fariz418740/status/1903525881395507337
рейтинг хостинг
| #
рейтинг хостинг
DonDonfaita
| #
darknet marketplace https://github.com/nexusdarkrtv1u/nexusdark – dark market
Omega282
| #
Как вам этот ресурс о финасах? https://avgust74.ru
Fang9837
| #
Пример https://zalognt.ru из практики: Финансы.
Kxyufaita
| #
dark markets 2025 https://cannahome-darkmarket-online.com/ – dark web markets
nvblfurn
| #
https://cofe.ru/auth/articles/igru_na_dvoih_onlayn_besplatno__vremya_dlya_ves_logo_dosuga.html игра сокровища пиратов скачать бесплатно на телефон без регистрации на русском
Eagle550
| #
Изучите этот материал о финансах: https://lombard-gold.ru
Hefozafem
| #
На сайте https://xn--90a1af.xn—-8sbkf3cqel4a6c.xn--p1ai/ закажите обратный звонок для того, чтобы больше узнать о таких полезных, нужных услугах, как: ДТФ-принты на заказ, ДТФ-печать на ткани, ДТФ-печать на спецодежде либо коже. Здесь же вы ознакомитесь с расценками на перечисленные работы. В любом случае они остаются на доступном уровне. Услуги подойдут самым разным сферам бизнеса, а также бизнесменам, которые только пытаются раскрутиться. ДТФ-печать подойдет артистам, актерам, а также дизайнерам.
Volodyanaf
| #
darkmarket 2025 https://darkfoxdarkweb.com/ – darknet drug market
Pingrutty
| #
darknet markets 2025 https://github.com/aresonioncq0a7/aresonion – dark market onion
Lazrvab
| #
Диплом ВУЗа РФ!
Без ВУЗа очень трудно было продвинуться по карьерной лестнице. Именно из-за этого решение о заказе диплома стоит считать целесообразным. Выгодно купить диплом института cristian.5nx.ru/viewtopic.php?f=49&t=331
Donaldlaf
| #
darknet market lists https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet drug links
StormHacker
| #
Мне самому помог этот https://ultra-kredit.ru ресурс по финасам.
spacenet one
| #
Aw, this was an extremely good post. Taking a few minutes and
actual effort to generate a great article… but what can I say… I hesitate a whole lot and never manage to get
anything done.
MarkNOlaf
| #
onion dark website https://kingdomdarkmarketplace.com – darknet drugs
gamagWaw
| #
Lolzteam Market предоставляет широкий ассортимент аккаунтов по привлекательным ценам. Здесь можете найти то, что вам необходимо. Каждый аккаунт подвергается тщательной проверке на подлинность. https://lzt.market/ – портал для тех, кто с гарантией безопасности хочет купить аккаунты. На платформе представлено множество категорий, включая Steam, Riot Games, Supercell, World of Tanks, Minecraft, Social Club, War Thunder, Instagram, Warface и др. Специалисты поддержки работают 24/7. Чтобы вам помочь, они все возможное сделают.
Toliknaf
| #
darkmarket link https://github.com/abacuslink4jjku/abacuslink – darknet drug links
Hacker2163
| #
Базовые принципы финансов для новичков: https://itccoop.ru
Storm_Guardian
| #
Преимущества разных https://audit-versia.ru подходов к финансам.
FNDaviditabe
| #
dark websites https://onion-dark-market.shop/ – dark market
WilliamIRrutty
| #
darknet site https://kingdommarketdarknet.com/ – darknet markets url
Eagle19
| #
Как работает механизм финасов? https://kredit-24h.ru.
http://leadwith.org/bbs/board.php?bo_table=free&wr_id=87071
| #
http://leadwith.org/bbs/board.php?bo_table=free&wr_id=87071
A Brief History of Elemental Analysis » Artemis Analytical
обзор хостинг-провайдеров
| #
обзор хостинг-провайдеров
Rabyitabe
| #
dark web markets https://github.com/darkwebsitesyhshv/darkwebsites – dark web market
Kxyufaita
| #
darkmarket link dark market list
DonDonfaita
| #
darknet markets 2025 https://github.com/nexusdarkrtv1u/nexusdark – dark market url
Iron_Striker
| #
https://zolotoy-pk.ru Предлагаю подискутировать о финансах.
OmegaFang
| #
https://l-lombard.ru Как монетизировать финасов в бизнесе?
1win_skol
| #
1win бк 1win824.ru .
Pingrutty
| #
darknet marketplace https://github.com/aresonioncq0a7/aresonion – darknet sites
Billymep
| #
https://imgtube.ru/
1win_jqKr
| #
1 win официальный сайт вход https://www.1win825.ru .
Guardian2016
| #
Как https://alfastrah54.ru применять финансы в бизнесе?
MarkNOlaf
| #
darknet market links darkmarket link
VictorSef
| #
Считаете, что качественный недорогой ремонт квартир в Москве — это миф? Мы предлагаем цены на 20% ниже рынка за счет прямых поставок материалов. Экономьте без ущерба для дизайна: паркетная доска «под старину» или мраморная крошка в ванной — всё реально.
1win_bjol
| #
1wi https://1win824.ru .
Volodyanaf
| #
dark market 2025 dark websites
1win_bkKr
| #
1вин бет официальный сайт 1вин бет официальный сайт .
AlbertHef
| #
vegasy casino рабочее зеркало
1win_yrOa
| #
one win one win .
Donaldlaf
| #
dark market link https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark websites
GufapeHoold
| #
Сайт https://joinwork.ru/ представляет собой фриланс биржу, на которой вы сможете найти специалистов по дизайну, разработке, на тему SEO. Также получится воспользоваться услугами из сферы рекламы, социальных сетей, аудио, видео. Есть возможность воспользоваться безопасной сделкой, которая очень простая. Предложения от вас увидит огромное количество посетителей, а потому они захотят воспользоваться ими. Вы сможете оформить свой профиль так, будто это магазин. А в блоге вы найдете много полезных статей.
Master1833
| #
https://igk-invest.ru Инфографика по финасам.
WilliamIRrutty
| #
darknet drug links darknet market list
1win_nsOa
| #
1win казино http://www.1win811.ru .
Toliknaf
| #
tor drug market https://github.com/abacuslink4jjku/abacuslink – darkmarkets
DarkByte
| #
https://kpkrahinsky.ru Подкаст о финансах.
1win_amol
| #
1win официальный сайт скачать http://1win824.ru/ .
1win_poKr
| #
1вин партнерка http://www.1win825.ru .
Ghost_Overlord
| #
Мой опыт показывает, что по финансам лучше всего смотреть здесь: https://biznes-zrelost.ru
FNDaviditabe
| #
dark markets darknet market
Kxyufaita
| #
darkmarket link darkmarket link
Ghost_Slayer
| #
Как избежать проблем с финасами? https://ptbleasing.ru
AlbertHef
| #
https://www.vegasy-casino.net/
Rabyitabe
| #
dark web sites https://github.com/darkwebsitesyhshv/darkwebsites – dark web markets
Pingrutty
| #
darknet marketplace https://github.com/aresonioncq0a7/aresonion – dark market link
1win_dcOa
| #
1 win 1win811.ru .
DonDonfaita
| #
dark market list https://github.com/nexusdarkrtv1u/nexusdark – dark market 2025
Iron562
| #
Лучшие инструменты для работы с финансами: https://atg71.ru
MarkNOlaf
| #
darknet market links darknet websites
Reaper5130
| #
Какие технологии использовать для финасов? https://svobodnieludi.ru
Omega_Nomad
| #
Новые подходы в https://lombard-afina.ru финансах.
Sazrvgs
| #
купить диплом в исгз
hMustafaVep
| #
Какой аромат! Пионы — это всегда восторг!
Доставка цветов Томск
Volodyanaf
| #
dark web marketplaces darknet market links
WilliamIRrutty
| #
darknet markets onion dark market list
Donaldlaf
| #
darknet drug store https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – onion dark website
WolfPioneer
| #
Реальные примеры https://abb-kronos.ru применения финасов.
nvblfurn
| #
https://www.amnis.ru/ajax/articles/?igru_risovat_onlayn__tvorchestvo_na_konchikah_palcev.html игры 24 года на пк скачать
StormV2
| #
Нюансы работы с финансами: https://primarifinance.ru
Toliknaf
| #
dark web drug marketplace https://github.com/abacuslink4jjku/abacuslink – best darknet markets
FNDaviditabe
| #
darknet markets darkmarket 2025
Kxyufaita
| #
darknet market links dark web link
Natasesunice
| #
Ищете оригинальный мерч от любимых российских и зарубежных звезд, фестивалей и шоу? Посетите https://showstaff.ru/ – мы с 2002 года радуем наших клиентов качественной продукцией, доставкой по всей России и поддержкой талантливых артистов. Заходите на наш сайт и выбирайте свой стиль! ShowStaff – это магазин оригинального мерча, официально и качественно. Посетите каталог, и вы обязательно найдете для себя уникальные вещи!
Pingrutty
| #
darknet markets onion https://github.com/aresonioncq0a7/aresonion – bitcoin dark web
Vortex_Spark
| #
Лично мне помог https://22buh.ru этот ресурс по финансам.
Fang7961
| #
Простой язык https://buronational.com о финасах.
Rabyitabe
| #
darknet markets https://github.com/darkwebsitesyhshv/darkwebsites – dark market list
MarkNOlaf
| #
dark market list tor drug market
CyberMaster
| #
Где смотреть статистику https://mir-plitki68.ru о финансах?
DonDonfaita
| #
darknet market links https://github.com/nexusdarkrtv1u/nexusdark – onion dark website
PhantomZ5
| #
Эксклюзивное интервью с экспертом по финасам: https://skupkann.ru
ruzimSinue
| #
Телеграм-канал t.me/official_izzicasino будет, несомненно, вам полезен. Здесь рассказываем, как выиграть в онлайн казино. Вы сможете узнать, что делать, когда не загружаются автоматы. Подготовили несколько рекомендаций, которые дадут возможность вам на выигрыш в Izzi Casino увеличить шансы. Играйте в завлекательные игры уже сегодня. Ищете izzi casino официальный сайт? T.me/official_izzicasino – канал, подпишитесь скорее на него, чтобы в курсе последних новостей быть и ни одной интересной акции не пропустить. Иззи – казино с современным дизайном и удобной навигацией. Удостоверьтесь в этом лично!
JamesLah
| #
navigate to this site
www backpack
WilliamIRrutty
| #
tor drug market darkmarket link
Slayer2978
| #
Полезные сервисы для работы с финансами: https://mcn-partner.ru
Donaldlaf
| #
onion dark website https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark web marketplaces
Nomad8410
| #
Реальные примеры применения финансов: https://prmzc.ru
Volodyanaf
| #
dark market 2025 darknet market
Shadow608
| #
Заинтересовался финасами. Решение https://vamdengy.ru здесь.
FNDaviditabe
| #
dark web market urls tor drug market
Kxyufaita
| #
darkmarket link best darknet markets
Pingrutty
| #
darknet markets 2025 https://github.com/aresonioncq0a7/aresonion – darknet site
Toliknaf
| #
darkmarket 2025 https://github.com/abacuslink4jjku/abacuslink – dark market url
где торгуют биткоинами
| #
где торгуют биткоинами
Fang3457
| #
Лучшие инструменты для работы с финансами: https://avtofinexpert.ru
BlueV3
| #
https://v100krat.ru Шутки про финасов.
MarkNOlaf
| #
tor drug market dark market 2025
Omega683
| #
Обзор популярных ресурсов по финансам: https://am-harvey.ru
Rabyitabe
| #
onion dark website https://github.com/darkwebsitesyhshv/darkwebsites – darknet links
DonDonfaita
| #
darknet drug store https://github.com/nexusdarkrtv1u/nexusdark – darknet markets
erotik film
| #
Yetişkinler için en iyi çevrimiçi sinemalardan biri olan ilginç ve
heyecan verici bir porno sitesindesiniz. Video kütüphanesi sıcak
videolar, uzun metrajlı filmler, müstehcen fotoğraflar ve harika seks
hikayeleriyle dolu. Bütün bunlar çevrimiçi olarak kayıt olmadan ve kaliteli
olarak mevcuttur. Bir resim seçmenin kolaylığı için tüm filmler kategorilere
ayrılmıştır; Ne izleneceği konusunda daha detaylı bir araştırma
için her zevke uygun çok sayıda seçim oluşturuldu.
Sinemamızın en popüler videosunu ayrı bir bölümde izleyebilirsiniz.
Aynı şekilde iki bölüm daha oluşturuldu: Modeller ve Stüdyolar,
bu da elbette aramayı daha kolay ve hızlı hale getiriyor.
Porno filmlerin yanı sıra erotik filmleri de çevrimiçi olarak ücretsiz izleyebilirsiniz.
Yetişkinlere yönelik kısa videolar, büyüleyici
anlar, keskin pozlar ve sonsuz orgazmlarla sizi memnun edecek.
Uzun metrajlı bir film izlemek ve seksin yanı sıra hikayeyi görmek isteyenler için ayrı bir
kategori oluşturuldu. Video kütüphanemizdeki en popüler olanı elbette Rusça tercümesi olan yetişkinlere
yönelik filmler. Beden dili herkes tarafından anlaşılabilir
ancak karakterlerin konuşmalarını anladığınızda film daha ilgi
çekici oluyor. Tizam.tv sadece ücretsiz içerik değil aynı zamanda sınırsızdır.
Çok sayıda videoya rağmen site her gün sıcak filmlerle güncellenmektedir.
Bizimle kal! Her birinize minnettarız! Erotik film
WilliamIRrutty
| #
darknet websites dark web drug marketplace
ThunderWalker
| #
Профессионалы рекомендуют эти ресурсы по финасам: https://finexpert40.ru
BlainePalry
| #
Названа дата, когда привычная всем жизнь на Земле станет невозможной https://x.com/Fariz418740/status/1903674512593359008
BlueMaster
| #
Что нельзя делать с финансами? https://zaimy-reshenie.ru
Pingrutty
| #
dark markets https://github.com/aresonioncq0a7/aresonion – dark markets 2025
Ghost213
| #
Инфографика по финансам: https://kpkmoscow.ru.
FNDaviditabe
| #
dark market url darkmarket url
Donaldlaf
| #
darknet links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark web sites
Kxyufaita
| #
dark market 2025 onion dark website
doyarNuple
| #
Ищете жби изделия? Сайт global-gbi.ru Завода ЖБИ в СПб и вы найдете широкий ассортимент продукции по выгодным ценам. Глобал ЖБИ – компания, которая является крупнейшим в регионе изготовителем изделий из железобетона и бетона. У нас новейшее оборудование и большая номенклатура. Мы работаем со строительными фирмами, частными лицами и иными организациями. Посмотрите каталог товаров и вы обязательно найдете для себя необходимую продукцию.
Volodyanaf
| #
darknet markets url dark websites
casino-eldorado-klub.com
| #
Мечтаете о крупных выигрышах? Тогда вам в Эльдорадо Казино! https://casino-eldorado-klub.com/ На этой платформе представлены лучшие слоты, щедрые бонусы и быстрый кэш-аут!
Почему именно Эльдорадо Казино?
Коллекция игр от известных разработчиков.
Щедрые награды при первом депозите.
Постоянные акции с крупными призами.
Оперативный вывод средств без ожиданий.
Удобная навигация без ограничений.
Круглосуточная поддержка отвечает мгновенно.
Регистрируйтесь прямо сейчас и испытайте азарт уже сегодня!
AlphaX9
| #
Современные методы по финасам: https://refinansmfc.ru
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://bd-rielt.ru/
Toliknaf
| #
dark market 2025 https://github.com/abacuslink4jjku/abacuslink – darkmarket 2025
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://bdrsu-2.ru/
MarkNOlaf
| #
onion dark website best darknet markets
check my site
| #
Hi there! I know this is somewhat off topic but I was wondering which blog platform are
you using for this website? I’m getting fed up of WordPress because I’ve had problems with hackers and I’m
looking at options for another platform. I would be fantastic if you could point me in the direction of a good platform.
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://belaya-dacha-park.ru/
ShadowWarden
| #
Как монетизировать https://garant-mebel38.ru финансы в бизнесе?
RobertFal
| #
https://helpdesk-test.zcu.cz/wiki/Rolex_57w
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные рекомендации — https://belebey-beton.ru/
Rabyitabe
| #
darknet links https://github.com/darkwebsitesyhshv/darkwebsites – darknet market
Wolf633
| #
Как https://mybroker-spb.ru не сдаться при изучении финасов.
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы — https://best-kuzminki.ru/
WilliamIRrutty
| #
dark web link darknet drug links
Pingrutty
| #
darknet market list https://github.com/aresonioncq0a7/aresonion – best darknet markets
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности https://beton911.ru/
топ хостингов
| #
топ хостингов
nvblfurn
| #
https://tj-service.ru/news/?igru_onlayn_besplatno__gde_poigrat_.html игры онлайн огонь и вода на 1 человека
Jasonquinc
| #
Курьер позвонил заранее, всё чётко и аккуратно.
доставка цветов
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://betonika38.ru/
FNDaviditabe
| #
darknet market list darknet markets
SteelProtocol
| #
https://flowers-nvrsk.ru Практическое применение финансов.
Kxyufaita
| #
darknet markets darknet links
CyberZ3
| #
Давайте обсудим о финасах: https://prosto-fa.ru.
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности https://bgberger.ru/
Donaldlaf
| #
dark market url https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark markets 2025
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы https://bhs39.ru/
Toliknaf
| #
darknet drugs https://github.com/abacuslink4jjku/abacuslink – darknet market links
Billymep
| #
https://shvejnye.ru/
Singlweirm
| #
Магазин «Столица Швейных Машин» в Челябинске https://shveichel.ru/ — это надежный поставщик качественной швейной техники: швейные машинки, оверлоки, промышленные швейные машины, гладильное оборудование, все для вышивки от мировых брендов: Juki, Bernina, Aurora, Janome, Leader. Профессиональные консультации: сотрудники помогут выбрать швейную технику под ваши задачи. Гарантия и сервис: товары сопровождаются гарантией, а сервисный центр обеспечивает ремонт и обслуживание. Удобное расположение: магазин находится по адресу Молодогвардейцев 64.
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы https://bitarel.ru/
Volodyanaf
| #
dark web marketplaces darknet drug market
MarkNOlaf
| #
best darknet markets darknet site
Markcen
| #
Новостройки или вторичка? Узнайте подробности https://bizkonta.ru/
Light698
| #
Лучшие инструменты для работы с финансами: https://fsr-holding.ru
Cyber_Slayer
| #
Полное руководство https://2-expert.ru по финансам от А до Я.
IronZ2
| #
Как применять финасов в бизнесе? https://vip-163.ru
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://bizneskuvshinka.ru/
Rabyitabe
| #
dark markets https://github.com/darkwebsitesyhshv/darkwebsites – darkmarket link
Pingrutty
| #
darknet links https://github.com/aresonioncq0a7/aresonion – darknet drug store
WilliamIRrutty
| #
darknet drugs darknet site
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://blok-beton.ru/
DonDonfaita
| #
darknet market links https://github.com/nexusdarkrtv1u/nexusdark – darknet drug store
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://bor-klimat.ru/
nvblfurn
| #
https://sorvachev.com/code/pages/igru_io__zahvatuvaushie_srazgheniya_za_vuzghivanie.html 44 котенка кис кис коты на задании
FNDaviditabe
| #
dark web market darknet drug links
Kxyufaita
| #
darkmarkets darknet markets url
Thunder430
| #
Эксклюзивное интервью с экспертом по финасам: https://kpk-kristall.ru
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности https://bortehsnab.ru/
ThunderSpark
| #
Какой вариант предпочесть https://amega-center.ru в финансах?
mostbet_qkKl
| #
мостбет скачать http://mostbet785.ru .
GhostByte
| #
Какие изменения в финансах? https://3karat.ru
Jariorxyw
| #
Где купить диплом по необходимой специальности?
Наши специалисты предлагают максимально быстро приобрести диплом, который выполняется на оригинальной бумаге и заверен печатями, штампами, подписями. Документ способен пройти лубую проверку, даже с применением специально предназначенного оборудования. Решайте свои задачи максимально быстро с нашими дипломами.
Купить диплом о высшем образовании diplomus-spb.ru/kupit-diplom-inzhenera-elektrika-8/
1win_vjEl
| #
1 вин про 1 вин про .
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://brigadaptz.ru/
Donaldlaf
| #
dark websites https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet links
игры с реальными призами
| #
игры с реальными призами
A Brief History of Elemental Analysis » Artemis Analytical
mostbet_rxKl
| #
скачать mostbet на телефон скачать mostbet на телефон .
Вован демо-игры
| #
Вован демо-игры
A Brief History of Elemental Analysis » Artemis Analytical
Sazrhab
| #
Мы изготавливаем дипломы любых профессий по выгодным тарифам.– asmc55418.com
1win_evEl
| #
1win win https://www.1win826.ru .
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://brizproekt.ru/
1win_svSl
| #
1vin kg https://1win812.ru/ .
RobertFal
| #
https://earthpedia.wiki/index.php/Rolex_4i
skapa ett binance-konto
| #
I don’t think the title of your article matches the content lol. Just kidding, mainly because I had some doubts after reading the article. https://accounts.binance.com/id/register?ref=GJY4VW8W
Toliknaf
| #
dark market url https://github.com/abacuslink4jjku/abacuslink – darkmarket 2025
MarkNOlaf
| #
darkmarket url dark market list
that site
| #
Hmm is anyone else experiencing problems with the pictures on this blog loading?
I’m trying to find out if its a problem on my end or if it’s
the blog. Any suggestions would be greatly appreciated.
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://bti-nsk.ru/
Hunter4033
| #
Ответы на частые https://roszaym.com вопросы о финасах.
1win_tuSl
| #
1 вин вход https://www.1win812.ru .
Pingrutty
| #
bitcoin dark web https://github.com/aresonioncq0a7/aresonion – darknet markets url
Byte8233
| #
Эксперты рекомендуют эти https://trade-matica.ru материалы по финансам.
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы https://build34.ru/
Volodyanaf
| #
darknet markets dark web market
mostbet_bhKl
| #
mostbet kg скачать mostbet785.ru .
RedFang
| #
Мне самому помог https://blackdiamond-val.ru этот ресурс по финансам.
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации — https://bynker63.ru/
1win_bzEl
| #
1 win.pro 1 win.pro .
WilliamIRrutty
| #
bitcoin dark web darknet markets links
Rabyitabe
| #
darknet market lists https://github.com/darkwebsitesyhshv/darkwebsites – dark market onion
RobertFal
| #
https://menuaipedia.menuai.id/index.php/Rolex_6q
Markcen
| #
Как выбрать надёжного застройщика? Читайте проверенные советы https://cemstroy-t68.ru/
DonDonfaita
| #
onion dark website https://github.com/nexusdarkrtv1u/nexusdark – dark market list
Вован казино официальный
| #
Вован казино официальный
A Brief History of Elemental Analysis » Artemis Analytical
StormProtocol
| #
Как монетизировать финасов https://finansdepo.ru в бизнесе?
1win_brSl
| #
1win rossvya 1win rossvya .
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности https://center-usadba.ru/
FNDaviditabe
| #
dark web market urls dark web link
Kxyufaita
| #
dark web market urls dark markets
Eagle478
| #
Примеры https://mapadom.ru успеха в финансах.
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://centrgilya.ru/
EagleStriker
| #
Как монетизировать финансы в бизнесе? https://centinvest.ru
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы https://centro-kraska.ru/
Donaldlaf
| #
dark market link https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet drug market
eco-sistem.com
| #
Утилизировать отходы в полевых условиях — больше не проблема. Передвижной инсинератор от ЭКОСИСТЕМЫ легко транспортируется, быстро запускается и работает автономно. Подходит для удалённых объектов, военных баз, стройплощадок и экстренных ситуаций.
RobertFal
| #
https://isekaiwiki.com/index.php?title=User:EfrenBooth33
nvblfurn
| #
https://offroadmaster.com/rotator/articles/?igru_na_dvoih_onlayn___veselo__uvlekatelno_i_udobno.html леди баг и супер кот целуются
Dnrteoq
| #
Где купить диплом специалиста?
Мы готовы предложить дипломы психологов, юристов, экономистов и прочих профессий по приятным тарифам. Мы предлагаем документы техникумов, расположенных в любом регионе РФ. Можно купить качественный диплом от любого учебного заведения, за любой год, указав актуальную специальность и оценки за все дисциплины. Дипломы и аттестаты выпускаются на “правильной” бумаге самого высшего качества. Это дает возможность делать государственные дипломы, не отличимые от оригинала. Документы заверяются необходимыми печатями и штампами. Стараемся поддерживать для заказчиков адекватную ценовую политику. Для нас важно, чтобы документы были доступны для большого количества граждан. 10000diplomov.ru/kupit-attestati-za-11-2-7
Dark_Overlord
| #
https://kpk-pv.ru Продвинутые техники финасов.
Pingrutty
| #
dark market 2025 https://github.com/aresonioncq0a7/aresonion – darknet markets links
Markcen
| #
Новостройки или вторичка? Узнайте подробности https://ceramodecol.ru/
Toliknaf
| #
darkmarket https://github.com/abacuslink4jjku/abacuslink – dark web market urls
Billymep
| #
https://voenoboz.ru/
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы https://chelstg.ru/
get-x скачать
| #
get-x скачать
A Brief History of Elemental Analysis » Artemis Analytical
Phantom581
| #
Эксклюзивное интервью с экспертом по финансам: https://luxstoneinvest.ru
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные статьи и советы — https://cheshire-apart.ru/
WilliamIRrutty
| #
dark market list dark web markets
Rabyitabe
| #
darkmarkets https://github.com/darkwebsitesyhshv/darkwebsites – dark market
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы https://citadel-ca.ru/
LightSlayer
| #
https://aktiv93.ru Реальные примеры применения финансов.
Вован казино онлайн
| #
Вован казино онлайн
A Brief History of Elemental Analysis » Artemis Analytical
Hunter3732
| #
Лично мне помог этот https://sdengi.com ресурс по финасам.
Volodyanaf
| #
darknet marketplace dark web sites
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://cks-vrn.ru/
Kxyufaita
| #
dark web drug marketplace darknet markets links
FNDaviditabe
| #
darknet markets best darknet markets
DonDonfaita
| #
darknet drugs https://github.com/nexusdarkrtv1u/nexusdark – dark web sites
RobertFal
| #
http://investorswiki.com/index.php/Rolex_7x
Markcen
| #
Новостройки или вторичка? Полезные рекомендации — https://cmy33.ru/
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы — https://concept-floor.ru/
Protocol2239
| #
Обзор популярных ресурсов по https://www-binarium.ru финансам.
Pingrutty
| #
darknet drug links https://github.com/aresonioncq0a7/aresonion – darkmarket 2025
CyberSpark
| #
Как достичь успеха в финасах? https://4kapital.ru
TawewhJog
| #
На сайте http://gorodnsk63.ru вы сможете почитать интересные, любопытные новости, посвященные городу Самаре и не только. К примеру, жителей ожидает ретроградный Меркурий, который продлится вплоть до 7 апреля текущего года. Также есть новости о заводе «Балтика» и о том, что он больше не будет сотрудничать с крафтовым брендом. Кроме того, в Самаре будут проводиться дорожные работы в 2025 году. А с 19 марта ожидаются ночные заморозки. Интересные, любопытные и увлекательные новости выкладываются регулярно, чтобы с ними ознакомились и вы.
Donaldlaf
| #
dark web link https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet markets 2025
Markcen
| #
Новостройки или вторичка? Узнайте подробности https://construct-metall.ru/
LightV2
| #
Секреты профессионалов https://avtozaim16.ru в финансах.
MarkNOlaf
| #
dark market dark web market urls
suprememasterchinghai.net
| #
suprememasterchinghai.net
A Brief History of Elemental Analysis » Artemis Analytical
MichaelMiz
| #
Привет любителям свободы!
Чат GPT 4 — современное решение для профессионалов в области анализа данных и бизнес-аналитики. Автоматизируйте составление отчетов, изучайте тренды и оптимизируйте рабочие процессы с помощью чат GPT для бизнеса, а чат GPT перевод обеспечит качественную работу с документами на разных языках.
Подробнее здесь: https://talkchatgpt.com
как chat gpt ответы на вопросы бот скачать chatgpt apk чат gpt онлайн бесплатно
нейросеть для написания постов для соцсетей бесплатно
Преобразуйте знания в действия!
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://credomir.ru/
Лев казино
| #
Лев казино
blog topic
Trefgri
| #
Приобрести документ о получении высшего образования можно в нашем сервисе. diplomgorkiy.com/kupit-diplom-kolledzha-4-52
Click here
| #
Great article. I am going through a few of these issues
as well..
Toliknaf
| #
onion dark website https://github.com/abacuslink4jjku/abacuslink – dark web market list
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://crema-kuhni.ru/
JamesGow
| #
https://medium.com/@winsystem.online/%D1%83%D1%81%D0%BB%D1%83%D0%B3%D0%B0-%D1%80%D0%B0%D0%B7%D0%B2%D0%B0%D0%BB%D0%B0-%D1%81%D1%85%D0%BE%D0%B6%D0%B4%D0%B5%D0%BD%D0%B8%D1%8F-%D0%BD%D0%B0-%D0%B0%D0%B2%D1%82%D0%BE-%D0%BF%D0%BE%D1%87%D0%B5%D0%BC%D1%83-%D1%8D%D1%82%D0%BE-%D0%B2%D0%B0%D0%B6%D0%BD%D0%BE-%D0%B8-%D0%B3%D0%B4%D0%B5-%D1%81%D0%B4%D0%B5%D0%BB%D0%B0%D1%82%D1%8C-%D0%BA%D0%B0%D1%87%D0%B5%D1%81%D1%82%D0%B2%D0%B5%D0%BD%D0%BD%D0%BE-36910cd57819
LowigtNot
| #
На сайте https://petersburglife.ru/ ознакомьтесь с зеркалом популярного онлайн-заведения «Arkada». Это удивительная и уникальная платформа, которая предназначена для разнообразных игр, интересного времяпрепровождения. Гемблер по достоинству оценит понятный, простой в управлении интерфейс, сориентируется в выборе. Всем новичкам за регистрацию положен бонус. Здесь вы найдете все необходимое, включая классические игры, а также удивительные новинки. Также есть казино с живыми дилерами.
WilliamIRrutty
| #
darknet markets links bitcoin dark web
Iron_Nomad
| #
Что нового https://shlapny-dvorik.ru в финансах?
Markcen
| #
Покупка квартиры без рисков? Полезные статьи и советы — https://crimeaservice.ru/
RedZ6
| #
https://soglasiekpk.ru История становления финасов.
RobertFal
| #
https://caersidiwiki.com:443/index.php/Rolex_62l
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные рекомендации — https://crimsipstroy.ru/
Rabyitabe
| #
darkmarket url https://github.com/darkwebsitesyhshv/darkwebsites – darknet site
LightX7
| #
Как выбрать лучшее https://binance-registration.ru решение для финансов.
Kxyufaita
| #
dark markets bitcoin dark web
FNDaviditabe
| #
darknet drugs darknet sites
Markcen
| #
Новостройки или вторичка? Полезные рекомендации — https://crystalit34.ru/
Volodyanaf
| #
darknet drugs dark websites
DonDonfaita
| #
dark web sites https://github.com/nexusdarkrtv1u/nexusdark – darknet market links
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://cspkaliningrad.ru/
nvblfurn
| #
https://billionnews.ru/templates/artcls/index.php?igru_na_74.html пасьянс паук4масть играть онлайн бесплатно и без регистрации на весь экран
RobertFal
| #
https://joshblock.wiki/index.php?title=Rolex_28R
Pingrutty
| #
dark market url https://github.com/aresonioncq0a7/aresonion – tor drug market
Billymep
| #
https://my-caffe.ru/
StormSpark
| #
Факты и вымысел о финансах: https://o-mu.ru
AlphaZ4
| #
Читал прогнозы, что Финасы будет https://irkschooloflife.com главным трендом. Возражения?
wogonbum
| #
Baliyants.com – ваш верный источник знаний в области маркентинга. В работе с абсолютно каждым клиентом детально погружаемся в бизнес. Регулярно анализируем данные, чтобы ничего не упустить. Воспользуйтесь нашими услугами! Ищете дорожная карта? Baliyants.com – здесь представлены уникальные авторские проекты. Они созданы для того, чтобы помочь действенно применять рекламные кампании, оптимизировать бюджеты и повысить рентабельность ваших маркетинговых усилий. Специалисты – драгоценность агентства Baliyants. Мы работаем только на результат!
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы https://cspstroy.ru/
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности https://ctromm34.ru/
MarkNOlaf
| #
darknet site darknet marketplace
RobertFal
| #
https://earthpedia.wiki/index.php/Rolex_59L
Donaldlaf
| #
darknet site https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark web sites
Steel_Spark
| #
Коллеги, поделитесь опытом по финансам! https://alombard22.ru
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности https://cuppro-style.ru/
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации — https://cvetkrovli.ru/
Toliknaf
| #
dark web sites https://github.com/abacuslink4jjku/abacuslink – darknet markets onion address
Silver_Master
| #
Эксперты https://sibir-invest.com рекомендуют эти материалы по финасам.
Spark9549
| #
Где смотреть актуальные https://s-pudra.ru данные о финансах?
Markcen
| #
Новостройки или вторичка? Узнайте подробности https://dar-dereva.ru/
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы https://dc-svd.ru/
RobertFal
| #
https://www.wikiliad.it/index.php?title=Rolex_91h
Storm_Blade
| #
Согласно исследованиям, лучший источник о финансах – https://parik72.ru
Rabyitabe
| #
bitcoin dark web https://github.com/darkwebsitesyhshv/darkwebsites – dark market link
Pingrutty
| #
dark websites https://github.com/aresonioncq0a7/aresonion – bitcoin dark web
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://dekor-alfa.ru/
Red386
| #
Новые подходы в https://nalogservis-44.ru финасах.
DonDonfaita
| #
dark markets 2025 https://github.com/nexusdarkrtv1u/nexusdark – dark web market urls
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://deluxe-avenue.ru/
Robertraisk
| #
площадка для продажи аккаунтов покупка аккаунтов соц сетей
Kevinvep
| #
биржа аккаунтов маркетплейс готовых аккаунтов
WilliamFed
| #
где можно продать аккаунт биржа аккаунтов соц сетей
ShadowZ8
| #
Полное руководство по финансам от А до Я: https://sockapital.ru
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации — https://deluxe-ceramica.ru/
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы — https://dema26.ru/
yiyigoyThush
| #
Цель студии MARBL DESIGN – создать интерьер, который даст вам возможность находиться в ресурсном состоянии и поддерживать ритм жизни. С вами исключительно лучшие специалисты в сфере создания комфорта и уюта будут работать. Сотрудничаем с достойными поставщиками материалов и мебели. Ищете заказать дизайн интерьера? Marbl.ru – здесь можете ознакомиться с портфолио проектов. Также тут указаны цены на наши услуги. Выполним по недорогой цене красивый дизайн квартиры. Можете оставить заявку на сайте. Мы в ближайшее время перезвоним вам. С удовольствием все вопросы обсудим.
Red903
| #
Эксклюзивное https://carfamily63.ru интервью с экспертом по финансам.
eyotoAcaxy
| #
Cargo Jet – компания, которая помогает с закупками китайских товаров оптом. Начните свой бизнес с нами. Мы предлагаем широкий выбор продукции. Обладаем достаточным опытом и умеем находить уникальные решения для ваших логистических задач. Горды тем, что нас больше 3000 клиентов выбрали. Ищете карго доставка из Китая в Россию? Cargo-jet.ru – здесь представлена более детальная информация о нас. Точно знаем, вы качеством и результатом наших услуг останетесь довольны. С Cargo Jet доставка из Китая в РФ становится выгоднее и проще. Начните уже сейчас экономить!
Donaldlaf
| #
darknet markets onion address https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet drug links
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы https://deon-stroy.ru/
Thunder_Warden
| #
Сравнительный https://nomos38.ru анализ финасов.
bankrotstvo v moskve_uuKi
| #
банкротство
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы https://design-lis.ru/
Phantom_Code
| #
Оптимизация процессов через финансы: https://kpkprogress.ru
Toliknaf
| #
dark web drug marketplace https://github.com/abacuslink4jjku/abacuslink – dark web market links
Cazrxjh
| #
Приобрести диплом ВУЗа по невысокой стоимости вы можете, обращаясь к проверенной специализированной фирме. Мы предлагаем документы высших учебных заведений, которые находятся в любом регионе России. diplomt-v-chelyabinske.ru/gosudarstvennij-diplom-kupit
Sazrjoa
| #
Мы предлагаем дипломы психологов, юристов, экономистов и любых других профессий по приятным тарифам. Плюсы приобретения документов в нашей компании
Вы приобретаете диплом в надежной и проверенной компании. Такое решение сэкономит не только денежные средства, но и ваше время.
Плюсов намного больше:
• Дипломы печатаются на настоящих бланках со всеми печатями;
• Можно приобрести дипломы любого ВУЗа РФ;
• Цена в разы меньше той, которую довелось бы заплатить за обучение в университете;
• Быстрая доставка в любые регионы России.
Приобрести диплом ВУЗа– http://dachkanews.ru/dokumentyi-dlya-uspeshnoy-kareryi-bez-lishnih-voprosov/ – dachkanews.ru/dokumentyi-dlya-uspeshnoy-kareryi-bez-lishnih-voprosov
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://design70.ru/
RobertFal
| #
https://joshblock.wiki/index.php?title=Rolex_35n
Джет тон казино
| #
Добро пожаловать в Jetton Casino – место, где каждый спин может принести крупный выигрыш. В нашем казино представлены только лучшие игровые автоматы, покер, рулетка и многое другое. Присоединяйтесь прямо сейчас, чтобы испытать удачу и воспользоваться выгодными бонусами.
https://ligapark.ru/ – это не просто казино, а целая вселенная для любителей азартных игр. Для наших игроков доступны регулярные турниры, программа лояльности, а также специальные бонусы для активных пользователей. Гарантируем безопасные транзакции, а также удобные способы пополнения счета и вывода выигрышей.
Огромный ассортимент развлечений для всех любителей азартных игр.
Ежедневные подарки, кэшбэк и выгодные предложения для новых и постоянных игроков.
Честные и прозрачные условия, быстрые транзакции и полная защита данных.
Эксклюзивные турниры с высокими призовыми фондами и шанс сорвать джекпот.
Начните игру в Jetton Casino и сделайте первый шаг к крупным победам.
Eanrlws
| #
Для эффективного продвижения вверх по карьерной лестнице потребуется наличие диплома о высшем образовании. Купить диплом о высшем образовании у проверенной компании: diplom4you.com/kupit-starij-diplom/
Pingrutty
| #
dark web market urls https://github.com/aresonioncq0a7/aresonion – darkmarket link
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности https://deutz-parts.ru/
nvblfurn
| #
http://khomus.ru/lib/pages/igru_dlya_malchikov_onlayn_besplatno_bez_registracii.html игра ежики онлайн бесплатно без регистрации
DarkWalker
| #
Может кто знает ориентируется в финасах? https://pawnshopnugget.com Куда зайти?
NightX4
| #
Давайте https://andreeva36.ru обсудим о финансах.
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы — https://diamantgeo.ru/
Rabyitabe
| #
darknet market lists https://github.com/darkwebsitesyhshv/darkwebsites – darknet sites
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние возможности.
Наша продукция:
Мишень для распыления сплава AlCu
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
поставляемая продукция:
Заготовка из конструкционной стали листовая 65 мм 20пс ОСТ 3-1686-90 Выбирайте конструкционные заготовки высокого качества для вашего проекта на Редметсплав.рф. Наш ассортимент включает прочные и долговечные материалы, идеально подходящие для различных областей применения, включая строительство и машиностроение.
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их качество. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы по мере того как находить ответы под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Проволока висмутовая N26055 – UNS Проволока висмутовая N26055 – UNS предназначена для применения РІ различных сферах, включая электронику Рё медицинские технологии. Рзготавливается РёР· высококачественного висмута, что гарантирует отличные физико-химические свойства. Проволока обладает высокой РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью Рё хорошей проводимостью, что делает ее идеальным выбором для специализированных проектов. Если РІС‹ ищете надежный Рё качественный материал, купите Проволока висмутовая N26055 – UNS Рё получите РїСЂРѕРґСѓРєС‚, который удовлетворит ваши потребности. Доступна РІ различных диаметрах Рё длинах.
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы — https://dilerskiy-tsentr-baumit.ru/
Vortex876
| #
https://red-crystal.ru Ошибки новичков в финансах.
DonDonfaita
| #
darknet market links https://github.com/nexusdarkrtv1u/nexusdark – darknet markets
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы https://disc-kbr.ru/
Vortex274
| #
Лично мне https://tenderland24.ru помог этот ресурс по финасам.
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы https://disk-pila.ru/
1win_xsPi
| #
1 вин войти 1 вин войти .
1win_bpen
| #
1win moldova download 1win moldova download .
Billymep
| #
https://kaizen-tmz.ru/
Markcen
| #
Как выбрать надёжного застройщика? Читайте проверенные советы https://dmitrov-klimat.ru/
Donaldlaf
| #
dark markets 2025 https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet drug store
Thunder_Master
| #
Нюансы работы с https://al-biz.ru финансами.
1win_plPi
| #
1вин сайт официальный https://1win827.ru/ .
mostbet_zyel
| #
скачат мостбет http://mostbet786.ru .
1win_zten
| #
1win aplicația http://www.1win5003.ru .
Pingrutty
| #
darknet drug links https://github.com/aresonioncq0a7/aresonion – darkmarkets
Vortex_Warden
| #
Пошаговая инструкция по финансам: https://tradematika.ru
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://dom-na-kosmonavtov.ru/
ShadowHacker
| #
Мифы и правда о финасах: https://achirok.com
Toliknaf
| #
darknet websites https://github.com/abacuslink4jjku/abacuslink – dark market list
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://dom-techno161.ru/
mostbet_fzel
| #
мостбет казино войти http://mostbet786.ru/ .
Markcen
| #
Хотите сдать квартиру в аренду? Читайте проверенные советы https://dom-vasilevo.ru/
RobertFal
| #
https://dynastyascend.com/wiki/User:FelipaWaechter
1win_cgPi
| #
1win. pro https://1win827.ru .
1win_dqen
| #
1win http://www.1win5003.ru .
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы https://domloko.ru/
Rabyitabe
| #
darknet markets links https://github.com/darkwebsitesyhshv/darkwebsites – darknet markets links
DarkSentinel
| #
Мифы и правда о https://ac-fors.ru финансах.
Markcen
| #
Как выбрать надёжного застройщика? Читайте проверенные советы https://domnabakalinskoy.ru/
Byte4353
| #
Профессионалы рекомендуют эти материалы по финансам: https://mamatobolsk.ru
Ghost_Striker
| #
Обзор https://doavansa.com популярных ресурсов по финасам.
RobertFal
| #
https://sbwiki.davnit.net/index.php/User:WilbertBriley
mostbet_jwel
| #
motbet mostbet786.ru .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние возможности.
Наши товары:
Высокочистые плитки алюминия
Pingrutty
| #
darkmarket list https://github.com/aresonioncq0a7/aresonion – darknet markets url
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://dompodkluch33.ru/
NightX3
| #
Современные методы https://jasendeluxe.ru по финансам.
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://domzenit.ru/
Donaldlaf
| #
darknet market list https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark markets 2025
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их качество. Превосходное обслуживание – наш стандарт – мы на связи, чтобы улаживать ваши вопросы а также адаптировать решения под специфику вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наша продукция:
Магний MAG 121 – BS 3370 Магний MAG 121 – BS 3370 – это высококачественная добавка, предназначенная для поддержки Р·РґРѕСЂРѕРІСЊСЏ Рё активного образа жизни. РћРЅ способствует нормализации обмена веществ, улучшает работу мышц Рё нервной системы. РџСЂРѕРґСѓРєС‚ активно используется спортсменами Рё людьми, стремящимися поддерживать СЃРІРѕР№ уровень энергии. Каждый элемент формулы тщательно подобран для максимального эффекта. Р’С‹ можете купить Магний MAG 121 – BS 3370 для достижения оптимальных результатов РІ вашем режиме тренировок Рё восстановления. Рнвестируйте РІ СЃРІРѕРµ Р·РґРѕСЂРѕРІСЊРµ СЃ поддержкой этого уникального комплекта!
Vortex_Sentinel
| #
Как монетизировать финансы в бизнесе? https://bankrot-penza.ru
RobertFal
| #
https://telegra.ph/Fake-cartier-watches-03-20
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности https://don-profil.ru/
Toliknaf
| #
darkmarket link https://github.com/abacuslink4jjku/abacuslink – darkmarket
Markcen
| #
Новостройки или вторичка? Полезные рекомендации — https://donskoy-alians.ru/
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://doors-pervouralsk.ru/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Мишень для распыления скандия
SteelOverlord
| #
Как реализовали финансов в проекте: https://megalodonsantekhnika.ru
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://ds-krsk.ru/
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наша продукция:
РўСЂСѓР±Р° нержавеющая электросварная 30×2 РјРј 08РҐ18Рќ10 шлифованная Купить трубу нержавеющую электросварную РїРѕ выгодной цене. РЁРёСЂРѕРєРёР№ выбор нержавеющих электросварных труб для применения РІ промышленности, пищевой Рё медицинской сферах. Гарантированное качество Рё надежность. Доставка РїРѕ всей Р РѕСЃСЃРёРё.
Hacker9422
| #
Новые подходы в финансах: https://alexprom-invest.ru
Rabyitabe
| #
dark web market links https://github.com/darkwebsitesyhshv/darkwebsites – dark web drug marketplace
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://dvemarket.ru/
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы по мере того как находить ответы под особенности вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наши товары:
Тантал 5Р° Тантал 5Р° – это высококачественный материал, обладающий выдающимися свойствами. РћРЅ широко используется РІ различных отраслях, включая электронику Рё атомную энергетику, благодаря своей отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкости Рё стабильности. Приобретая Тантал 5Р°, РІС‹ получаете надежный Рё долговечный РїСЂРѕРґСѓРєС‚. Ртот материал РїРѕРґС…РѕРґРёС‚ для создания деталей, которые требуют высокой прочности Рё термостойкости. РќРµ упустите возможность купить Тантал 5Р° РїРѕ подтвержденной цене Рё обеспечить вашу продукцию необходимыми характеристиками. Убедитесь РІ выгоде Рё качестве данного материала.
DonDonfaita
| #
darkmarket 2025 https://github.com/nexusdarkrtv1u/nexusdark – darknet market links
nvblfurn
| #
http://tiflos.ru/content/pags/igru_so_slovami___treniruem_um_i_poluchaem_udovolstvie.html игры для пк три в ряд
Pingrutty
| #
darkmarket list https://github.com/aresonioncq0a7/aresonion – darknet markets onion
RobertFal
| #
https://telegra.ph/Rolex-bluesy-submariner-03-20
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности https://dveripraktika.ru/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Мишень для распыления скандия
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://dverito.ru/
Alpha905
| #
Подборка https://skpk-avantag.ru ресурсов по финансам.
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наша продукция:
Латунная полоса 1.3×100 РјРј Р›68 ГОСТ 931-90 Покупайте латунную полосу различных размеров Рё толщин РЅР° сайте Редметсплав.СЂС„. РЈ нас РІС‹ найдете высококачественный материал СЃ высокой прочностью, стойкостью Рє РєРѕСЂСЂРѕР·РёРё Рё возможностью легкой обработки. Рспользуйте латунную полосу для создания декоративных элементов, мебели, украшений Рё РґСЂСѓРіРёС… изделий. Доступны различные размеры Рё толщины.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние достижения.
Наши товары:
Медно-никелевая поверхность для распыления
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы https://dveritrik.ru/
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их качество. Опытная поддержка – наш стандарт – мы на связи, чтобы улаживать ваши вопросы и находить ответы под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
Проволока висмутовая H57Bi3A – JIS Z 3282 Проволока висмутовая H57Bi3A – JIS Z 3282 представляет СЃРѕР±РѕР№ высококачественный материал, предназначенный для использования РІ различных промышленных сферах. Благодаря отличной электропроводности Рё РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкости, данный РїСЂРѕРґСѓРєС‚ идеально РїРѕРґС…РѕРґРёС‚ для производства компонентов, требующих надежности Рё долговечности. Если РІС‹ ищете надежное решение, чтобы улучшить качество СЃРІРѕРёС… изделий, вам стоит купить Проволока висмутовая H57Bi3A – JIS Z 3282. Рнвестируйте РІ этот товар, Рё получите стабильные результаты РІ своей работе.
Donaldlaf
| #
dark markets 2025 https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – onion dark website
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наша продукция:
Труба из драгоценных металлов золотая 500х3х0.7 мм Зл99.9 ТУ Гарантированное качество и изысканный дизайн – купите драгоценные трубы для ювелирных украшений, машиностроения и архитектуры. Широкий выбор и высокое качество! Доставка по России.
DarkZ3
| #
Топовый ресурс по теме https://buhgalter-ekspert.ru финансов.
TommyCeT
| #
Назван простой напиток, который спасает от повышения сахара в крови https://x.com/Fariz418740/status/1904006505147629966
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности https://dveritula71.ru/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наша продукция:
Гранулы силикона 4N
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог высококачественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их качество. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы и адаптировать решения под требования вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
поставляемая продукция:
РљСЂСѓРі магниевый MAG 131 – BS 3370 Фольга магниевая MAG 131 – BS 3370 представляет СЃРѕР±РѕР№ высококачественный материал, используемый РІ различных отраслях, включая строительства Рё автомобильное производство. РћРЅР° отличается отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ устойчивостью Рё легкостью, что делает ее идеальной для применения РІ условиях повышенной влажности. Благодаря СЃРІРѕРёРј физико-химическим свойствам фольга надежна Рё долговечна. Купить Фольга магниевая MAG 131 – BS 3370 можно, обратившись Рє нашим специалистам. РќРµ упустите возможность использовать этот товар для СЃРІРѕРёС… проектов!
Markcen
| #
Покупка квартиры без рисков? Полезные статьи и советы — https://dverivoevoda.ru/
RobertFal
| #
https://caersidiwiki.com:443/index.php/User:ShalandaBeardsmo
Light_Master
| #
Как не сдаться при изучении финансов: https://lombard-alpha22.ru
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://dwenertec.ru/
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их происхождение. Опытная поддержка – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы по мере того как находить ответы под специфику вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наша продукция:
Проволока висмутовая 811 – EN ISO 9453 Проволока висмутовая 811 – EN ISO 9453 является высококачественным материалом, подходящим для различных сварочных работ. РћРЅР° обеспечивает отличные механические свойства Рё РєРѕСЂСЂРѕР·РёРѕРЅРЅСѓСЋ стойкость, что делает её идеальной для применения РІ специализированных отраслях. РћСЃРЅРѕРІРЅРѕРµ преимущество данной проволоки заключается РІ её способности минимизировать нежелательные реакции РїСЂРё сварке. Если РІС‹ ищете надежный материал для СЃРІРѕРёС… проектов, вам стоит купить Проволока висмутовая 811 – EN ISO 9453. Ртот РїСЂРѕРґСѓРєС‚ гарантирует стабильность Рё эффективность, необходимые для успешного выполнения работ.
Markcen
| #
Как выбрать надёжного застройщика? Читайте проверенные советы https://eco-construct.ru/
Pingrutty
| #
onion dark website https://github.com/aresonioncq0a7/aresonion – dark market list
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы https://ecodom-svetlograd.ru/
Rabyitabe
| #
darknet markets onion address https://github.com/darkwebsitesyhshv/darkwebsites – dark web drug marketplace
WolfV2
| #
По данным аналитиков, лучший источник https://lombard-yalta.ru о финансах.
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы — https://ecofaiber.ru/
Вулкан Платинум мобильная версия
| #
Vulkan Platinum — это платформа для игроков, которые ценят качественный азарт и надежность. Каждая игра здесь — это шанс погрузиться в мир развлечений и высоких выигрышей. Вулкан Платинум предоставляет надежную платформу с быстрыми выплатами и полной прозрачностью.
Что отличает https://clubvulkan24-slots.com/ от других казино? Каждый новый игрок может рассчитывать на приятные бонусы, а постоянные клиенты получают доступ к эксклюзивным предложениям. Мы заботимся о безопасности наших игроков и о том, чтобы процесс игры был максимально комфортным.
Когда начать играть? Чем раньше, тем лучше! Регистрация займет всего несколько минут, а вы сможете сразу же получить доступ ко всем бонусам и акциям. Вот что вас ждет в Vulkan Platinum:
Мгновенные выплаты и надежность.
Вам доступны не только приветственные бонусы, но и специальные предложения для наших постоянных клиентов.
В Vulkan Platinum вы найдете все, от любимых слотов до захватывающих игр с живыми дилерами.
Vulkan Platinum — это место, где каждый момент может стать выигрышным.
DonDonfaita
| #
darknet links https://github.com/nexusdarkrtv1u/nexusdark – darknet markets links
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://eniseyburvod.ru/
IronX8
| #
Где смотреть актуальные данные о финансах? https://spectehcredit.ru
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Высокочистые гранулы молибдена
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы https://eniseynev.ru/
RobertFal
| #
https://telegra.ph/Rolex-submariner-mens-03-20
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://enrubattery.ru/
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
поставляемая продукция:
Прецизионная труба для гидравлических и пневматических энергосистем 28х2 мм E355 EN 10305-4 Выбирайте прецизионные трубы для гидравлических и пневматических систем в Редметсплав для гарантированной надежности и эффективности промышленного оборудования. Широкий выбор высококачественных труб для различных отраслей промышленности. Применение в гидравлических системах машин и оборудования, в пневматических системах управления и в других технических системах, обеспечивая надежную передачу энергии.
Donaldlaf
| #
darknet drugs https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark market onion
SteelMaster
| #
Особенности реализации финансов: https://buh-biznes29.ru
Markcen
| #
Новостройки или вторичка? Полезные рекомендации — https://etalon-voda.ru/
Donaldwhago
| #
Holgura mecanica
Dispositivos de ajuste: clave para el rendimiento suave y óptimo de las dispositivos.
En el ámbito de la tecnología actual, donde la productividad y la fiabilidad del dispositivo son de suma significancia, los equipos de calibración tienen un función crucial. Estos sistemas adaptados están concebidos para balancear y asegurar componentes giratorias, ya sea en dispositivos industrial, medios de transporte de transporte o incluso en electrodomésticos caseros.
Para los especialistas en mantenimiento de aparatos y los profesionales, operar con sistemas de equilibrado es importante para promover el rendimiento uniforme y confiable de cualquier dispositivo móvil. Gracias a estas soluciones tecnológicas innovadoras, es posible disminuir considerablemente las oscilaciones, el estruendo y la presión sobre los rodamientos, aumentando la longevidad de partes costosos.
Asimismo trascendental es el función que desempeñan los sistemas de calibración en la atención al cliente. El soporte profesional y el reparación continuo utilizando estos aparatos permiten brindar servicios de gran nivel, incrementando la contento de los consumidores.
Para los titulares de proyectos, la financiamiento en unidades de balanceo y medidores puede ser fundamental para optimizar la rendimiento y desempeño de sus aparatos. Esto es particularmente relevante para los emprendedores que dirigen reducidas y intermedias negocios, donde cada aspecto cuenta.
Por otro lado, los sistemas de ajuste tienen una gran aplicación en el sector de la prevención y el supervisión de nivel. Posibilitan encontrar posibles defectos, impidiendo intervenciones caras y averías a los aparatos. También, los datos extraídos de estos equipos pueden emplearse para maximizar procedimientos y incrementar la reconocimiento en sistemas de consulta.
Las áreas de utilización de los dispositivos de equilibrado incluyen numerosas áreas, desde la manufactura de ciclos hasta el control ambiental. No influye si se considera de importantes producciones de fábrica o reducidos establecimientos hogareños, los sistemas de calibración son necesarios para proteger un desempeño óptimo y sin presencia de detenciones.
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их соответствие стандартам. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы по мере того как адаптировать решения под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
Фольга молибденовая МР-10 Фольга молибденовая МР-10 – это высококачественный материал, предназначенный для применения в различных отраслях, включая электронику и металлургию. Она обладает отличной теплопроводностью и устойчивостью к высоким температурам, что делает её идеальным выбором для термоизолирующих решений. Данный продукт гарантирует надежность и долговечность в эксплуатации. Приобретая фольгу молибденовую МР-10, вы получаете гарантированное качество и отличные технические характеристики. Не упустите возможность купить Фольга молибденовая МР-10 для ваших нужд!
Guardian9751
| #
Как выбрать лучшее решение для финансов: https://zoo380.ru
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://fabrikavoshod.ru/
Toliknaf
| #
darkmarket url https://github.com/abacuslink4jjku/abacuslink – dark market 2025
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Высокочистые гранулы молибдена
Pingrutty
| #
best darknet markets https://github.com/aresonioncq0a7/aresonion – dark web marketplaces
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности https://fantek-nn.ru/
RobertFal
| #
https://joshblock.wiki/index.php?title=Rolex_61n
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наша продукция:
Кольцо РёР· драгоценных металлов золотое 25С…6С…2.5 РјРј Р—Р»99.9 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://fasad-alex.ru/
Rabyitabe
| #
dark web market https://github.com/darkwebsitesyhshv/darkwebsites – darkmarkets
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы https://fasadtut.ru/
Light_Hunter
| #
Нестандартный подход к финансам: https://2ndfl-doc.ru
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена требуемыми документами, подтверждающими их происхождение. Опытная поддержка – наш стандарт – мы на связи, чтобы ответить на ваши вопросы и адаптировать решения под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Фольга магниевая ISO-MgAl5Mn – ISO 16220 РўСЂСѓР±Р° магниевая ISO-MgAl5Mn – ISO 16220 представляет СЃРѕР±РѕР№ современный легкий материал, используемый РІ различных отраслях благодаря своей высокой прочности Рё РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкости. РћРЅР° идеально РїРѕРґС…РѕРґРёС‚ для авиационной Рё автомобильной промышленности, Р° также РІ сферах, РіРґРµ важна минимизация веса. Приобретая трубу магниевую ISO-MgAl5Mn – ISO 16220, РІС‹ получаете РїСЂРѕРґСѓРєС‚, который демонстрирует выдающиеся характеристики РІ условиях эксплуатации. Заказывая Сѓ нас, РІС‹ обеспечите надежность Рё долговечность СЃРІРѕРёС… изделий. РќРµ упустите возможность купить трубу магниевую ISO-MgAl5Mn – ISO 16220 РїСЂСЏРјРѕ сейчас для повышения эффективности ваших проектов!
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы https://feniks31.ru/
Click here
| #
Good write-up. I certainly appreciate this website.
Keep writing!
NightV3
| #
Пример из практики: Финансы – https://autofinance14.ru
DonDonfaita
| #
darknet links https://github.com/nexusdarkrtv1u/nexusdark – darknet market
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации — https://fenix-14.ru/
xonuCunny
| #
Looking for prediccion del precio de la plata? Тhetradable.com/es/ es un poderoso portal de informacin que le trae a su atencin las ltimas noticias del mundo de los negocios, las criptomonedas, todo sobre Forex y la economa global. Aqu encontrar informacin actualizada sobre criptomonedas, bolsas de valores, metales preciosos como el oro y otros activos clave.
gorgeous-squash-08b.notion.site
| #
HorsePower Brands Omaha
2525 N 117tһ Ave #300,
Omaha, NE 68164, United Ⴝtates
14029253112
franchising opportunities һome; gorgeous-squash-08b.notion.site,
nedodjorse
| #
Koch Market гарантирует индивидуальный подход к каждому клиенту. Специализируемся на услугах дооснащение, полировка, тюнинг, детейлинг, оклейка. Обеспечиваем высокое качество работы. Расскажем подробнее о действующих акциях и ответим на вопросы. Ищете оклейка кабины грузовика пленкой? Koch-market.ru – тут размещаем для автолюбителей полезные статьи. Цель Koch Market – сократить издержки, увеличить обороты и доходность вашего бизнеса. Оставьте заявку на сайте, и мы перезвоним в ближайшее время. Обсудим задачи, найдем наилучшее решение и запланируем работы.
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы — https://ferrumclean.ru/
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы https://fguesv.ru/
Donaldlaf
| #
darknet site https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet drug store
Pingrutty
| #
dark web marketplaces https://github.com/aresonioncq0a7/aresonion – best darknet markets
EagleX9
| #
Финансы https://avtodengy.ru для компаний.
Markcen
| #
Новостройки или вторичка? Полезные рекомендации — https://findombani.ru/
Dragon_Warden
| #
Видел прогнозы, что Финансы будет главным трендом. Соглашусь? https://555400.ru
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Мишень гафния
RobertFal
| #
https://telegra.ph/Replica-watch-best-03-20-2
Toliknaf
| #
onion dark website https://github.com/abacuslink4jjku/abacuslink – darknet markets onion address
Markcen
| #
Хотите сдать квартиру в аренду? Читайте проверенные советы https://findomfundament.ru/
nvblfurn
| #
https://trial-tour.ru/wp-content/themes/tri_v_ryad_igrat_besplatno__polnue_versii_bez_registracii_1.html экзамен в гибдд онлайн как в гаи
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наша продукция:
Труба из драгоценных металлов золотая 150х3х1 мм Зл99.9 ТУ Гарантированное качество и изысканный дизайн – купите драгоценные трубы для ювелирных украшений, машиностроения и архитектуры. Широкий выбор и высокое качество! Доставка по России.
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы — https://fine-art12.ru/
Davidcenly
| #
Где смотреть прямые трансляции матчей? Полный список доступных площадок — https://gallerix.ru/pnews/202102/yavlyaetsya-li-futbol-v-bolshey-stepeni-iskusstvom-ili-arkadoy/
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные статьи и советы — https://finkom74.ru/
Rabyitabe
| #
onion dark website https://github.com/darkwebsitesyhshv/darkwebsites – darkmarket 2025
Dark_Blade
| #
Полное руководство по финансам от А до Я: https://lombardgoldmine.ru
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог высококачественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их качество. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы а также находить ответы под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
Порошок вольфрамовый WC Порошок вольфрамовый WC — это высококачественный материал, широко используемый в промышленности. Он отличается высокой прочностью и температурной стойкостью, что делает его идеальным для производства тугоплавких сплавов. Вольфрамовый порошок незаменим в машиностроении, металлообработке и в производстве инструментов. Если вы ищете надежный и долговечный компонент, который поможет улучшить характеристики ваших изделий, то покупка Порошка вольфрамового WC станет идеальным решением. Не упустите возможность купить Порошок вольфрамовый WC уже сегодня!
Williamseals
| #
Магазин https://4766.ru/ предлагает лучшие цены на Webasto EVO, Air Top, а также надежные автомобильные холодильники Alpicool. Качество на высоте!
Markcen
| #
Новостройки или вторичка? Узнайте подробности https://fisher-kp.ru/
Sentinel4052
| #
Особенности реализации финансов: https://capital-invest26.ru
Walker2683
| #
Креативное решение к финансам: https://cnp55.ru
DonDonfaita
| #
best darknet markets https://github.com/nexusdarkrtv1u/nexusdark – onion dark website
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://flagman102.ru/
Harrisvah
| #
казино вулкан
Markcen
| #
Новостройки или вторичка? Узнайте подробности https://floor-ashton.ru/
Pingrutty
| #
darknet markets onion address https://github.com/aresonioncq0a7/aresonion – darknet market
DragonX7
| #
Согласно https://dostale.ru исследованиям, лучший источник о финансах.
TommyCeT
| #
Назван простой напиток, который спасает от повышения сахара в крови https://x.com/Fariz418740/status/1904006505147629966
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://fm70.ru/
Donaldlaf
| #
darknet market links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark web market urls
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://forma-doma.ru/
StormZ5
| #
Простой язык о финансах: https://ipoteka-rassrochka.ru
DragonZ3
| #
Использование на практике https://7sevenauto.ru финансов.
Avrora casino
| #
Avrora casino
A Brief History of Elemental Analysis » Artemis Analytical
Toliknaf
| #
darkmarket https://github.com/abacuslink4jjku/abacuslink – darkmarket link
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации — https://formatlight.ru/
Blade1366
| #
Как сделать финансы? https://korel-surgut.ru
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://forpost-ural.ru/
FaridBaile
| #
Наконец-то нашел идеальную кухню! “Эталон Кухни” – выполнили в срок, качественно. Звоните: 8-843-258-86-66 Посмотреть на карте
Rabyitabe
| #
dark web market https://github.com/darkwebsitesyhshv/darkwebsites – tor drug market
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы https://forstnpf.ru/
nvblfurn
| #
http://spbsseu.ru/page/pages/mir_onlayn_igr_dlya_malchikov__razvlechenie_i_razvitie.html игра противоположности для дошкольников 6 7
KeithCat
| #
iphone specifications iphone prices
Code3173
| #
Эксперты рекомендуют эти ресурсы https://remont-skupka.ru по финансам.
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности https://fort-pro.ru/
DragonZ6
| #
https://sapfir42.ru Как устроен механизм финансов?
DonDonfaita
| #
dark web link https://github.com/nexusdarkrtv1u/nexusdark – darknet drug store
NightV7
| #
Как выбрать лучшее решение для финансов: https://finpartners44.ru
Markcen
| #
Новостройки или вторичка? Полезные рекомендации — https://freski23.ru/
Philliptroli
| #
Прочные и удобные зип пакет опт и розница, разные размеры, надежная застежка. Для хранения, фасовки и перевозки. Оформите заказ онлайн с быстрой доставкой!
BediktUnlon
| #
На сайте https://orliman.shop вы найдете огромное количество ортопедических товаров, созданных для вашего здоровья. В каталоге представлены коленные, голеностопные ортезы, а также корсеты. В большом количестве находятся комфортные приспособления для фиксации. Также имеются бандажи, стельки, специальная обувь. Все это представлено в огромном ассортименте и по доступным ценам. Перед вами новинки, а также те товары, которые пользуются огромным спросом. В магазине постоянно проходят увлекательные акции.
Warrennapix
| #
Discover Trip to Montenegro crystal clear sea, mountain landscapes, ancient cities and delicious cuisine. We organize a turnkey trip: flight, accommodation, excursions.
RichardMog
| #
В Баку найдена одна из трех пропавших без вести несовершеннолетних подруг https://x.com/Fariz418740/status/1904141438733979773
RobertSania
| #
Индивидуалки Екатеринбурга Индивидуалки Екб ,Индивидуалки Екатеринбурга ,Индивидуалки Екатеринбург ,Проститутки Екб ,Проститутки Екатеринбурга ,Эскорт Екатеринбург ,Дешевые проститутки Екатеринбурга ,Дешевые проститутки Екб ,Шлюхи Екатеринбург ,Путаны Екатеринбург
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы — https://fsk16.ru/
Donaldlaf
| #
darknet markets onion https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet sites
Light_Master
| #
Список инструментов по финансам: https://to-click.ru
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://fuseitdecore.ru/
IronZ5
| #
Друзья, поделитесь опытом по финансам! https://avto-arenda38.ru
cisco
| #
The Cisco Nexus 9300 series is perfect for scalable, secure networks.
We’ve been working with similar technologies—check it out
at Visit US.
Markcen
| #
Как выбрать надёжного застройщика? Читайте проверенные советы https://gala-park.ru/
Toliknaf
| #
dark markets https://github.com/abacuslink4jjku/abacuslink – darknet marketplace
ThunderZ4
| #
Авторитетное https://artkuhna.ru мнение о финансах.
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://galastroy-sk.ru/
Code4228
| #
Всем привет! Подскажите, где посмотреть информацию о финансах? https://maks-granit.ru
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://gameliongroup.ru/
mtsekaterinburgsig
| #
мтс тв екатеринбург
https://teachersconsultancy.com/employer/157233/24inetrnet-domashnij-ekb-2
провайдер мтс
poyufpailk
| #
Beterex – компания, которая обладает приличным опытом в решении любых задач. Мы лучшую логистическую цепочку выстроим, и ваши расходы на перевозку грузов из Китая снизим. Прозрачно работаем и оперативнее конкурентов. Взятые на себя обязательства гарантируем. Ищете жд перевозки из Китая? Mybeterex.com – портал, где более подробная информация о компании Бэтэрэкс представлена, советуем ее посмотреть уже сейчас. Предоставляем только качественные услуги. Найдем нужных вам производителей и поставщиков в Китае. Любой запрос ваш будет на все 100% осуществлен.
Pingrutty
| #
darknet sites https://github.com/aresonioncq0a7/aresonion – darknet market list
Rabyitabe
| #
dark web sites https://github.com/darkwebsitesyhshv/darkwebsites – dark market onion
GeorgeCon
| #
check this link right here now surf wallet
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://gavrilov-sergey.ru/
EagleReaper
| #
Как думаете, https://finance-alliance.ru где быстрее всего изучить финансы?
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы https://gazuslugi.ru/
DonDonfaita
| #
bitcoin dark web https://github.com/nexusdarkrtv1u/nexusdark – darknet links
Light959
| #
Как думаете, где лучше всего понять финансы? https://nikiforov-finance.ru
Alpha_Walker
| #
Простой язык о финансах: https://avtolombardbryansk.ru.
Billymep
| #
https://artcet.ru/
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы https://geogroupeng.ru/
nvblfurn
| #
http://peling.ru/wp-content/pages/mir_priklucheniy__ogon_i_voda___igra_s_ogonkom.html сапер играть онлайн бесплатно без регистрации
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://geotech48.ru/
Donaldlaf
| #
tor drug market https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark web market list
RedStriker
| #
Давайте обсудим о финансах: https://skupka-52.ru
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные статьи и советы — https://geotop62.ru/
1win_pnMl
| #
1вин официальный сайт вход 1вин официальный сайт вход .
OmegaSpark
| #
Ответы на частые вопросы о финансах: https://solnce38.ru
1win_gySt
| #
1win зайти http://www.1win813.ru .
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы https://geovodservice.ru/
Toliknaf
| #
darknet market links https://github.com/abacuslink4jjku/abacuslink – dark markets 2025
Pingrutty
| #
darknet markets https://github.com/aresonioncq0a7/aresonion – darknet drug market
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы https://germes-alania.ru/
1win_vrMl
| #
1win kg скачать 1win6011.ru .
Thunder_Protocol
| #
Плюсы и минусы разных подходов к финансам: https://zaim-pod-zalogi.ru
IronGuardian
| #
Основы https://mdk-finance.ru финансов для новичков.
1win_xuSt
| #
1win rossvya https://1win813.ru/ .
WilliamIRrutty
| #
dark market onion dark market list
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации — https://ggs45.ru/
mostbet_caMa
| #
mostbet kg отзывы http://www.mostbet794.ru .
Rabyitabe
| #
darknet markets onion https://github.com/darkwebsitesyhshv/darkwebsites – dark web drug marketplace
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные рекомендации — https://ginloft.ru/
FNDaviditabe
| #
darknet websites dark web markets
Kxyufaita
| #
dark web market links darknet marketplace
ThomasVep
| #
Спасибо за красивые розы! Девушка была в восторге.
букет цветов томск
Warden1961
| #
Эксклюзивное интервью с экспертом по финансам: https://n-fanera.ru
1win_nbMl
| #
1win партнёрка http://www.1win6011.ru .
mostbet_keMa
| #
скачат мостбет https://mostbet794.ru/ .
Ghost_Nomad
| #
Доступно объяснено https://kpkperspektiva.ru о финансах.
DonDonfaita
| #
darkmarket 2025 https://github.com/nexusdarkrtv1u/nexusdark – darknet markets url
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://gismt72.ru/
1win_xrSt
| #
1wi https://www.1win813.ru .
Volodyanaf
| #
darkmarket link darkmarket list
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы https://gk-olympic.ru/
IronX4
| #
Подборка ресурсов по финансам: https://galatur-perm.ru
PabloTon
| #
Planning a vacation? Podgorica airport is a warm sea, picturesque beaches, cozy cities and affordable prices. A great option for a couple, family or solo traveler.
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://glaskek.ru/
MarkNOlaf
| #
darknet markets onion darknet websites
Donaldlaf
| #
darknet market lists https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – onion dark website
GhostV5
| #
Новые https://lombard-zaklad.ru подходы в финансах.
Michaelhum
| #
Holidays in Tivat are European comfort at an affordable price. Transparent sea, clean beaches, historical cities and warm climate all year round.
Pingrutty
| #
best darknet markets https://github.com/aresonioncq0a7/aresonion – darknet markets
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы — https://glass161.ru/
mostbet_cbMa
| #
motsbet mostbet794.ru .
DragonZ3
| #
Детальный анализ финансов: https://jaklin63.ru
Toliknaf
| #
dark markets https://github.com/abacuslink4jjku/abacuslink – onion dark website
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности https://glatt-nsk.ru/
WilliamIRrutty
| #
darknet drug market darknet markets url
GeorgeTom
| #
Source:
casinorushplay.org
Sazruun
| #
Мы изготавливаем дипломы любых профессий по приятным тарифам. Стоимость может зависеть от определенной специальности, года выпуска и университета. Стараемся поддерживать для покупателей адекватную политику тарифов. Важно, чтобы дипломы были доступны для большого количества граждан. купить диплом продавца
LightCode
| #
Подробное https://vash-lombard.ru руководство по финансам.
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности https://glavdecor-ekb.ru/
Bernieimist
| #
перенаправляется сюда http://maps.google.mk/url?q=https://www.google.com.na/url?sa=t&rct=j&q=&esrc=s&source=web&cd=11&ved=0ahUKEwin16O_he3KAhVDPBoKHXUcA8IQFghFMAo&url=https://iskprogress.ru/blog/geotehnicheskij-monitoring/
1win_liSa
| #
1win ваучер http://pboarders.borda.ru/?1-11-0-00000929-000-0-0-1742818701 .
SteelX1
| #
Эксклюзивное интервью с https://buhcentr-vlg.ru экспертом по финансам.
FNDaviditabe
| #
darknet market dark market list
Kxyufaita
| #
dark web link dark websites
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности https://glavsnab-gbi.ru/
Rabyitabe
| #
darkmarket 2025 https://github.com/darkwebsitesyhshv/darkwebsites – dark markets
Rezaimotcor
| #
Современные МФО предлагают займ без отказа под ПТС автомобиля без изъятия транспорта. Это удобно, если авто необходимо для работы или повседневных дел. Деньги выдаются быстро, без проверки кредитной истории.
1win_ddSa
| #
1win скачать kg https://pboarders.borda.ru/?1-11-0-00000929-000-0-0-1742818701/ .
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы https://glavtorgmsk.ru/
Night_Fang
| #
Практические советы для работы с https://kraft-leasing.ru финансами.
Wolf458
| #
Как сэкономить на финансах: https://24ufa-service.ru
DonDonfaita
| #
darknet market lists https://github.com/nexusdarkrtv1u/nexusdark – darknet site
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности https://globus-balakovo.ru/
mostbet_qroa
| #
мастбет https://shorts.borda.ru/?1-18-0-00000397-000-0-0 .
Gilbertdic
| #
код вилочного погрузчика
MarkNOlaf
| #
darknet drugs dark market list
Pingrutty
| #
dark web market list https://github.com/aresonioncq0a7/aresonion – bitcoin dark web
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы — https://glwin.ru/
Volodyanaf
| #
dark market list dark market onion
1win_rzSa
| #
1win. com 1win. com .
DarkV5
| #
https://astfin.ru Коллеги, поделитесь опытом по финансам!
Storm_Protocol
| #
Полезные сервисы для работы с https://leasing-box.ru финансами.
Donaldlaf
| #
darknet links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darkmarket link
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации — https://goncharoff-victory.ru/
mostbet_bdoa
| #
мостбет скачать казино мостбет скачать казино .
Gilbertdic
| #
экскаватор погрузчик jcb 4cx в россии
WilliamIRrutty
| #
dark market link darkmarket list
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы — https://gor-bur.ru/
CyberHunter
| #
Подборка ресурсов по финансам: https://sarros.ru
Toliknaf
| #
dark markets https://github.com/abacuslink4jjku/abacuslink – dark web sites
SilverX4
| #
Лучшие инструменты для работы https://gk-transstroy.ru с финансами.
Kxyufaita
| #
dark web drug marketplace dark market onion
hMustafaVep
| #
Пионы — это всегда стильно и элегантно!
пионы купить
Iariorusp
| #
Приобрести документ университета можно в нашем сервисе. advokatitrebinje.com
Jameswew
| #
Новруз может быть объявлен официальным праздником в Турции https://x.com/Fariz418740/status/1904268540363915402
WilliamSmamb
| #
профильные стальные трубы proftruba-moscow.ru с полимерным покрытием. защита от коррозии для использования в агрессивных средах.
mostbet_esoa
| #
мостбет скачать казино http://www.shorts.borda.ru/?1-18-0-00000397-000-0-0 .
Cazrpwe
| #
Добрый день!
Мы изготавливаем дипломы психологов, юристов, экономистов и любых других профессий по выгодным тарифам. Стоимость может зависеть от определенной специальности, года выпуска и образовательного учреждения: п»їtutdiploms.com/
Rabyitabe
| #
dark market link https://github.com/darkwebsitesyhshv/darkwebsites – darknet markets 2025
Uazrbny
| #
Приветствую!
Для некоторых людей, заказать диплом университета – это необходимость, удачный шанс получить достойную работу. Однако для кого-то – это понятное желание не терять время на учебу в университете. С какой бы целью вам это не понадобилось, наша компания готова помочь. Максимально быстро, профессионально и по доступной цене изготовим документ любого года выпуска на настоящих бланках со всеми необходимыми печатями.
Ключевая причина, почему многие люди покупают документ, – желание занять хорошую работу. Предположим, знания дают возможность кандидату устроиться на привлекательную работу, а подтверждения квалификации нет. Если для работодателя важно наличие “корочек”, риск потерять вакантное место довольно высокий.
Приобрести документ о получении высшего образования вы имеете возможность в нашем сервисе. Мы предлагаем документы об окончании любых ВУЗов России. Вы сможете получить необходимый диплом по любой специальности, включая документы СССР. Даем 100% гарантию, что при проверке документа работодателем, никаких подозрений не возникнет.
Ситуаций, которые вынуждают приобрести диплом о среднем образовании достаточно. Кому-то очень срочно потребовалась работа, и нужно произвести впечатление на начальство на протяжении собеседования. Некоторые желают устроиться в большую компанию, чтобы повысить собственный статус в обществе и в дальнейшем начать свое дело. Чтобы не тратить массу времени, а сразу начать удачную карьеру, используя имеющиеся навыки, можно заказать диплом в интернете. Вы станете полезным для социума, получите денежную стабильность максимально быстро и просто- аттестат купить за 9 класс
Overlord5772
| #
Полезные сервисы для работы с финансами: https://nogataprofi.ru
JeffryZeque
| #
Портал для любознательных https://vseduxi.ru/sekretnyy-yazyk-podiuma-chto-daet-angliyskiy-fashion-guru/ Полезная информация, тренды, обзоры, советы и много вдохновляющего контента. Каждый день — повод зайти снова!
Sazrqan
| #
Приобрести диплом о высшем образовании !
Покупка диплома ВУЗа РФ в нашей компании – надежный процесс, ведь документ заносится в реестр. При этом печать выполняется на специальных бланках, установленных государством. Заказать диплом о высшем образовании arenadiplom24.online/vuzy/uralskij-finansovo-yuridicheskij-institut
Sazremn
| #
Приобрести диплом университета по невысокой стоимости возможно, обратившись к надежной специализированной фирме. Мы оказываем услуги по производству и продаже документов об окончании любых университетов России. Заказать диплом о высшем образовании– diploma-groups24.ru/kupit-diplom-v-gorode/gelendzhik.html
Night160
| #
Видеоуроки по финансам: https://f-m-a.ru
Walker1729
| #
Предлагаю подискутировать о финансах: https://aspire-capital.ru
Pingrutty
| #
onion dark website https://github.com/aresonioncq0a7/aresonion – dark markets
DonDonfaita
| #
darknet markets 2025 https://github.com/nexusdarkrtv1u/nexusdark – tor drug market
MarkNOlaf
| #
dark market url tor drug market
Williemem
| #
Добрый день!
Один из популярных вопросов среди пользователей — как выбрать лучший ноутбук для работы или игр? На информационных сайтах можно найти подробные обзоры самых актуальных моделей. Здесь рассказывается о характеристиках ноутбуков, таких как процессор, память, графика и другие важные параметры. Также рассматриваются аспекты, которые важно учитывать при выборе устройства, будь то для учебы, работы или игр. Эти сайты помогут вам выбрать оптимальный ноутбук в зависимости от ваших нужд.
Как преодолеть страх публичных выступлений? Начните с практики перед зеркалом или перед друзьями. Чем больше вы будете тренироваться, тем уверенно будете себя чувствовать на сцене.
Больше информации по ссылке – https://asimutaero.ru/
как сделать черный краситель, видео интересные истории, лучшие недорогие наушники беспроводные
интересные факты фиджи, лучшие ироничные детективные сериалы, интересные факты про вулканы
Удачи!
SilverStriker
| #
Как вам этот ресурс о финансах? https://yuvelir-versal.ru
Volodyanaf
| #
darkmarkets darknet drug market
Donaldlaf
| #
dark markets https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark web markets
DragonZ9
| #
Простой язык о финансах: https://1centr-prodazhi.ru
EagleX3
| #
Секреты https://success-finance41.ru профессионалов в финансах.
Toliknaf
| #
dark web marketplaces https://github.com/abacuslink4jjku/abacuslink – darknet market list
Petuvhnig
| #
На сайте https://kometagaming.site/ мы собрали рейтинг официальных букмекерских контор с лицензией, которые позволяют легально делать ставки на спорт и где будет гарантирована честная игра и своевременные выплаты. Мы решили этот вопрос и собрали лучшие компании в топ букмекерских контор. Лучшие официальные букмекерские конторы для ставок на спорт – это полезная информация для тех, кто хочет начать играть, а также получать различные бонусы и легкий вывод денег.
PhantomStriker
| #
Коллеги, поделитесь опытом https://lombardpysh.ru по финансам!
Robertemele
| #
Интернет-магазин дисков и шин Disks Tyres занимается розничной продажей шин и дисков на легковые автомобили грузовые авто и внедорожники https://disks-tyres.ru/
Pingrutty
| #
dark market onion https://github.com/aresonioncq0a7/aresonion – best darknet markets
rokogokag
| #
Ресурс torentino.org скачать игры через торрент вам предлагает. Предлагаем целый ряд конкурентных преимуществ. Наши игры постоянно обновляются. Комфортная система поиска дает возможность быстро найти то, что нужно. Насладитесь возможностями новейшей индустрии развлечений. Ищете скачать игру 2015? Torentino.org – здесь представлены разные игры, которые помогут скоротать время. Мы распределили их по множеству категорий, чтобы вам было удобно отыскать и скачать игру. Каждый файл обязательно проверяется на наличие вирусов. Оперативных вам загрузок!
Cyber48
| #
Предлагаю подискутировать о финансах: https://winfindmd.ru
RichardFub
| #
В «Фабрике остекления» мы предлагаем широкий выбор оконных конструкций для любых нужд. Наши пластиковые окна предназначены для остекления балконов, лоджий, квартир и загородных домов. Мы работаем только с высококачественными профильными системами, что позволяет создавать окна, которые идеально подходят для местных климатических условий. Наши окна обеспечивают отличную защиту от холода и шума, создавая комфортную атмосферу в вашем доме.
Master9344
| #
Шутки про https://spb-lombard.ru финансы.
Harryskync
| #
Добрый день!
Как создать здоровые привычки? Начните с маленьких шагов и делайте их постоянными. Используйте принцип 21 дня, чтобы новая привычка стала частью вашей жизни. Помните, что ключ к успеху — это последовательность и упорство.
Как стать более продуктивным на работе? Ставьте четкие цели, оптимизируйте рабочие процессы и избегайте отвлечений на посторонние вещи.
Больше информации по ссылке – https://asimuthaero.ru
как сделать разрешение свое, фейк паспорт как сделать, поздравления интересные
интересные бары москвы, как сделать викторину презентацию, как сделать сердце
Удачи!
Silver_Slayer
| #
Примеры успеха в финансах: https://ikmir.ru.
DonDonfaita
| #
dark web marketplaces https://github.com/nexusdarkrtv1u/nexusdark – dark markets 2025
Dragon827
| #
Какие платформы https://etcenrt.ru использовать для финансов?
Donaldlaf
| #
darknet drug market https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet websites
Matthewadare
| #
сервисы для покупки аккаунтов http://social-accounts-marketplace.ru
ArchieMog
| #
Добрый день!
Как повысить свою самооценку? Работайте над собой, ставьте реальные цели и добивайтесь их. Положительные аффирмации и благодарность за то, что вы уже достигли, также помогут улучшить самооценку.
Для начинающих растяжка — это отличная возможность улучшить гибкость и подготавливать тело к интенсивным тренировкам. Растяжка для начинающих поможет предотвратить травмы и улучшить подвижность. Упражнения на растяжку подходят для всех, кто хочет улучшить свое здоровье. Занимаясь растяжкой, вы можете заметно повысить уровень своей физической подготовки.
Больше информации по ссылке – https://ptello.ru/
интересные факты про шолохова, как сделать содовый раствор, как пишется поинтересней
задачи на логику интересные, как сделать лайфхак, рейтинг лучших вкладышей беспроводных наушников
Удачи!
Edwardadusy
| #
биржа для продажи аккаунтов маркетплейс готовых аккаунтов
Striker7014
| #
Лучшие инструменты для работы с финансами: https://eurekainvest.ru
Franktaf
| #
zooma casino зеркало
Blue420
| #
Всем привет! Подскажите, где https://limonykt.ru посмотреть информацию о финансах?
Toliknaf
| #
dark market 2025 https://github.com/abacuslink4jjku/abacuslink – dark web market list
RedGuardian
| #
Как устроен механизм финансов? https://head-bk.ru
Pingrutty
| #
darkmarket url https://github.com/aresonioncq0a7/aresonion – darknet market
Rabyitabe
| #
bitcoin dark web https://github.com/darkwebsitesyhshv/darkwebsites – dark web market links
Cyber_Walker
| #
Глубокий анализ https://sermusmax.ru финансов.
RedReaper
| #
Приветствую. Подскажите, где найти https://ic-gpp.ru информацию о финансах?
ojepaket
| #
Портал настроим-яндекс-директ.рф предоставляет квалифицированные услуги. Мы глубокими знаниями платформы Яндекс Директ располагаем. Предлагаем лучшую стратегию и учитываем особенности вашего бизнеса. Настроены на долгосрочные взаимоотношения. Ищете настройка и ведение яндекс директ? Xn—–6kcrbecsfrepkel5alfjkq8y.xn--p1ai – здесь представлены отзывы клиентов, ознакомиться с ними можете в любое время. Воспользуйтесь достоинствами ведения контекстной рекламы в Яндекс Директ с нашей командой. Поможем с максимальной эффективностью достичь ваших бизнес-целей!
Pioneer4514
| #
Лично мне помог этот https://cpk-garant.ru ресурс по финансам.
DonDonfaita
| #
darknet markets links https://github.com/nexusdarkrtv1u/nexusdark – dark market 2025
RichardFub
| #
«Фабрика остекления» предоставляет полный спектр услуг по остеклению квартир, коттеджей и коммерческих объектов. Мы используем только современные профильные системы и стеклопакеты, чтобы гарантировать тепло, уют и долговечность. Наши окна обеспечат отличную шумоизоляцию и защиту от внешних факторов.
StormWarden
| #
Какие платформы использовать для финансов? https://fs60.ru
kusegpor
| #
Компания «Отопление и водоснабжение» предоставляет профессиональные услуги по монтажу отопления. Мы предлагаем доступные цены. Сотрудничаем с ведущими изготовителями оборудования. Готовы создать теплый дом для вас и вашей семьи. Ищете монтаж водоснабжения? Santex-uslugi.ru – портал, где вы заявку можете оставить. Работаем с новейшим оборудованием и передовыми технологиями. Используем высококачественные материалы, чтобы гарантировать долговечность вашей системы отопления. Вы можете проконсультироваться с нами по интересующим вопросам.
Pingrutty
| #
darknet markets https://github.com/aresonioncq0a7/aresonion – darknet markets url
DragonV9
| #
Простой язык о финансах: https://ligaexpert.ru
Toliknaf
| #
darkmarket 2025 https://github.com/abacuslink4jjku/abacuslink – darkmarket url
Steel10
| #
Памятка https://larecgold.ru для работы с финансами.
Franktaf
| #
zooma casino официальный
Rabyitabe
| #
darkmarket list https://github.com/darkwebsitesyhshv/darkwebsites – darkmarket list
BlueWalker
| #
Преимущества разных https://naabinvest.ru подходов к финансам.
Vortex70
| #
Полное руководство по финансам от А до Я: https://ipoteka-sf.ru
DonDonfaita
| #
dark web market urls https://github.com/nexusdarkrtv1u/nexusdark – darkmarkets
Eagle_Slayer
| #
Лично мне помог https://rozalya.ru этот ресурс по финансам.
Samuellok
| #
Здравствуйте!
Купить сигареты Кент Блю 1000 (мрц200) — это выбор для тех, кто предпочитает мягкие и легкие сигареты с свежим вкусом. Сигареты Кент Блю 1000 подарят вам освежающее курение с легким и приятным ароматом. Заказывайте сигареты Кент Блю 1000 (мрц200) с доставкой на дом. Эти сигареты идеально подходят для любителей легких и свежих ароматов. Сделайте покупку и наслаждайтесь курением!
Купить сигареты Кент кристал синий – 950 (мрц169) — это выбор для тех, кто ищет более мягкие сигареты с необычным вкусом. Сигареты Кент кристал синий 950 подарят вам невероятно гладкое курение с легкими нотками свежести. Вы можете купить сигареты Кент кристал синий – 950 (мрц169) с быстрой доставкой в любой уголок города. Они подойдут для тех, кто хочет наслаждаться качественными сигаретами каждый день. Закажите Кент кристал синий прямо сейчас!
Лучшие сигареты по ссылке – https://t.me/sigaretikupit_ru/, канал в telegram – @sigaretikupit_ru
chapman сигареты classic купить, Купить сигареты Минск нано 850, мальборо сигареты зеленые купить
Сигареты Ротманс нано 950 (мрц185), купить сигарету электронную в украине, Купить сигареты Минск компакт син/красн – 900
Удачи за сигареткой!
BillyWes
| #
Добрый день!
Вопросы выбора и использования умных часов часто обсуждаются на информационных сайтах. Здесь можно найти обзоры популярных моделей, таких как Apple Watch, Samsung Galaxy Watch и других. Сайты подробно рассказывают, как выбрать умные часы в зависимости от их функций и совместимости с другими устройствами. Также рассматриваются вопросы о том, как использовать умные часы для мониторинга здоровья и фитнес-активности. Эти ресурсы помогут вам выбрать идеальные умные часы для вашего стиля жизни.
На информационных сайтах можно найти много полезной информации о различных гаджетах для путешественников. В статьях рассматриваются устройства, которые помогут вам в поездках, такие как портативные зарядные устройства, наушники с шумоподавлением и другие аксессуары. Сайты предлагают советы по выбору гаджетов, которые облегчат вашу жизнь в дороге. Также рассматриваются вопросы о том, как выбрать гаджеты для комфортного путешествия и работы в пути. Эти ресурсы помогут вам собрать идеальный набор гаджетов для путешествий.
Больше информации по ссылке – https://onello.ru/
как сделать вкусно макароны, фильмы комедии интересные, как сделать повышенное давление
самый страшный фильм психологический ужасов, почему телефон стал быстро разряжаться, как маникюр сделать дома
Удачи!
Gilbertdic
| #
обзор фронтального погрузчика
Pingrutty
| #
darknet websites https://github.com/aresonioncq0a7/aresonion – darkmarket
EaglePioneer
| #
Какие изменения в финансах? https://dominvestment.ru
Donaldlaf
| #
darknet markets https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet market lists
Robertemele
| #
Интернет-магазин дисков и шин Disks Tyres занимается розничной продажей шин и дисков на легковые автомобили грузовые авто и внедорожники https://disks-tyres.ru/
Jeffreymak
| #
Добрый день!
Как научиться планировать бюджет? Определите свои доходы и расходы, следите за расходами и ставьте цели на экономию, чтобы управлять своими финансами эффективно.
Как научиться быть терпеливым? Работайте над собой, примите, что не все можно получить сразу, и старайтесь сохранять спокойствие в сложных ситуациях.
Больше информации по ссылке – https://tyrtsia.ru
гендер пати как сделать, айти профессии самые высокооплачиваемые, интересные русские сериалы новые
как сделать кудри африканские, как сделать правильно тефтели, как сделать поджарку
Удачи!
DarkBlade
| #
https://arenda-lizing.ru Коллеги, поделитесь опытом по финансам!
GeorgeTom
| #
Source:
– casinorushplay.org
Toliknaf
| #
darkmarket url https://github.com/abacuslink4jjku/abacuslink – dark markets 2025
EagleV3
| #
Нюансы https://kpk-mbk.ru работы с финансами.
OmegaV6
| #
https://teknos59.ru Чек-лист для работы с финансами.
KurersSnums
| #
На сайте http://umk-trade.ru изучите новости автомира, ознакомьтесь с тест-драйвами автомобилей различных марок, чтобы принять правильное решение о покупке. Вы узнаете обо всех технических характеристиках, особенностях каждой модели. Есть тест-драйв автозапчастей Hordano, шин и дисков, которые крайне важны в эксплуатации автомобиля. Кроме того, есть информация по поводу того, стоит ли брать Мерседес в аренду, какие преимущества вы получите от этого. Любопытные, увлекательные статьи появляются регулярно.
Rabyitabe
| #
darknet sites https://github.com/darkwebsitesyhshv/darkwebsites – darknet markets 2025
FNDaviditabe
| #
darknet markets links dark market
Phantom30
| #
Полное руководство по финансам от А до Я: https://debetum-online.ru
Pingrutty
| #
dark market 2025 https://github.com/aresonioncq0a7/aresonion – darknet links
Phantom161
| #
Как считаете, где лучше всего изучить финансы? https://tandemkreditnn.ru
DonDonfaita
| #
darknet market https://github.com/nexusdarkrtv1u/nexusdark – darknet markets onion
AlphaX9
| #
https://buhusfin.ru Перспективы финансов.
Overlord4086
| #
Кто-нибудь ориентируется в финансах? https://finansist11.ru Куда зайти?
MarkNOlaf
| #
dark markets 2025 tor drug market
MartyRow
| #
Здравствуйте!
На информационных сайтах для любителей технологий можно найти подробные руководства по сборке и настройке ПК. В статьях пошагово объясняется, как выбрать компоненты для компьютера, а также как собрать его самостоятельно. Сайты предлагают советы по выбору процессора, видеокарты, материнской платы и других деталей. Также представлены рекомендации по оптимизации работы ПК для игр или профессиональной деятельности. Это полезная информация для тех, кто хочет создать персональный компьютер, подходящий под его нужды.
Если вы увлекаетесь фотографией, информационные сайты предлагают советы по выбору гаджетов для фотосъемки. В статьях рассматриваются камеры, объективы, дронов и другие устройства, которые помогут вам делать качественные фотографии. Сайты предлагают рекомендации по выбору подходящего оборудования для разных типов съемки, будь то пейзаж, портрет или макросъемка. Эти ресурсы помогут вам создать лучшие снимки с помощью новейших технологий.
Больше информации по ссылке – https://azimuhtaero.ru
как сделать самодельную бомбочку, как сделать атласную розу, как сделать объемную
интересные книги скачать бесплатно, как сделать колонку блютуз, как сделать авито про
Удачи!
Donaldlaf
| #
dark market url https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark market list
WilliamIRrutty
| #
darknet market list tor drug market
SilverX9
| #
Шутки про финансы: https://jet-ready.ru
MichaelMep
| #
Пластиковые окна от «Окна Мастер СПб» – это максимум комфорта и минимум хлопот. Мы используем лучшие профильные системы и исключаем любые недочёты на производстве.
Toliknaf
| #
darknet sites https://github.com/abacuslink4jjku/abacuslink – onion dark website
RobertDaw
| #
Non Gamstop casinos no deposit deals are a game-changer—wow!
non gamstop casinos 2020
Voyager4412
| #
Обязательно посмотрите этот материал о финансах: https://cf-mfo.ru
ShadowV1
| #
https://afk-zalog.ru Технические нюансы финансов.
Volodyanaf
| #
dark market list dark market
FNDaviditabe
| #
darknet markets links dark market list
Xafonunors
| #
На сайте https://strah.shop/ почитайте информацию на тему финансов, страховки и многого другого. Вы узнаете о том, как простимулировать семьи к тому, чтобы они повысили рождаемость и захотели родить второго, третьего ребенка. Есть советы по поводу того, как защитить себя от карманников. Есть публикации на тему того, как правильно и грамотно управлять своими долгами. Здесь постоянно появляются свежие записи, которые будут интересны всем. На сайте также имеются статьи на другие темы, интересные россиянам.
HintoBor
| #
Looking for the best cryptocurrency exchange rates? Discover the Coinxes cryptocurrency exchange https://coinxes.io/ – the best rates, the lowest commissions. The Coinxes platform is a digital exchange that allows users to buy and sell cryptocurrencies from anywhere in the world. Unlike a regular exchange, it boasts a large number of useful features.
Pingrutty
| #
darkmarket list https://github.com/aresonioncq0a7/aresonion – darknet drug store
1win_ovor
| #
1win официальный сайт вход http://boardwars.forum24.ru/?1-10-0-00000406-000-0-0 .
Rabyitabe
| #
dark websites https://github.com/darkwebsitesyhshv/darkwebsites – dark web drug marketplace
Silver677
| #
Давайте обсудим о финансах: https://lombard26.ru.
1win_baor
| #
1win войти https://boardwars.forum24.ru/?1-10-0-00000406-000-0-0 .
DonDonfaita
| #
darknet market https://github.com/nexusdarkrtv1u/nexusdark – bitcoin dark web
Hunter6748
| #
Последние обновления по https://zaympts42.ru финансам.
MarkNOlaf
| #
dark web drug marketplace darknet drug store
1win_tjsr
| #
1вин кыргызстан 1win6012.ru .
1win_vwSn
| #
1вин войти http://1win814.ru/ .
Omega_Warden
| #
https://bkrural.ru Забавные моменты в финансах.
1win_cfor
| #
сайт 1win официальный сайт вход http://www.boardwars.forum24.ru/?1-10-0-00000406-000-0-0 .
mostbet_ampr
| #
мостбет казино войти http://tagilshops.forum24.ru/?1-4-0-00000205-000-0-0/ .
DonnellAlock
| #
Крайне советую сантехник Краснодар
MichaelMep
| #
Компания «Окна Мастер» из Санкт-Петербурга работает по принципу: “качество прежде всего”. Мы не допускаем брака и гарантируем точную геометрию каждого окна.
Steel932
| #
Практическое https://triumfi.ru применение финансов.
1win_rysr
| #
1вин вход с компьютера https://1win6012.ru .
1win_qfSn
| #
1win букмекер http://www.1win814.ru .
Donaldlaf
| #
darknet markets url https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark market onion
WilliamIRrutty
| #
dark web market dark market
gejufdlycle
| #
Ищете магазин лепнины? Piterra.ru/catalog/lepnina/ можно для разного стиля интерьера. Современная лепнина органично вписывается в пространство, оживляя стены, потолки, скрывая провода и электрические розетки, а также придавая выразительности помещению с помощью различных оттенков краски.
mostbet_chpr
| #
most bet http://www.tagilshops.forum24.ru/?1-4-0-00000205-000-0-0 .
mostbet_fnol
| #
mostbet официальный сайт mostbet официальный сайт .
FNDaviditabe
| #
dark markets 2025 dark web marketplaces
Kxyufaita
| #
tor drug market dark markets
уборка после ремонта спб
| #
Безупречный клининг в Санкт-Петербурге: Ваш неизменный партнер по наведению порядка!
Наша компания более 10 лет оказывает услуги клининга в Санкт-Петербурге и зарекомендовала себя как надежный партнер. Мы гордимся опытом нашей команды, которая состоит из высококвалифицированных специалистов, прошедших обучение и имеющих все необходимые сертификаты. Для нас важна не только оперативность, но и наивысшее качество выполняемых услуг.
Не ждите, пока грязь и беспорядок станут катастрофой. Дайте нам шанс вернуть вашему пространству чистоту и порядок! Узнайте всё о наших услугах и оставьте заявку на сайте : https://uberu21.ru/
Toliknaf
| #
dark web market list https://github.com/abacuslink4jjku/abacuslink – dark market link
Nomad9184
| #
https://avtolombard-odin24.ru Доступно объяснено о финансах.
Pingrutty
| #
dark market https://github.com/aresonioncq0a7/aresonion – darknet markets onion address
mostbet_eyol
| #
мостбет скачать бесплатно mostbet795.ru/ .
BlueZ9
| #
Интерактивный курс по финансам: https://spbbulat.ru.
1win_lysr
| #
1 win pro http://1win6012.ru/ .
Michaelkep
| #
https://rowe-av.ru/
1win_ofSn
| #
1win ракета https://1win814.ru/ .
mostbet_pmpr
| #
мостбет вход http://www.tagilshops.forum24.ru/?1-4-0-00000205-000-0-0 .
Omega601
| #
https://alfa55.ru Креативное решение к финансам.
Rabyitabe
| #
dark market list https://github.com/darkwebsitesyhshv/darkwebsites – darkmarket 2025
OrlandoVor
| #
Доброго!
Какие шаги помогут повысить свою продуктивность? Начинайте день с составления плана, выделяя время на важные задачи. Разбивайте крупные задачи на более мелкие и не забывайте делать перерывы, чтобы избежать перегрузки. Технологии, такие как тайм-трекинг, также могут помочь вам оставаться организованным.
Для того чтобы сделать скриншот на айфоне, достаточно нажать две кнопки одновременно. Это простой способ запечатлеть информацию или кадр с экрана. Как на айфоне сделать скриншот — это вопрос, который многие задают. Но ничего сложного в этом нет, и вы сможете легко научиться делать снимки экрана.
Больше информации по ссылке – https://akhobeda.ru
интересный русский детектив, полис омс как сделать, крокодил игра слова интересные
российские сериалы список лучших детективные, как фото сделать прозрачным, овсяное молоко как сделать
Удачи!
hacobBar
| #
Ищете купить обои с доставкой? piterra.ru/catalog/oboi – широкий выбор обоев в СПб и МСК. В нашем каталоге вы отыщите обои на кухню, обои под покраску, обои на стену. Вы можете купить обои в интернет-магазине или выбрать в салонах по адресам. У нас на обои приемлемые цены, широкий выбор и удобная доставка. Не знаете, где найти магазин обоев в МСК либо СПб? Ознакомьтесь с нашим каталогом, сравните цены и подберите для вашего интерьера идеальные обои!
Vortex885
| #
Эксклюзивное интервью с https://exclusivebrn.ru экспертом по финансам.
mostbet_qmol
| #
мастбет mostbet795.ru/ .
Stewartiniva
| #
мобильная версия риобет зеркало riobet
DonDonfaita
| #
darkmarkets https://github.com/nexusdarkrtv1u/nexusdark – dark markets
Albertcoile
| #
продажа аккаунтов социальных сетей маркетплейсы для покупки аккаунта
казино с моментальными выводами
| #
казино с моментальными выводами
A Brief History of Elemental Analysis » Artemis Analytical
IronWarden
| #
Список инструментов по финансам: https://nacburo.ru
Jariorhje
| #
Где заказать диплом по нужной специальности?
Наша компания предлагает быстро и выгодно приобрести диплом, который выполнен на бланке ГОЗНАКа и заверен печатями, водяными знаками, подписями. Данный документ пройдет лубую проверку, даже с использованием специального оборудования. Решите свои задачи максимально быстро с нашим сервисом. Приобрести диплом ВУЗа! paning.flybb.ru/viewtopic.php?f=7&t=1853
Mazrwss
| #
Мы предлагаем дипломы любой профессии по доступным ценам. Стараемся поддерживать для заказчиков адекватную политику цен. Для нас важно, чтобы дипломы были доступными для большого количества граждан.
Приобретение диплома, подтверждающего обучение в университете, – это выгодное решение. Купить диплом любого ВУЗа: okdiplom.ru/kupit-diplom-novogo-obraztsa-9/
Sazrolx
| #
Студенческая жизнь прекрасна, пока не приходит время писать диплом, как это случилось со мной. Не стоит отчаиваться, ведь существуют компании, которые помогают с написанием и защитой диплома на высокие оценки!
Сначала я искал информацию по теме: купить дипломы в ставрополе недорого, купить оригинальность диплома, купить программное обеспечение для заполнения дипломов спо, куплю диплом высшего образования форум, куплю диплом о высшем образовании читать, а потом наткнулся на diplomybox.com/diplom-magistra
ShadowVoyager
| #
Доступно объяснено о финансах: https://dengiyest.ru
Alpha_Sentinel
| #
Мнение https://pravohelp23.ru специалистов о финансах.
Donaldlaf
| #
dark websites https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark web markets
Timothyner
| #
Zalogowalem sie na strone nayttely i od razu tego pozalowalem! Partie bez przerwy zacinaja sie, menu jest koszmarnie nieintuicyjny, a reklamy sa wszedzie. Mozna pomyslec, jak gdyby serwis byla zaniedbana – zero wsparcia, usterki na kazdej stronie, a dzialanie jest koszmarnie wolne. Informacji praktycznie brak, a sa one nieaktualne. Jesli chcesz bez problemow grac w brydza, wybierz alternatywne serwisy – tu nie znajdziesz nic wartosciowego!
Vortex_Slayer
| #
Повышение эффективности через финансы: https://yamal-stroy-invest.ru
Edwardwef
| #
Exponent Finance is redefining DeFi lending by providing secure, transparent, and high-yield investment solutions. Through smart contract-powered lending pools, Exponent Finance DeFi platform allows users to borrow and lend crypto assets with optimal efficiency and minimal risk. Whether you’re looking to earn passive income through staking or access instant liquidity, Exponent Finance offers a decentralized, non-custodial, and user-friendly solution to meet all your financial goals in the crypto ecosystem. https://exponent.ink
Dark_Walker
| #
Давайте обсудим о финансах: https://mikfinance.ru
Scottaxono
| #
Flaunch is the leading blockchain gaming launchpad, designed to help game developers and investors thrive in the Web3 gaming ecosystem. By offering secure token launches, NFT integrations, and decentralized crowdfunding, Flaunch enables game creators to fund, develop, and scale their projects with full transparency and community-driven support. Whether you’re a developer or an investor, Flaunch provides the tools to connect and grow in the blockchain gaming space. https://flaunch.tech
nvblfurn
| #
https://www.snabco.ru/fw/inc/geometry_dash__pogruzghenie_v_mir_geometricheskih_priklucheniy.html обвести рисунок по точкам для детей 7 8 лет
DonaldPauch
| #
Купить бонг
KennethGab
| #
Cytonic is revolutionizing blockchain security with advanced cybersecurity solutions tailored for Web3 applications. By integrating decentralized encryption, AI-powered threat detection, and smart contract auditing, Cytonic ensures maximum protection against cyber threats. Whether you’re securing DeFi protocols, NFTs, or enterprise blockchain systems, Cytonic’s cutting-edge security technology provides the highest level of data integrity and protection. https://cytonic.cc
Rabyitabe
| #
darknet drug store https://github.com/darkwebsitesyhshv/darkwebsites – dark markets 2025
Blue142
| #
Секреты профессионалов в финансах: https://biznes-curs.ru
DonDonfaita
| #
darknet markets onion address https://github.com/nexusdarkrtv1u/nexusdark – dark market url
DragonBlade
| #
Вебинар о финансах: https://stgold.ru.
AlbertSom
| #
Откровенно, сначала было ощущение, — ну, типичное казино. А потом незаметно.
Интерфейс удобный, всё реактивное, не нужно путаться в кнопках, чтобы запустить игру.
Темы очень разнообразные — от знакомых до тех, где глаза разбегаются.
Финансы заливал — зашли сразу, забирал — получил быстро, аж удивился, что всё чётко.
Техподдержка тоже не подводят: обратился, через короткое время уже всё решили.
В целом, казино Вулкан — неожиданно хорошая площадка. Не идеально, но честно, и местами даже весело. Главное — держать в голове, что всё должно быть в меру.
Pingrutty
| #
darknet market links https://github.com/aresonioncq0a7/aresonion – darkmarket link
GhostV9
| #
Малоизвестные фишки финансов: https://otdelresheniy.ru
ChrisWrard
| #
Elara Finance is transforming decentralized lending by offering secure, transparent, and flexible crypto loan solutions. Built on blockchain technology, Elara Finance enables users to borrow and lend digital assets seamlessly without intermediaries. With low-interest rates, automated smart contracts, and a permissionless DeFi environment, Elara Finance is making crypto lending accessible and profitable for investors worldwide. https://elara.ink
ZavuhgoW
| #
Топ букмекерских контор для ставок на сайте https://pkrdomgaming.site/ – это лучшие официальные букмекерские конторы для ставок на спорт. Гарантирована честная игра и своевременные выплаты. Мы решили этот вопрос и собрали лучшие компании в топ букмекерских контор. Лицензия, отличные коэффициенты, простота ввода и вывода денег.
Donaldlaf
| #
darknet market links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark market url
Iron415
| #
https://logalk.ru Как сэкономить на финансах.
RafaelFer
| #
DEQ Finance is revolutionizing decentralized trading by offering a seamless, secure, and efficient crypto exchange experience. Built with cutting-edge blockchain technology, DEQ Finance provides traders with fast transaction speeds, deep liquidity, and a transparent trading environment. Whether you’re a beginner or a professional trader, DEQ Finance delivers high-performance DeFi solutions tailored to modern trading needs. https://deq.li
Dragon_Guardian
| #
Видел прогнозы, что Финансы будет главным трендом. Соглашусь? https://a-nalogi-a.ru
AllenTaulk
| #
Ремонт помещений https://msksfera.ru/remont-gostinicz/ под ключ: офисы, отели, склады. Полный комплекс строительно-отделочных работ, от черновой отделки до финишного дизайна.
Toliknaf
| #
dark web drug marketplace https://github.com/abacuslink4jjku/abacuslink – dark web market urls
Light838
| #
Эксперты рекомендуют эти ресурсы https://moslomb.ru по финансам.
Light_Overlord
| #
Эксклюзивное интервью с https://char21.ru экспертом по финансам.
Rabyitabe
| #
dark web drug marketplace https://github.com/darkwebsitesyhshv/darkwebsites – darknet markets onion
Iron_Spark
| #
Как устроен механизм финансов? https://pts59.ru
Pingrutty
| #
onion dark website https://github.com/aresonioncq0a7/aresonion – darkmarkets
DonDonfaita
| #
dark markets https://github.com/nexusdarkrtv1u/nexusdark – dark websites
ThomasVep
| #
Букет выше всех похвал! Очень доволен.
заказ цветов томск с доставкой
сайт Вован казино
| #
сайт Вован казино
A Brief History of Elemental Analysis » Artemis Analytical
burexTroky
| #
С t.me/mvavada вам скучать не придется. Официальный канал проекта Вавада предлагает атмосферу настоящего казино почувствовать. В VAVADA абсолютно каждый найдет игру по душе. Скорее к нам присоединяйтесь. Участвуйте в акциях и получайте шанс на дополнительные выигрыши. Испытайте удачу! https://t.me/mvavada – здесь всегда есть что-то интересное. Следите за обновлениями канала. В Вавада вас слоты с захватывающим геймплеем и неповторимым дизайном ждут. Играйте уже сегодня. Побеждайте вместе с VAVADA. Меняйте свою жизнь к лучшему!
RedX6
| #
Полное руководство по финансам от А до Я: https://kreditorium21.ru
SilverHacker
| #
Оптимизация https://light-gold.ru процессов через финансы.
Donaldlaf
| #
dark markets https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet links
ocenka-zagorod
| #
сбербанк оценка земельного участка http://ocenka-zagorod.ru
Storm94
| #
Полное руководство по финансам от А до Я: https://posuda-vo-dvore.ru
RobertDaw
| #
This non Gamstop casino has unreal graphics—totally blown away!
boku casino no gamstop
Toliknaf
| #
dark web market https://github.com/abacuslink4jjku/abacuslink – darknet marketplace
Kxyufaita
| #
darknet drug links tor drug market
WilliamIRrutty
| #
dark web sites dark web market urls
FNDaviditabe
| #
darknet sites dark markets 2025
Walker2835
| #
Советую качественный ресурс по https://proauto42.ru финансам.
GeorgeTom
| #
Source:
luckyspincasino.org
Master7006
| #
Как https://komisionka26.ru не сдаться при изучении финансов.
DavidAnync
| #
типография печать заказ https://tipografiya-poligrafiya2.ru
Aaronovexy
| #
типография печатной типография цены
Pingrutty
| #
dark web link https://github.com/aresonioncq0a7/aresonion – darknet sites
Volodyanaf
| #
darknet market darkmarkets
Rabyitabe
| #
darknet markets https://github.com/darkwebsitesyhshv/darkwebsites – onion dark website
fuel pressure sensor
| #
Great goods from you, man. I’ve understand your stuff previous to and you’re just extremely magnificent.
I really like what you have acquired here, certainly like what you’re stating
and the way in which you say it. You make it enjoyable and
you still care for to keep it sensible. I can’t wait to read far more from you.
This is actually a tremendous website.
Ghost_Walker
| #
Топовый ресурс по теме финансов: https://mat-kapital124.ru
DonDonfaita
| #
darknet drug links https://github.com/nexusdarkrtv1u/nexusdark – darkmarket list
GhostOverlord
| #
Подкаст о финансах: https://pvh-tyumen.ru
RobertDaw
| #
Non Gamstop casinos UK are my new go-to—super fun!
non uk casinos not on gamstop
Dark_Spark
| #
Ошибки новичков в финансах: https://taxandlex.ru
NewRetroCasino_bael
| #
New Retro Casino https://newretromirror.ru/ .
Kxyufaita
| #
dark web drug marketplace darknet markets links
WilliamIRrutty
| #
darkmarket url darknet market
Donaldlaf
| #
darknet markets url https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet drug links
LightZ6
| #
Эксклюзивное интервью с экспертом по финансам: https://lombardiago.ru
FNDaviditabe
| #
darknet site darknet sites
GilbeelUNLAT
| #
На сайте https://advelma.ru изучите огромное количество объявлений на самую разную тематику, включая автотранспорт, растения и животные, мобильные телефоны, мебель, интерьер и многое другое. Здесь также вы сможете и познакомиться с мужчиной либо женщиной. Всего на сайте представлено почти 150 рубрик. Если и вам нужно что-то продать, то выложите объявление на этом портале. Регулярно здесь добавляются новые, интересные предложения. Для того чтобы воспользоваться полным функционалом, нужно зарегистрироваться.
Toliknaf
| #
dark websites https://github.com/abacuslink4jjku/abacuslink – dark web link
Betfew
| #
من شروع کردم به بازی کردن کازینوی آنلاین Takbet و تا الان مشکل خاصی نداشتم.
ناوبری ساده و کاربرپسنده و بازیها سریع لود میشن.
من بیشتر از همه از بازیهای لایو کازینو خوشم اومد — آدرنالین رو بالا میبره.
همچنین یه جامعه پشتیبانی در تلگرام دارن اینجا: takbetplay](https://t.me/takbetplay) — برای دریافت بونوس و پشتیبانی خوبه.
چرخشهای رایگانش رو دوست داشتم.
کسی دیگه اینجا امتحانش کرده؟.
اگه راهنمایی یا تجربهای دارید، بگید.
Pingrutty
| #
darknet site https://github.com/aresonioncq0a7/aresonion – dark market onion
Voyager7188
| #
Ответы на частые вопросы о финансах: https://artcredit124.ru
Guardian2833
| #
https://nn7lombard.ru Преимущества разных подходов к финансам.
OmegaFang
| #
Продвинутые техники финансов: https://region-omsk.ru
Rabyitabe
| #
darknet market list https://github.com/darkwebsitesyhshv/darkwebsites – darknet markets 2025
Wubucelrip
| #
На сайте https://xn--80abeehcvkgdf2bbeq5an.xn--p1ai/ почитайте всю полезную и актуальную информацию о популярном онлайн-заведении Casino Kent. Вас ожидает сервис высокого уровня, огромное количество динамичных развлечений, щедрые бонусы, приятная атмосфера. И самое важное, что ресурс обязательно понравится как новичкам, которые только осваивают подобные заведения, так и профессионалам, которые находятся в поисках идеального места. Оно радует безупречным уровнем сервиса, можно задать вопросы, когда захотите.
Premium SEO links for sale
| #
Many thanks for the awesome facts enclosed within your blog,
what follows is a little test for your blog page viewers. Who said the following quote?
. . . .A lie gets halfway around the world before the truth has a chance to get its pants on.
DonDonfaita
| #
dark web link https://github.com/nexusdarkrtv1u/nexusdark – darkmarket url
ErnestoNow
| #
ауф казино
Warden8770
| #
Эксперты https://kgu-invest.ru рекомендуют эти материалы по финансам.
DragonSlayer
| #
Как применять финансы в бизнесе? https://finansist-msk.ru
WilliamIRrutty
| #
darknet links darknet drug links
Kxyufaita
| #
darkmarkets best darknet markets
casino Aurora
| #
Aurora Casino — это ваш идеальный выбор для захватывающих и выгодных
игр. Здесь вы найдете самые популярные игры,
включая слоты, настольные
игры и живое казино с реальными крупье.
Не забывайте про бонусы и регулярные акции,
которые сделают вашу игру еще более увлекательной.
Почему казино с бесплатными
играми стоит выбрать? Мы предоставляем высококачественные игры, защищенные
платёжные системы и гарантируем быстрые выплаты.
Наши игроки всегда могут быть уверены в честности игры и прозрачности всех
условий.
Когда лучше начинать в Aurora Casino?
Зарегистрируйтесь, чтобы уже сегодня
получить доступ ко всем возможностям и
бонусам. Вот, что вас ждет:
Вам будут предложены большие бонусы, как
только вы зарегистрируетесь.
Ежедневные акции и турниры.
Большой выбор слотов и настольных игр.
В Aurora Casino ваши мечты о больших выигрышах становятся
реальностью. https://aurora-diamondcasino.quest/
DennisgaP
| #
дешево заказать грузчиков грузчики заказать
KevinGaito
| #
голд казино
FNDaviditabe
| #
darknet markets 2025 darknet markets onion
MarkNOlaf
| #
darknet markets onion address darknet drug market
TerrySouse
| #
грузчики заказать https://toguchin.standart-express.ru
AlbertDiubs
| #
нужны грузчики для переезда нанять грузчиков
Jerrysiz
| #
Elevate your car’s functionality and appearance with our remarkable tuning options. Find the genuine ability of your car through bespoke advancements. Our professionals of experts provides incomparable quality and ingenuity. Check out our webpage https://precisionautobodyfrederick.com/ to understand extra, and start your venture towards motoring superiority today.
Volodyanaf
| #
darknet marketplace darkmarket link
Donaldlaf
| #
dark markets 2025 https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark market onion
Pingrutty
| #
dark web market list https://github.com/aresonioncq0a7/aresonion – darknet drug store
RobertDaw
| #
Best non Gamstop casino I’ve played at is a gem—cool!
are non gamstop casinos safe
PhantomZ9
| #
Эволюция развития финансов: https://mikrozaimdv.ru
Toliknaf
| #
dark web drug marketplace https://github.com/abacuslink4jjku/abacuslink – darkmarket 2025
AlphaProtocol
| #
История внедрения https://cbu63.ru финансов в компании.
Robertfus
| #
Доброго!
Как снизить уровень стресса? Для этого полезны расслабляющие практики, такие как медитация, йога или простое дыхательное упражнение. Осознанность и регулярный отдых помогут вам восстановить баланс и снизить стресс.
Как научиться прощать? Прощение — это процесс, который начинается с вас. Принятие того, что не все можно изменить, и отпускание обид помогает освободить сердце и душу. Прощение — это не только для других, но и для вашего внутреннего спокойствия.
Больше информации по ссылке – https://piano-quartet.ru
как сделать бесконечный двигатель, как сделать физраствор, как сделать карту озон
какие недорогие беспроводные наушники хорошие, турецкие сериалы самые интересные, интересные факты про крым
Удачи!
Marioskach
| #
Glory Casino app
1win_zmoi
| #
1win скачать последнюю версию 1win скачать последнюю версию .
1win_husr
| #
1win kg скачать https://www.1win6014.ru .
mostbet_lzKr
| #
казино онлайн kg kharkovbynight.forum24.ru/?1-15-0-00003047-000-0-0-1742814422 .
1win_gskl
| #
1win официальный сайт скачать http://yamama.forum24.ru/?1-11-0-00000459-000-0-0-1742818616 .
JesusIceme
| #
Привет всем!
Как развить эмоциональный интеллект? Слушайте свои эмоции и учитесь их распознавать. Работайте над эмпатией и стараясь понять чувства других. Управляйте своими реакциями в различных ситуациях.
Для начинающих растяжка — это отличная возможность улучшить гибкость и подготавливать тело к интенсивным тренировкам. Растяжка для начинающих поможет предотвратить травмы и улучшить подвижность. Упражнения на растяжку подходят для всех, кто хочет улучшить свое здоровье. Занимаясь растяжкой, вы можете заметно повысить уровень своей физической подготовки.
Больше информации по ссылке – https://alcogolizmstop.ru
как сделать основным гугл, интересные факты о атмосфере, ужасы смотреть интересные
смотреть русские интересные детективы, как сделать свою клавиатуру, интересные факты дания
Удачи!
Hacker2948
| #
Давайте обсудим о финансах: https://kombodrayv.ru
Rabyitabe
| #
darkmarket https://github.com/darkwebsitesyhshv/darkwebsites – darkmarket list
1win_wmoi
| #
1win.kg 1win.kg .
mostbet_geei
| #
aviator mostbet aviator mostbet .
1win_lzsr
| #
1win официальный сайт регистрация https://1win6014.ru/ .
mostbet_ooKr
| #
скачат мостбет скачат мостбет .
1win_lmkl
| #
1win.kg http://www.yamama.forum24.ru/?1-11-0-00000459-000-0-0-1742818616 .
WilliamIRrutty
| #
darknet sites darknet markets
DonDonfaita
| #
dark web sites https://github.com/nexusdarkrtv1u/nexusdark – darkmarket list
FNDaviditabe
| #
darknet markets links darknet market lists
WarrenWak
| #
https://tur-kazan.ru/
mostbet_edei
| #
mostbets https://www.mostbet6001.ru .
Pingrutty
| #
dark web markets https://github.com/aresonioncq0a7/aresonion – dark market list
Overlord4154
| #
Полное руководство по финансам от А до Я: https://micro-car.ru
Blue_Fang
| #
Какие платформы использовать для финансов? https://billum-zaim.ru
MatthewPeeda
| #
Здравствуйте!
Как научиться работать в команде? Будьте открытыми к сотрудничеству, уважайте мнения других и поддерживайте коллег в совместной работе.
На информационных сайтах можно найти массу полезных рекомендаций по выбору гаджетов для интимной жизни. В статьях рассматриваются устройства для повышения либидо, улучшения эрекции и снижения уровня стресса. Сайты предлагают советы по выбору гаджетов, которые помогут улучшить качество сна и настроить гормональный фон. Эти ресурсы помогут вам наладить отношения с партнером и повысить сексуальную активность.
Больше информации по ссылке – https://art-novosibirsk.ru/
как сделать ко, как сделать глазурь белую, самые лучшие зарубежные детективные сериалы
как дома сделать караоке, мужские интересные имена, сериалы детективные hbo лучшие
Удачи!
1win_dsoi
| #
ванвин https://www.1win815.ru .
1win_rdsr
| #
1вин сайт официальный 1вин сайт официальный .
mostbet_yeKr
| #
мостбет авиатор https://kharkovbynight.forum24.ru/?1-15-0-00003047-000-0-0-1742814422 .
1win_ofkl
| #
1win вход на сайт http://yamama.forum24.ru/?1-11-0-00000459-000-0-0-1742818616/ .
Donaldlaf
| #
darknet markets 2025 https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet markets
mostbet_exei
| #
мостбет войти https://www.mostbet6001.ru .
Toliknaf
| #
dark web markets https://github.com/abacuslink4jjku/abacuslink – darknet drug market
SilverSentinel
| #
Эксперты рекомендуют эти ресурсы по финансам: https://zolotopenza.ru
gates of olympus bonus buy
| #
excellent issues altogether, you just won a new reader.
What could you recommend in regards to your put
up that you simply made some days in the past?
Any positive?
Reaper2046
| #
Конкретные шаги для работы с финансами: https://spasskomi.ru
zaimpodzalogptsekbsig
| #
займ под залог автоломбард
avtolombard-11.ru/ekb.html
взять займ под залог авто
RobertDaw
| #
Best non Gamstop casino for slots is my new favorite—sweet!
non gamstop paypal casino
Volodyanaf
| #
darknet market list dark web sites
WilliamIRrutty
| #
darknet drug market darknet markets links
Kxyufaita
| #
dark market link dark markets 2025
Rabyitabe
| #
darkmarket url https://github.com/darkwebsitesyhshv/darkwebsites – dark market onion
FNDaviditabe
| #
dark markets darknet markets url
MarkNOlaf
| #
darknet markets onion dark markets
SilverV4
| #
Оптимизация процессов через финансы: https://realtor27.ru
Geraldalory
| #
Добрый день!
Как научиться ценить свое время? Устанавливайте четкие приоритеты, избегайте прокрастинации и уделяйте время только тем вещам, которые действительно важны для вас.
Как справиться с трудными ситуациями? Применяйте гибкость, ищите решения, не зацикливайтесь на проблемах и помните, что трудности — это часть пути к успеху.
Больше информации по ссылке – https://relation1.ru/
корсет как сделать, греческий йогурт как сделать, игры в стиме интересные
как из майнкрафта сделать, как можно сделать валентинку, как сделать букву а
Удачи!
Foleyelova
| #
Ищете надежных мастеров и бригаду без посредников? Посетите сайт https://poisk-pro.ru/ – это бесплатный подбор надежных исполнителей в сфере ремонта, строительства и других видов услуг от частных лиц и организаций. Более 30000 исполнителей в сфере строительства и ремонта, дизайна, бытовых услуг, ремонта техники и прочих услуг. Подробнее на сайте.
Pingrutty
| #
darknet sites https://github.com/aresonioncq0a7/aresonion – darknet market lists
DonDonfaita
| #
dark web market https://github.com/nexusdarkrtv1u/nexusdark – dark web drug marketplace
WarrenWak
| #
казань йошкар ола автобус экскурсия
Cyber42
| #
Новые подходы в финансах: https://86148.ru
Warden4981
| #
Интерактивный курс по https://metrikacredit.ru финансам.
Donaldlaf
| #
dark market 2025 https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darkmarket url
Robertpug
| #
Добрый день!
Как развить силу воли? Работайте над самодисциплиной, ставьте перед собой четкие цели и преодолевайте трудности на пути к их достижению.
Как быть более внимательным к себе? Развивайте осознанность, слушайте свои потребности и чувства. Регулярно уделяйте время себе: отдыхайте, медитируйте и заботьтесь о своем теле и разуме.
Больше информации по ссылке – https://psihfak.ru/
интересные хобби, как сделать сыр косичку, хорошие наушники маленькие беспроводные
растяжка стретчинг для начинающих, как сделать драг клик, шукшин факты интересные
Удачи!
Toliknaf
| #
darknet sites https://github.com/abacuslink4jjku/abacuslink – dark web sites
WilliamIRrutty
| #
tor drug market darkmarket link
Kxyufaita
| #
darknet marketplace dark web sites
Red122
| #
Видел прогнозы, что Финансы будет главным трендом. Соглашусь? https://topazrussia.ru
GhostX6
| #
Что https://zaim-pensioner.ru нового в финансах?
LuzehCon
| #
На сайте https://xn—-7sbbstdmdlcdhy.xn--p1ai/ вы сможете ознакомиться со всей необходимой информацией, касающейся майнинга и криптовалюты. Этот ресурс является свободным информационным и предлагает ознакомиться со всеми полезными рекомендациями. Только на этом сайте представлены самые точные и любопытные сведения, касающиеся цифровой валюты, криптовалюты и всего, что с ней связано. Представленная информация будет полезна и новеньким. Вас обязательно заинтересует и крипто библиотека, в которой есть много всего полезного.
FNDaviditabe
| #
darkmarket 2025 darknet links
Pingrutty
| #
dark web market urls https://github.com/aresonioncq0a7/aresonion – darknet markets url
https://jetton-casinoempire.lol/
| #
Welcome to Jetton Casino – a world of entertainment and big winnings.
We offer the best slot machines, table games, and exclusive bonuses
for all users. Join us and start your journey into the world of gambling right now.
What makes Jetton login special? Our players not only get access to top-tier games but
also enjoy favorable winning conditions. Here, you’ll find regular tournaments,
cashback, free spins, and personalized bonus offers.
A huge selection of slot machines, roulette, poker, and live
games.
Regular promotions and personalized offers.
Fast withdrawals and secure transactions without delays.
Competitions for the most passionate players with valuable prizes.
Discover the best gaming opportunities with Jetton Casino. https://jetton-casinoempire.lol/
Rabyitabe
| #
darkmarket https://github.com/darkwebsitesyhshv/darkwebsites – darknet drugs
RobertDaw
| #
Non-Gamstop casinos offer the best escape—highly recommend!
casino free spins no deposit not on gamstop
Marioskach
| #
Glory Casino Spribe
GregoryClile
| #
Единственный знак зодиака, который является магнитом для несчастий и проблем https://x.com/Fariz418740/status/1904737515858174078
Fang1876
| #
Подробный разбор финансов: https://skupka-noutbukov-orenburg.ru
DonDonfaita
| #
darknet market https://github.com/nexusdarkrtv1u/nexusdark – dark web sites
GeorgeTom
| #
Source:
luckyspincasino.org
Volodyanaf
| #
darknet markets onion address darkmarket
Thunder_Code
| #
Будущее финансов: https://avtodengy-pts.ru
Vocapves
| #
На сайте https://exkavatornews.ru/ почитайте содержательные и полезные материалы на тему лесозаготовительной техники. Здесь вы найдете ответ на вопрос, стоит ли приобретать китайскую технику, какое оборудование считается максимально эффективным. Есть информация и о дорожно-строительной технике и о том, как ей лучше пользоваться. Ознакомьтесь с инновационными технологиями в сфере дорожного строительства. Изучите мнения экспертов на данную тему. Регулярно здесь появляются новые, любопытные материалы, которые будут полезны.
Sentinel1907
| #
https://pal58.ru Памятка для работы с финансами.
Donaldlaf
| #
darkmarket url https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darkmarket link
WilliamIRrutty
| #
darknet markets links darknet drugs
FNDaviditabe
| #
dark web drug marketplace darknet drug store
Toliknaf
| #
darkmarket 2025 https://github.com/abacuslink4jjku/abacuslink – darknet markets links
Pingrutty
| #
dark market 2025 https://github.com/aresonioncq0a7/aresonion – dark web market urls
RebeccaTaund
| #
The Aviator game on Betika (https://betika-aviator.readme.io/reference/betika-aviator-a-thrilling-ride-to-fortune) is a high-stakes arcade-style game where instinct and speed decide your win.
The rules are easy to follow: a plane rises higher and higher, and as it flies, the multiplier increases.
Hit the stop button before the flight ends
Be greedy — and the bet vanishes.
That’s why this game is about strategy too.
With Betika, you get:
– a user-friendly interface
– fast withdrawals
– 24/7 support
– play on any device
Play anytime, anywhere — at home, at work, or on the go.
Sign up on Betika and make your first takeoff.
Betika Aviator – your flight to big wins!
NightV2
| #
Ответы на частые вопросы https://cashufa102.ru о финансах.
VortexX4
| #
Как выбрать https://mnr-1.ru лучшее решение для финансов.
Rabyitabe
| #
darknet market lists https://github.com/darkwebsitesyhshv/darkwebsites – tor drug market
RobertDaw
| #
Casino non Gamstop sites have the best promos—super fun!
non gamstop casinos uk free spins
DonDonfaita
| #
dark market link https://github.com/nexusdarkrtv1u/nexusdark – dark web sites
Cyber_Master
| #
Технические нюансы финансов: https://radiolomdv.ru
Steel253
| #
https://expert42.ru Как сэкономить на финансах.
Kxyufaita
| #
darkmarket url dark web link
WilliamIRrutty
| #
dark web market urls dark web market urls
jecifuger
| #
Ikostum предлагает купить мужской костюм недорого. В нашем интернет магазине регулярно распродажи и акции проводятся. У нас найдется костюм на любой вкус. Поможем вам с фасоном, тканью и цветом определиться, предложим чехол и галстук подходящий, а также проконсультируем о последних тенденциях. https://ikostum.com/ – тут можете посмотреть условия доставки в любое время. Мы сотрудничаем с ведущими белорусскими и российскими производителями мужской одежды. Регулярно ассортимент обновляем. Отменное качество продукции гарантируем.
MarkNOlaf
| #
darknet market dark web link
Pingrutty
| #
darknet drugs https://github.com/aresonioncq0a7/aresonion – darkmarket url
Donaldlaf
| #
darknet marketplace https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark market list
IronSlayer
| #
Базовые принципы финансов для https://raiseauto.ru новичков.
Toliknaf
| #
tor drug market https://github.com/abacuslink4jjku/abacuslink – darknet markets
Volodyanaf
| #
darknet websites dark markets 2025
Davidslate
| #
спарк ру новости Мир азартных игр в интернете непрерывно эволюционирует, и портал Спарк.ру остается надежным источником актуальных новостей и аналитики в этой сфере. Казино онлайн переживают период бурного роста, привлекая все больше пользователей благодаря удобству, разнообразию игр и привлекательным бонусам.
xutuyDaw
| #
Чат-бот на базе ИИ ChatGPT – профессиональный помощник, который может понимать запросы, поддерживать диалог и предоставлять индивидуальные решения. Цепляющий контент создавайте, продажи оптимизируйте и в 10 раз оперативнее задачи решайте. Сосредоточьтесь на самом главном. Ищете бесплатный чат gpt? Ai-gptbot.ru – сайт, где представлены отзывы, ознакомьтесь с ними. Тут рассказываем, кому чат GPT нужен. Все работает быстро и удобно. Цены удивляют приятно своей гибкостью и демократичностью. Не упускайте шанс попробовать AI-GPTbot прямо сегодня!
rexejChask
| #
Магазин «Adept Apple» продает оригинальные качественные товары с гарантией. Мы большой ассортимент аксессуаров Apple и гаджетов предлагаем. Обладаем значительным опытом и стали для тысячи клиентов надежным партнером. Стремимся ваше обслуживание сделать максимально приятным и удобным. Ищете айфон в Туле? Adeptapple.ru – здесь можете найти что-то для себя. Делаем доступной для каждого технику Apple. С удовольствием поможем подобрать устройство или аксессуар, которые идеально подойдут, именно вам. Для нас крайне важно доставить от покупки клиентам удовольствие!
NightVoyager
| #
Подборка ресурсов по финансам: https://primadonna-ekb.ru.
KevinWoutt
| #
вентиляция в частном доме
Rabyitabe
| #
darkmarket list https://github.com/darkwebsitesyhshv/darkwebsites – darknet sites
DragonVoyager
| #
Что нового в финансах? https://gu72.ru
Richardodoge
| #
маркетплейс готовых аккаунтов онлайн магазин аккаунтов
CarmenPooky
| #
Доброго!
Скриншот на айфоне можно сделать за считанные секунды, просто нажав две кнопки одновременно. Это поможет вам сохранить важную информацию или момент, который вам нравится. Для того чтобы сделать скриншот на айфоне, одновременно нажмите кнопку включения и кнопку увеличения громкости. После этого вы увидите изображение экрана в галерее.
Как научиться управлять своим временем? Определите, что для вас действительно важно, и сфокусируйтесь на этих задачах. Разделите свой день на блоки времени, чтобы избежать отвлечений. Не забывайте выделять время для отдыха и восстановления.
Больше информации по ссылке – https://ffactor.ru/
интересный разбор слова, как сделать красивый шрифт, растяжка воронеж для начинающих
как сделать лаваш мягким, какой нибудь интересный факт, мелодрамы новые интересные
Удачи!
SteelProtocol
| #
Детальный анализ финансов: https://taxoparkinmsk.ru
Vortex37
| #
Обязательно посмотрите этот https://komissionkatula.ru источник о финансах.
Kxyufaita
| #
dark market url onion dark website
WilliamIRrutty
| #
darknet market links dark web drug marketplace
DonDonfaita
| #
dark market url https://github.com/nexusdarkrtv1u/nexusdark – darknet markets onion address
IronV9
| #
Креативное решение к https://podzalog34.ru финансам.
Pingrutty
| #
darknet markets 2025 https://github.com/aresonioncq0a7/aresonion – darknet markets links
FNDaviditabe
| #
dark markets dark websites
MarkNOlaf
| #
onion dark website darkmarket url
camp-centr
| #
Продажа путёвок https://camp-centr.com/camps/type/yazykovoy.html. Спортивные, творческие и тематические смены. Весёлый и безопасный отдых под присмотром педагогов и аниматоров. Бронируйте онлайн!
SilverX6
| #
Креативное решение к финансам: https://skupka-zalog-obmen.ru
Reaper7454
| #
Эксклюзивное интервью с экспертом по финансам: https://zelta-invest.ru
Michaelgaile
| #
печать визиток стоит двухсторонняя печать визиток
AlphaX2
| #
Примеры успеха в финансах: https://pba55.ru
Overlord7102
| #
Предлагаю подискутировать о финансах: https://nadezhnii-kredit.ru
BlueX1
| #
Авторитетное мнение о финансах: https://standart-audit.ru
Donaldlaf
| #
dark market https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark market 2025
tokorunorp
| #
Бэтэрэкс – компания, предоставляющая консалтинговые, финансовые, а также юридические услуги. Поможем вам снизить затраты с Китаем на ведение бизнеса. Принимаем заказы на доставку, как от частных лиц, так и от компаний. Сопроводим в деловых поездках и на переговорах. https://mybeterex.com – тут можете форму заполнить, и мы в ближайшее время с вами свяжемся. Найдем и предложим на товар в Китае лучшую оптовую цену. С удовольствием открыть производство вам поможем. Договоримся о выпуске продукции и проведем контроль качества. Обращайтесь!
Andrewcaf
| #
Комплексное проектирование Услуги по аренде техники и строительству с логотипом компании – На протяжении многих лет компания “ТрансАвто№7” предоставляет в аренду дорожно-строительную технику, а также выполняет широкий спектр строительных и земляных работ. Мы работаем как с крупными корпорациями, так и с малым и средним бизнесом, обеспечивая высокое качество услуг по строительству дорог, благоустройству территорий и прокладке инженерных сетей.
Hunter3219
| #
Может кто знает ориентируется в https://goldmaster1.ru финансах? Куда зайти?
zaimpodptskazancoN
| #
автоломбард птс
avtolombard-11.ru/kazan.html
займ под залог автомобиля
StormNomad
| #
Как https://rassbery.ru преуспеть в финансах?
JeremyPycle
| #
https://vodkacoin.online/
Spark9061
| #
Может кто знает ориентируется в финансах? https://sarlombard.ru Куда зайти?
MicheleMouck
| #
Добрый день!
Как быть уверенным в себе? Развивайте самоуважение, принимайте себя с вашими сильными и слабыми сторонами. Каждый шаг к уверенности — это шаг к самосознанию и осознанию своей ценности.
Как научиться прощать себя? Признание своих ошибок и понимание, что все люди совершают их, помогает избавиться от чувства вины. Прощение себя — это путь к внутреннему покою.
Больше информации по ссылке – https://lala-express.ru
интересные мультики про машинки, короткие и интересные аниме, как сделать костер дейз
лучшие советские детективные сериалы, как сделать филадельфия роллы, фантастика самые интересные фильмы
Удачи!
1win_bkst
| #
скачать 1win с официального сайта 1win816.ru .
//zdjciaporno.com/
| #
Co To Jest Seks Oralny? Jaka laughter różnica
E ‘ en Seks Oralny, Fellatio, i drobniejsze Tums? Seks oralny wymaga partnera i niezależnie dd
tego, która pozycja działa najbardziej brutalnie.
Jeden z partnerów używa ust, warg lub języka, aby uformować penisa, pochwę lub orchid
arenosus partnera. Wiele sync używa move jako
sposobu na rozgrzanie się lub zjedzenie przekąski podczas stosunku, brew stymulacja ustna może zawsze odgrywać rolę w trakcie lub op, też.
Jak Działa Seks Oralny? Seks oralny daje Tobie i Twojemu partnerowi zwinny sposób na wklęsłodruk, z wyjątkiem regularnych harmonicznych san marinese.
Seks oralny joke tylko wtedy, gdy część phrase perform from nothing fishing registration. Jak
Działa Seks Oralny? Powszechnym aktem seksualnym wród sync w kadym wieku i kadej pci powszechnym aktem seksualnym
wród. Określany również jako giovanni boccaccio lub fellatio,
obejmuje ustną linię sukcesji upakowanych
komórek lub odbytu partnera. Może być dapat sama dotykowy,
jak samodzielny akt. https://git.uucloud.top/corinne49w1398
1win_bvmi
| #
1win кыргызстан http://1win6015.ru/ .
mostbet_kmKi
| #
мост бет http://maksipolinovtsu.forum24.ru/?1-1-0-00000194-000-0-0-1742815870/ .
Rabyitabe
| #
dark web sites https://github.com/darkwebsitesyhshv/darkwebsites – best darknet markets
Wolf_Blade
| #
Базовые принципы финансов для новичков: https://idance-school.ru
AlfredoFeend
| #
Odwiedzilem na witryne muodossa i przezylem szok. Szata graficzna sugeruje, jakby pochodzil sprzed dekady, dziala to dramatycznie, a merytorycznych danych prawie nie ma. Niektore sekcje sa puste, linki prowadza donikad, a wsparcie nie odpowiada. Falszywe informacje na stronie, serwis jest kompletnie bezuzyteczny! Jesli potrzebujesz sprawdzonych tresci o biegach, nie trac czasu na ten portal – ten serwis nic ci nie da.
1win_vjEn
| #
1 vin 1 vin .
Wolf_Master
| #
Изучите этот источник о финансах: https://cicred.ru
Kxyufaita
| #
darkmarket link dark market url
Iron578
| #
Факты и вымысел о финансах: https://armavir-avtolombard.ru
MatthewBoods
| #
Noon Capital is revolutionizing real estate crowdfunding by offering secure, decentralized, and high-yield investment opportunities. Through blockchain-powered property financing, Noon Capital investment platform allows investors to participate in fractional real estate ownership, earn passive income, and diversify their portfolios with full transparency and security. Whether you’re an institutional investor or an individual seeking real estate-backed DeFi solutions, Noon Capital provides a seamless and profitable investment experience. https://noon.ad
FrancisSah
| #
Kyros Finance is redefining the DeFi investment landscape by offering secure, scalable, and high-yield crypto solutions. With a focus on decentralized financial tools, Kyros Finance provides users with staking, lending, and automated yield farming strategies to maximize returns. Whether you’re a retail investor or an institutional participant, Kyros Finance ensures efficient, transparent, and secure access to the world of decentralized finance. https://kyros.ink
1win_qqst
| #
1win букмекер https://www.1win816.ru .
Volodyanaf
| #
darknet drugs darknet drugs
Pingrutty
| #
darkmarket url https://github.com/aresonioncq0a7/aresonion – darknet market list
1win_gqmi
| #
1win кейсы http://1win6015.ru/ .
mostbet_wmKi
| #
mostber http://maksipolinovtsu.forum24.ru/?1-1-0-00000194-000-0-0-1742815870 .
Ghost_Voyager
| #
Подробный разбор финансов: https://gipart52.ru
Thomasvak
| #
Официальный веб-сайт казино Рояль предлагает изысканную обстановку для поклонников азартных игр. С его элегантным дизайном и элитным выбором игр, Рояль является превосходным местом для тех, кто обожает качественное азартное развлечение. Исследуйте многочисленные игровых автоматов и настольных игр на основном веб-сайте royalcasino-slots.com/ru/.
Казино Рояль обеспечивает первоклассное обслуживание клиентов и поддержку. Внимательная команда поддержки доступна круглосуточно, оборудована отреагировать на любые вопросы и урегулировать проблемы, обеспечивая непрерывную игру и удовлетворение.
1win_dhEn
| #
1win на телефон http://mymoscow.forum24.ru/?1-6-0-00026928-000-0-0 .
FNDaviditabe
| #
darknet markets url darknet drugs
DonDonfaita
| #
darkmarket list https://github.com/nexusdarkrtv1u/nexusdark – dark web drug marketplace
MichaelZef
| #
GUD Tech is at the forefront of blockchain innovation, providing secure, scalable, and efficient decentralized technology solutions. Whether you’re looking for blockchain infrastructure, smart contract development, or enterprise-grade decentralized applications, GUD Tech offers cutting-edge solutions to enhance security and efficiency. Designed for businesses and developers, GUD Tech ensures seamless integration of blockchain technology into real-world applications. https://gudchain.net
Blue_Code
| #
Какой вариант предпочесть в финансах? https://999-lombard.ru
Joshuaimami
| #
Ямобуры для бурения Аренда и услуги спецтехники в Омске – Мы предоставляем услуги по аренде и эксплуатации спецтехники для различных сфер деятельности, включая строительство, дорожные работы и транспортировку грузов. Наши услуги охватывают аренду автокранов, экскаваторов, самосвалов и других машин для больших и малых предприятий, а также для частных клиентов. Спецтехника для вашего бизнеса с гарантией надежности и эффективности — это наш приоритет.
Guardian6610
| #
Подробный разбор финансов: https://clombard.ru
mostbet_bcon
| #
мостбет мобильная версия скачать https://www.mostbet6002.ru .
JamesUtile
| #
Read football news here! https://footballbritian.uk/
mostbet_jbon
| #
мостбет официальный сайт mostbet6002.ru .
1win_ksst
| #
казино 1win http://1win816.ru/ .
Nomad1510
| #
Подборка ресурсов по финансам: https://nkonsul21.ru
EagleHunter
| #
https://kziss.ru Будущее финансов.
1win_ummi
| #
1 вин про http://1win6015.ru .
mostbet_shKi
| #
мостбет кг http://maksipolinovtsu.forum24.ru/?1-1-0-00000194-000-0-0-1742815870/ .
Donaldlaf
| #
darknet websites https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark websites
1win_ipEn
| #
1win rossvya 1win rossvya .
AlphaHunter
| #
Кто-нибудь ориентируется в финансах? Где искать? https://zoom-finance.ru
CyberV7
| #
Какие изменения в финансах? https://mfo-orbita.ru
OmegaCode
| #
https://ipoteka11.ru Коллеги, поделитесь опытом по финансам!
LouisStulk
| #
Заправка кондиционера Mercedes-Benz Ремонт и обслуживание автомобилей Mercedes-Benz – Мы уже много лет предоставляем профессиональные услуги по ремонту и техническому обслуживанию автомобилей Mercedes-Benz как для крупных автопарков, так и для владельцев автомобилей малого и среднего бизнеса. Наши специалисты предлагают диагностику, кузовной ремонт, замену масла и другие услуги с использованием только оригинальных запчастей и современного оборудования.
Howardham
| #
Looking for high-quality slots to play? AbeBet Casino offers the best online slots for all players!
At AbeBet abe-casino.com/tr/games/demo/fire_joker, you’ll find popular slots from leading providers.
Join now to get exclusive offers.
Dive into an atmosphere of thrill with Abe Bet!
Toliknaf
| #
darkmarket list https://github.com/abacuslink4jjku/abacuslink – darknet drug store
cogebnaf
| #
Ищете форекс брокер для скальпинга? Time-forex.com/brokery-dly-skalpinga тут вы отменные для скальпинга брокеры отыщите. Форекс брокер для скальпинга это самая прибыльная стратегия трейдинга в данное время считается краткосрочный трейдинг, она предполагает открытие большого количества сделок в течении одной сессии. Посмотрите весь список надежных для скальпинга брокеров, которых мы внимательно отобрали и информацию структурировали.
Eagle_Fang
| #
Где учиться финансам? https://999gt.ru
Kxyufaita
| #
darknet websites darknet drug store
WilliamIRrutty
| #
darknet drug store dark markets
mostbet_mbon
| #
aviator mostbet mostbet6002.ru .
BlueByte
| #
Типичные промахи в финансах: https://kom1shop.ru.
Pingrutty
| #
dark market list https://github.com/aresonioncq0a7/aresonion – darknet markets onion
Rabyitabe
| #
dark market https://github.com/darkwebsitesyhshv/darkwebsites – darkmarket 2025
Edwardorex
| #
Натко фарма официальный сайта Natco Pharma Limited, широко известная как Натко фарма, зарекомендовала себя как один из лидеров индийской фармацевтической индустрии. Компания, представленная на своем официальном сайте natcopharma.co.in, специализируется на разработке, производстве и маркетинге широкого спектра фармацевтических препаратов, охватывающих различные терапевтические области.
Code4719
| #
Что лучше выбрать в финансах? https://credit-cheboksary.ru
Dragon_Hacker
| #
Глубокий анализ финансов: https://zol56.ru
MarkNOlaf
| #
darkmarket 2025 best darknet markets
FNDaviditabe
| #
dark web sites darkmarket
whatsapp group blaster
| #
I don’t even know how I ended up here, but I thought
this post was good. I do not know who you are but definitely you’re going to a famous blogger if you aren’t already 😉 Cheers!
WhatsApp hash channels supplier
| #
Genuinely no matter if someone doesn’t understand after that its up to other people
that they will help, so here it happens.
Buy WhatsApp hash channels
WhatsApp hash channels for sale
Purchase WhatsApp hash channels
WhatsApp marketing hash channels
WhatsApp hash channel provider
Affordable WhatsApp hash channels
WhatsApp hash channels bulk purchase
WhatsApp hash channels online store
Best WhatsApp hash channels
WhatsApp hash channels for businesses
WhatsApp hash channels for marketing
WhatsApp hash channels supplier
WhatsApp hash channels pricing
WhatsApp hash channels reseller
WhatsApp hash channels wholesale
WhatsApp hash channels service
WhatsApp hash channels shop
WhatsApp hash channels deals
WhatsApp hash channels packages
WhatsApp hash channels solutions
Blue155
| #
https://rafinad-lip.ru Как применять финансы в бизнесе?
Pioneer1133
| #
Видел прогнозы, что Финансы будет главным https://bazaferrit.ru трендом. Соглашусь?
DonDonfaita
| #
dark web drug marketplace https://github.com/nexusdarkrtv1u/nexusdark – darknet market list
DavidCreag
| #
Looking for exciting online casino games? MasalBet is the place to find new slot hits from renowned providers.
At masalbet-official.com/tr/, you’ll find exclusive promotions and simple gameplay.
Register now and get exclusive offers.
Play true excitement with MasalBet!
Thunder_Voyager
| #
Кто-нибудь ориентируется в финансах? Куда зайти? https://malganis.ru
crownsoft whatsapp github
| #
It’s awesome in favor of me to have a site, which is valuable in support
of my experience. thanks admin
Feel free to visit my site crownsoft whatsapp github
клининговые услуги
| #
Кристальная чистота в Санкт-Петербурге: Ваш надежный партнер по наведению порядка!
Наша компания более 10 лет оказывает услуги клининга в Санкт-Петербурге и зарекомендовала себя как стабильный партнер. Мы гордимся опытом нашей команды, которая состоит из высококвалифицированных специалистов, прошедших курс и имеющих все необходимые сертификаты. Для нас важна не только оперативность, но и наивысшее качество выполняемых услуг.
Не ждите, пока грязь и беспорядок станут проблемой. Подарите нам шанс вернуть вашему пространству чистоту и порядок! Узнайте всё о наших услугах и оставьте заявку на сайте : http://xn--bb0bw4mo1l2wn.shop/bbs/board.php?bo_table=free&wr_id=1645910
Guardian3349
| #
Как https://smartlombard24.ru сделать финансы?
Hepigfoown
| #
Ищете где купить бонсай? Зайдите на сайт https://bonsay.ru/ – мы уже 15 лет занимаемся японскими карликовыми деревьями бонсай с быстрой доставкой. Посмотрите наш каталог – он самый большой на рынке, а в наших магазинах в Москве и Санкт-Петербурге выбор еще больше. Купить бонсай в подарок или для себя, настоящее ухоженное дерево! Подробнее на сайте.
zaimpodptskemerovosig
| #
кредит под залог птс кемерово
avtolombard-11.ru/kemerovo.html
кредит под залог птс кемерово
Dark_Guardian
| #
Инфографика по финансам: https://rospremierinvest.ru
Donaldlaf
| #
darkmarket url https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet markets 2025
Volodyanaf
| #
tor drug market darknet market links
Red_Slayer
| #
Свежие тренды в финансах: https://center-zayma.ru
KevinGaito
| #
либет казино
StormX5
| #
Столкнулся с проблемой финансами. https://azimutfinancegroup.ru Ответ здесь.
CyberNomad
| #
https://mosautoinvest.ru Смешные случаи в финансах.
StormV1
| #
Секреты https://deneg-zaimi.ru профессионалов в финансах.
Essay Writing Service
| #
Eѵery paper and cᥙstomer іs Ԁifferent.
Seasoned writers guarantee aan individualized Essay Writing Service approɑch to all your paⲣers and essays.
It’s ɑ easy course of to get a quality paper wkth its convenient sеrvices.
Customer heⅼp that is out there 24/7.
Toliknaf
| #
darkmarket list https://github.com/abacuslink4jjku/abacuslink – dark market url
Josephglave
| #
Привет всем!
Информационные сайты помогают людям улучшить интимную жизнь с помощью новейших технологий. В статьях рассматриваются гаджеты для повышения либидо, улучшения настроения и снижения уровня стресса. Сайты предлагают советы по выбору устройств, которые могут помочь улучшить качество сна и гармонизировать отношения. Эти ресурсы помогут вам раскрыть новые возможности для улучшения сексуальной жизни.
Как справиться с прокрастинацией? Разделите большие задачи на маленькие шаги, установите реальные сроки и мотивируйте себя за каждое выполненное задание. Используйте методы визуализации результатов.
Больше информации по ссылке – https://nikita-bywalino.ru/
урбеч как сделать, рейтинг лучших беспроводных наушников вкладышей, интересные каналы в телеграм
как сделать воздушный крем, интересные факты атака титанов, как правильно сделать проект
Удачи!
Pingrutty
| #
darknet site https://github.com/aresonioncq0a7/aresonion – dark market list
ThunderX8
| #
Где учиться финансам? https://mam-kap.ru
Eagle514
| #
Преодоление трудностей при изучении финансов: https://dipom.ru
MarkNOlaf
| #
dark market link darknet markets url
FNDaviditabe
| #
darknet drug links dark market link
Nuhuwvap
| #
How to exchange Monero in the cryptocurrency exchanger quickex are Buy or sell XMR? In this https://www.youtube.com/watch?v=zXDvoUnvK8Y detailed guide, we will show you how to buy Monero (XMR) on the quickex platform in 5 easy steps. You will learn how to use a reliable service without registration and verification, getting access to the best rates from leading liquidity providers.
ErnestoNow
| #
битзамо казино
Rabyitabe
| #
dark web marketplaces https://github.com/darkwebsitesyhshv/darkwebsites – dark web markets
WolfZ3
| #
Что лучше выбрать в финансах? https://region-kapital.ru
https://presslibrary.wiki/index.php?title=User:Corazon22S
| #
В медицинских клиниках Москвы уютно, как дома — это редкость.
https://playmobilinfo.com/index.php/User:TammieMcGuigan
NightSlayer
| #
Сравнительный анализ финансов: https://kredit-konsalt.ru.
AaronCycle
| #
Гемблинг-клуб Вулкан – это захватывающий мир азарта, который поражает с первого взгляда. Благодаря прекрасному дизайну и интуитивно понятному интерфейсу, пользователи быстро оказываются в этом игровом пространстве.
Среди положительных моментов казино Вулкан казино следует отметить обширный выбор игр, включая слоты и настольные развлечения, которые подойдут каждому игроку. Качество графики и анимационного исполнения делает игровой процесс дополнительно увлекательным и захватывающим.
Однако больше всего привлек внимание подход казино к безопасности игроков. Они используют передовые технологии шифрования, чтобы обеспечить приватность личных и финансовых данных пользователей. Это делает казино Вулкан лучшим выбором для онлайн-игры.
https://kreosite.com/index.php/User:EarleMarcotte9
| #
Какую клинику выбрать, чтобы записаться к врачу в Москве без проблем?
http://fingado.ch/mw/index.php/User:DustyHayworth4
Silver_Walker
| #
Топовый ресурс по https://mos-balans.ru теме финансов.
DonDonfaita
| #
dark web sites https://github.com/nexusdarkrtv1u/nexusdark – dark web market urls
Iron_Hunter
| #
Памятка для работы с финансами: https://lemur-perm.ru
Storm_Warden
| #
Финансы для https://arenda-kapital.ru компаний.
Warden3953
| #
Практическое применение финансов: https://mebelex11.ru
IronV9
| #
Типичные промахи https://zalog-585.ru в финансах.
Yuxefllacark
| #
Ищете производителя качественных и современных мангалов? Посетите https://steelonfire.ru/ – ознакомьтесь с нашим ассортиментом. Мы производим: мангалы, костровые чаши, печи для сжигания мусора, буржуйки, учаги под казан, коптильни, чаны для бани и другие товары. Посмотрите каталог, воспользуйтесь доставкой в Москве, Санкт-Петербурге и по всей России или заберите самовывозом. Качественная продукция по отличным ценам!
Pingrutty
| #
dark market https://github.com/aresonioncq0a7/aresonion – darkmarket url
Alpha_Walker
| #
Забавные моменты в финансах: https://podzalogi.ru
Donaldlaf
| #
onion dark website https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – tor drug market
RobertDaw
| #
Casino non Gamstop sites are packed with action—great!
non gamstop boku casino sites
Kxyufaita
| #
darknet site onion dark website
AlphaHacker
| #
Примеры успеха в финансах: https://autolombard626.ru
energopto
| #
Выполняем качественное https://energopto.ru/raboty/proektirovanie/proektirovanie-itp-tstp/ под ключ. Энергоэффективные решения для домов, офисов, промышленных объектов. Гарантия, соблюдение СНиП и точные сроки!
DarkX4
| #
По данным аналитиков, лучший источник о финансах – https://icg-company.ru
Toliknaf
| #
dark market https://github.com/abacuslink4jjku/abacuslink – dark market
MarkNOlaf
| #
dark web marketplaces dark market 2025
FNDaviditabe
| #
darknet market list darknet markets 2025
Overlord4155
| #
Какие изменения в финансах? https://skupkamz.ru
SteelZ2
| #
Нужны реальные кейсы по финансам. https://elisey11.ru
Volodyanaf
| #
darknet markets url dark web marketplaces
BryanSpery
| #
read the article jaxx wallet
Jesusuniny
| #
печать фирменных бланков печать бланков писем
Storm585
| #
Приветствую. Подскажите, где https://chercft.ru посмотреть информацию о финансах?
SteveSuect
| #
заказать печать папок печать на пластиковых папках
Rabyitabe
| #
dark web market https://github.com/darkwebsitesyhshv/darkwebsites – darknet market
Red_Guardian
| #
Финансы для https://mm-consult.ru компаний.
RedZ6
| #
Пошаговая инструкция https://lombard-krim.ru по финансам.
Dark377
| #
Финансы для компаний: https://yuf74.ru
Night_Master
| #
Нестандартный подход к финансам: https://lombard-khamovniki.ru
DonDonfaita
| #
darknet marketplace https://github.com/nexusdarkrtv1u/nexusdark – darknet markets url
vodka kazino_kipr
| #
водка казино водка казино .
Nomad5611
| #
Ищу реальные кейсы по финансам. https://mk-tehnika.ru
gates of olympus slot
| #
I just like the valuable information you supply on your articles.
I will bookmark your blog and check once more right here
regularly. I am rather sure I’ll be informed a lot
of new stuff right here! Best of luck for
the following!
Pingrutty
| #
darkmarket list https://github.com/aresonioncq0a7/aresonion – dark web market links
Protocol8022
| #
Базовые принципы https://avto-finance24.ru финансов для новичков.
Virgillieta
| #
смотреть здеськупить хайп проект под ключ
JerryAspib
| #
онлайн казино В мире азартных игр онлайн казино заняли прочное место, предлагая игрокам широкий выбор развлечений и возможность испытать удачу, не выходя из дома. От классических слотов до настольных игр с живыми дилерами – разнообразие поражает воображение. Стратегии в казино играют важную роль, позволяя игрокам повысить свои шансы на успех. Однако стоит помнить, что казино – это прежде всего игра случая, и никакая стратегия не гарантирует выигрыш.
zaimpodptsmsksig
| #
деньги в залог авто
avtolombard-11.ru
взять кредит залогом авто
SteelX4
| #
Эволюция развития финансов: https://chelgrad74.ru
unlim-777-casino.homes
| #
Unlim Casino is a unique platform offering incredible conditions for gaming and an amazing experience for all gambling enthusiasts.
Here, you’ll find a large selection of slots, roulette, as well as many tournaments
and promotions that can significantly improve your chances of winning.
We are happy to offer a convenient interface, a vast collection of slots, and new table games.
Gambling with generous bonuses and regular promotions will make your gaming
experience even more exciting.
How to become part of our community?
Simple registration to start playing — quick profile setup, and you
are set to begin.
Fantastic bonuses for new players — we give you bonuses on your first deposit, providing
a great start to your gaming journey.
Daily promotions and tournaments — for all players who want to
boost their chances of winning and earn additional
prizes.
24/7 support ready to assist you with any questions or
issues related to gaming.
Games available on any device, so you can enjoy the gameplay, whether on your PC or smartphone.
Join us now! Exciting adventures await you at Unlim Casino, offering
waves of emotions and the chance to win big prizes. Join us and start winning today! https://unlim-777-casino.homes/
Donaldlaf
| #
darknet market lists https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark websites
Iron_Master
| #
Как сделать финансы? https://invdvlp.ru
Light562
| #
Новые подходы в финансах: https://zlatagems.ru
Hacker9890
| #
https://financial-house.ru Предлагаю подискутировать о финансах.
бнанс реферальна програма
| #
Can you be more specific about the content of your article? After reading it, I still have some doubts. Hope you can help me.
WilliamIRrutty
| #
darknet site best darknet markets
Toliknaf
| #
darknet markets onion address https://github.com/abacuslink4jjku/abacuslink – darknet markets onion address
Kxyufaita
| #
darknet drug market dark market 2025
hMustafaVep
| #
Какой шикарный букет! Заказала себе домой.
Доставка цветов Томск
AndreDap
| #
Спасибо за оперативность и качество!
101 роза
PhantomBlade
| #
Обзор https://investor21veka.ru популярных ресурсов по финансам.
Dark_Sentinel
| #
Повышение https://kam-trade.ru эффективности через финансы.
StormPioneer
| #
https://allrekords.ru Технические нюансы финансов.
Silver_Fang
| #
Изучите этот источник о финансах: https://myfxbot.ru
RedStriker
| #
Кто-нибудь ориентируется в финансах? Куда зайти? https://otchetp.ru
pocowalasy
| #
Компания YIT стремительно развивается и предлагает вам амортизаторы, которые отлично зарекомендовали себя. Гарантируем прекрасные условия для сотрудничества. Предоставляем профессиональные консультации по телефону. Доставку по всей РФ выполняем. Ваше время сэкономим. Ищете амортизаторы для stels guepard? Yit-shocks.com – здесь представлена продукция компании YIT. Ознакомиться с каталогом можете в любое удобное время. Можно оплатить заказ в офисе магазина. Свяжитесь с менеджером для совершения безналичного расчета. Мы рады вам!
MarkNOlaf
| #
best darknet markets darkmarket url
Rabyitabe
| #
dark web market links https://github.com/darkwebsitesyhshv/darkwebsites – darknet markets
Volodyanaf
| #
bitcoin dark web tor drug market
Free adult site list
| #
I need to to thank you for this fantastic read!!
I definitely loved every little bit of it.
I’ve got you book marked to look at new stuff you post…
HenryBance
| #
печать на холсте с подрамником печать на холсте недорого
Pingrutty
| #
dark market link https://github.com/aresonioncq0a7/aresonion – darknet drug links
Nomad6616
| #
Советую проверенный ресурс по https://buch-pro.ru финансам.
RobertJes
| #
dtf печать на текстиле https://dtf-pechat-spb.ru
DanielBuh
| #
струйная широкоформатная печать широкоформатная печать на заказ
DonDonfaita
| #
darknet drug store https://github.com/nexusdarkrtv1u/nexusdark – dark web drug marketplace
Vortex995
| #
Полное руководство по финансам от https://zalogspecteh.ru А до Я.
GeorgeTom
| #
Source:
royaljackpotcasino.org
Warden5464
| #
Успешные кейсы применения финансов: https://uis-finance.ru
Lesterweive
| #
грузовой техосмотр Москва – мегаполис, где техосмотр (ТО) является обязательной процедурой для большинства транспортных средств. Найти аккредитованный пункт техосмотра в Москве несложно, достаточно воспользоваться онлайн-картами или поисковыми системами. Важно помнить, что приобретение техосмотра без фактического осмотра автомобиля – незаконно. Стоимость прохождения ТО зависит от категории транспортного средства. Проверить легитимность диагностической карты ТО можно по базе ЕАИСТО (Единая автоматизированная информационная система техосмотра). Это позволяет убедиться, что данные внесены в официальную базу и соответствуют требованиям. Вопрос о необходимости техосмотра для конкретного типа транспорта регламентируется законодательством, и его отсутствие может повлечь штраф.
Night_Protocol
| #
Что нельзя делать с финансами? https://avtolombard-armavir.ru
https://t.me/officialonion
| #
Психология игры в онлайн казино: как не проиграть все
Иногда желание ощутить адреналин приводит к непродуманным шагам в виртуальных клубах. Участие в азартных мероприятиях может поглотить внимание и затянуть в водоворот эмоций. При этом важно уметь контролировать свои действия и осознавать риски, связанные с ставками.
Статистика показывает, что большинство пользователей сталкиваются с трудностями в управлении своим бюджетом. Исследования свидетельствуют, что возникновение азартного поведения часто связано с эмоциями и восприятием шансов на выигрыш. Четкое понимание своих целей и лимитов способно снизить риск неудач, минимизируя ущерб для финансов. Ставьте перед собой реалистичные ожидания и не позволяйте эмоциям принимать верх.
Использование стратегий при выборе ставок может существенно помочь сохранить интерес и при этом защитить средства. Например, выделение заранее оговоренной суммы для развлечения и строгая приверженность к этому лимиту позволяет избежать ненужных потерь. Разделение бюджета на части, например, фиксированные суммы для каждой сессии, может стать надежным вариантом для ответственного участия. Хорошая привычка – вести учет своих трат, что даст возможность лучше оценивать удачны ли были проведенные сессии.
Научитесь расслабляться и отвлекаться от процесса. Периодические перерывы в игре помогут оценить происходящее более трезво и предотвратят развитие зависимости. Помните: осознанный подход к азартным развлечениям может сделать их более приятными, сокращая при этом вероятность финансовых потерь.
Управление банкроллом
Контроль денежных средств – важный аспект активного участия в азартных развлечениях. Определите для себя фиксированную сумму, которую готовы инвестировать. Это должен быть предел, превышение которого приведет к нежелательным последствиям.
Рекомендуется выделить отдельную сумму для каждой сессии, чтобы избежать размытых границ между ставками и повседневными расходами. Поделите банкролл на несколько частей. Применение правила 1-5% от общего капитала на каждую ставку снизит риск быстрого исчерпания средств.
Запись успешных и неудачных ставок поможет анализировать свои действия, выявлять паттерны и обучаться на ошибках. Вместе с тем, выделяйте время на отдых, это предотвратит эмоциональное переутомление и поспешные решения.
Использование стратегий управления банкроллом, таких как Martingale или Fibonacci, может сулить определенные преимущества. Тем не менее, не забывайте об их рисках и возможностях потери. Старайтесь избегать ставок в погоне за удвоением средств – это может привести к еще большим потерям.
И, наконец, никогда не забывайте о важности развлечения. Подходите к этому процессу с умом, чтобы получать удовольствие от каждого момента, а не испытывать давление из-за финансовых потерь.
Эмоциональный контроль
Для успешного участия в азартных мероприятиях необходимо уметь управлять своими чувствами. Эмоции, такие как радость, разочарование или страх, могут существенно повлиять на решения участника. Поэтому стоит выделить несколько подходов к контролю своих эмоциональных состояний.
Первое, регулярно практикуйте осознанность. Умение находиться в настоящем моменте поможет снизить уровень стресса и предотвратить импульсивные реакции. Попробуйте медитацию или дыхательные упражнения, они способствуют улучшению концентрации и эмоциональной стабильности.
Второе, устанавливайте четкие лимиты. Заранее определите время и сумму, которую готовы потратить. Это создаст рамки, в которых вам будет легче принимать взвешенные решения, а не действовать под влиянием внезапных эмоций.
Третье, анализируйте свои эмоции. Записывайте, что именно вызывает у вас радость или разочарование. Это поможет выявить закономерности в своем поведении и выявить триггеры, которые ведут к негативным реакциям.
Четвертое, ознакомьтесь с принципами управления финансами. Осознание сути управления капиталом может снизить уровень тревожности. Чем лучше вы понимаете, как работают ваши деньги, тем меньше вероятность, что эмоции возьмут верх над разумом.
Пятое, используйте технику “временной паузы”. Если вы чувствуете прилив эмоций, делайте перерыв. Выходите на прогулку, выпейте воды или рефлексируйте над происходящим. Такой подход часто помогает привести в порядок мысли и восстановить уровень самообладания.
https://vk.com/topic-5580078_53468042
WilliamIRrutty
| #
dark market darknet drug links
Donaldlaf
| #
dark market onion https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darkmarket url
Kxyufaita
| #
darkmarkets darknet drugs
Dragon768
| #
Перспективы финансов: https://gktavr.ru
Warden6224
| #
Исследование финансов: https://deekat96.ru
https://t.me/onion_promokod
| #
Онлайн казино: обзор самых популярных игр с высоким RTP
Современные развлечения, связанные с азартом, стали доступнее благодаря технологиям. Игроки могут наслаждаться захватывающими сессиями, не покидая своих уютных уголков. Увлекательные автоматы, карты и другие варианты развлечений привлекают внимание благодаря увлекательным механикам и шансам на успех.
Выбор игровых автоматов с высокой вероятностью выигрышных выплат позволяет пользователям лучше управлять своими ставками. Некоторые из этих разработок предлагают возврат игроку, превышающий 96%, что дает шанс на более продолжительную игровую сессию. Среди таких предложений есть как классические, так и инновационные варианты, каждый из которых обладает уникальной атмосферой и тематикой.
Понимание механики работы различных развлечений поможет найти те, которые подходят именно вам. Разнообразие тем, а также наличие бонусов и фри-спинов в определённых слотах увеличивают шансы на получение выигрыша. Поэтому, анализируя доступные игры, стоит уделять внимание не только визуальному оформлению, но и математической модели каждого варианта.
Топ слотов с высоким коэффициентом возврата
Выбор игровых автоматов с хорошими показателями возврата позволяет увеличить шансы на выигрыш. Вот несколько слотов, которые выделяются среди остальных благодаря своим характеристикам.
Blood Suckers – этот автомат известен своим коэффициентом возврата в 98%. Тематика вампиров и множество бонусных функций делают его интересным выбором. Сочетание бесплатных вращений и умножителей повышает возможность получения значительных выплат.
Book of 99 предлагает игрокам коэффициент 99%, что делает его одним из самых прибыльных слотов на рынке. Тематика древнего Египта и возможность активации множителей увеличивают шансы на ощутимый выигрыш. Бесплатные раунды предоставляют дополнительный шанс на успех.
Jackpot 6000 – классический автомат с возвратом 98,8%, который сочетает в себе простоту и высокую доходность. Уникальная функция “Knock Out” позволяет игрокам увеличить свои выигрыши, используя стратегию рискованного выбора.
Starmania выделяется коэффициентом в 97,87%. Космическая тема и возможность выигрыша множителей делают игры увлекательными. Бонусный раунд с бесплатными вращениями предоставляет возможность для значительных выигрышей.
Persian Nights – слот с возвратом 97,22%, который переносит игроков в мир восточных сказок. Привлекательные символы и функции с бесплатными вращениями и умножителями увлекают и обнадеживают пользователей на хорошие выплаты.
Выбор слота с высоким коэффициентом возврата может значительно повысить шансы на успех, учитывая, что они предлагают тщательно продуманные механики и увлекательные тематики. Сравние различных игровых автоматов и их особенностей поможет сделать правильный выбор.
Настольные игры для выигрыша
Настольные развлечения давно завоевали популярность среди игроков благодаря стратегическому элементу и взаимодействию. По сравнению с автоматами, такие виды досуга предлагают больше возможностей для принятия решений и применения разнообразных тактик.
Блэкджек является одним из самых актуальных выборов. Игра предполагает минимизацию преимущества заведения за счёт грамотной стратегии управления картами. Опытные участники используют стратегии подсчета карт, что позволяет значительно повысить шансы на успешную игру. Важно учитывать, что некоторые заведения применяют несколько колод, что усложняет подсчёт.
Рулетка привлечет ценителей азарта. Игроки могут ставить на конкретные числа, группы, цвета или четность. Чтобы увеличить шансы, рекомендовано использовать стратегии, такие как Д’Аламбер или Мартингейл. При этом следует поддерживать разумный банкролл, чтобы избежать значительных потерь.
Покер – это не только удача, но и способности. Разные варианты, такие как Техасский Холдем или Омаха, требуют отличного понимания математики и психологии. Важно учиться читать соперников и агрессировать в подходящие моменты. Участие в турнирах с доступными бай-инами поможет развить навыки и познакомиться с другими игроками.
Крепкая комбинация стратегий, наблюдательности и контроля эмоционального состояния способна превратить настольные развлечения в прибыльный опыт. Осмысленный подход и постоянная практика приведут к долгосрочным успехам.
https://t.me/levclubx
PhantomX4
| #
Как работает https://sarlombard64.ru механизм финансов?
Omega_Overlord
| #
Нюансы работы с финансами: https://radiodetalitorg.ru
OmegaZ1
| #
Глубокий анализ https://rashpil54.ru финансов.
Toliknaf
| #
dark web link https://github.com/abacuslink4jjku/abacuslink – darknet drugs
MarkNOlaf
| #
darkmarket bitcoin dark web
DavidUnory
| #
steam desktop authenticator github
Pingrutty
| #
darknet markets links https://github.com/aresonioncq0a7/aresonion – dark websites
ThunderZ3
| #
Инфографика по финансам: https://abbeville-msk.ru
Shadow_Blade
| #
Как сэкономить на https://kabinet-lime-zaim.ru финансах.
LesterGeori
| #
заключить контракт на военную службу Процесс поступления на военную службу по контракту начинается с изъявления гражданином желания заключить соответствующий контракт. Это может быть как первичное поступление на службу, так и перезаключение контракта после окончания предыдущего. Основным документом, регулирующим данный процесс, является Федеральный закон “О воинской обязанности и военной службе”. Для заключения контракта необходимо обратиться в военный комиссариат по месту жительства или в пункт отбора на военную службу по контракту. Там гражданин получит подробную консультацию о требованиях, предъявляемых к кандидатам, и перечне необходимых документов. Важно учитывать, что к кандидатам предъявляются определенные требования по возрасту, образованию, состоянию здоровья и физической подготовке.
Rabyitabe
| #
darknet markets onion address https://github.com/darkwebsitesyhshv/darkwebsites – dark market list
Red459
| #
ТОП-10 ресурсов по финансам: https://papatender.ru
EagleGuardian
| #
Оптимизация процессов через финансы: https://ufa585.ru
DavidSaili
| #
Агентство оказывает премиальные услуги полного цикла в Москве по поддержке и SEO-оптимизации корпоративного портала группой квалифицированных экспертов с 2010 года. Ключевое направление нашей студии — https://www.obsluzhivanie-sajta.ru/ с фиксированной предсказуемой тарифом. Настроим техподдержку, миграцию на HTTPS, адаптацию под мобильные устройства, ускорение загрузки и бесперебойную работу интернет-представительства.
Реализуем: PWA-разработку, настройку API, оптимизацию Core Web Vitals, регулярный аудит. Разрабатываем под ключ популярные CMS для корпоративных сайтов: HostCMS, для интернет-магазинов: OpenCart, для блогов и лендингов: Tilda, для SaaS-платформ: Strapi. Предусмотрено 12+ индивидуальных решений с прозрачными расценками.
Shadow_Code
| #
Ищу практические https://slobodchikova-design.ru советы по финансам.
RaymondKag
| #
огнезащитный лак КМ1 для стен В мире современных интерьеров, где дерево занимает почетное место, особое внимание уделяется безопасности. Огнезащитный лак для деревянного паркета становится не просто декоративным элементом, а жизненно важным средством защиты. Лак КМ1, специально разработанный для огнезащиты паркетных полов, обеспечивает надежный барьер против распространения огня. Этот лак для паркетных покрытий с огнезащитой создает на поверхности древесины слой, способный замедлить или предотвратить возгорание. Огнезащитное покрытие для паркетных досок, такое как паркетный лак КМ1 с огнезащитными свойствами, позволяет не только сохранить эстетику натурального дерева, но и повысить пожарную безопасность помещения. Лак для паркета КМ1 – это гарантия спокойствия и уверенности в защите вашего дома.
zaimpodptsnovosibsig
| #
кредит в залог автомобиля
avtolombard-11.ru/nsk.html
авто под залог
TisajesDeats
| #
На сайте https://psihologist-spb.ru/uslugi-psikhologa/semeynaya-psikhoterapiya/ почитайте интересную и полезную информацию, которая касается того, чем вам поможет семейный психолог. Этот специалист поможет разрешить конфликт, улучшить отношения между супругами и сделать так, чтобы появилось доверие в паре. Кроме того, вы узнаете и о том, в каких случаях лучше всего обращаться к семейному психологу. Он поможет избежать недопонимания, стрессов, противоречий, поможет в воспитании детей и романтических отношениях.
DonDonfaita
| #
darknet links https://github.com/nexusdarkrtv1u/nexusdark – darkmarket url
WilliamIRrutty
| #
darknet markets onion address darknet drugs
Nujitesowl
| #
На сайте https://ai-gptbot.ru вы сможете воспользоваться всеми возможностями уникального и инновационного чата GPT, который предлагается на русском языке. При этом воспользоваться им вы сможете даже без VPN. Все ежедневные рутинные дела вы сможете доверить ИИ. У вас получится создать увлекательный, интересный контент. Теперь вы сможете решить все вопросы максимально быстро и точно. Чат позволит сосредоточиться на важных процессах. Он необходим разработчикам, менеджерам, которые раскручивают социальные сети, школьникам, студентам, которые готовятся к экзаменам.
Phantom_Master
| #
Перспективы финансов: https://k1business.ru
Kxyufaita
| #
darknet drug links dark web marketplaces
DavidUnory
| #
steam desktop authenticator github
Ghost561
| #
https://24matkap.ru С чего начать финансов?
Nomad6660
| #
Коллеги, поделитесь опытом по https://srnalog.ru финансам!
Eagle613
| #
https://lombard-orsk.ru Что лучше выбрать в финансах?
Volodyanaf
| #
darkmarket list darkmarket 2025
Omega_Pioneer
| #
Секреты профессионалов в финансах: https://9986978.ru
MarkNOlaf
| #
darknet drug market best darknet markets
1win_yppi
| #
1win ваучер 1win817.ru .
FNDaviditabe
| #
dark web market list darknet markets onion
Ghost_Warden
| #
Топовый ресурс по теме финансов: https://tradein39.ru
Donaldlaf
| #
tor drug market https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark web market links
1win_lyPi
| #
1вин официальный сайт вход https://dogzz.forum24.ru/?1-10-0-00000155-000-0-0-1742818537/ .
mostbet_hxSa
| #
mostbet игры http://corgan.borda.ru/?1-0-0-00000265-000-0-0 .
Curtisjef
| #
Когда ты встречаешь самую красивую девушку, первое впечатление всегда захватывает. Но настоящее знакомство начинается с понимания, что красота — это лишь малая часть её привлекательности, и каждый разговор с такой дамой приносит всё больше открытий https://t.me/devki_spb1
Samuelhox
| #
На официальном сайте Kent Casino вас ждёт огромный выбор игровых автоматов, лотерей и турниров с крупными призами. Регистрация займёт всего пару минут, а выплаты обрабатываются мгновенно, без задержек и скрытых комиссий: Кент казино вход
CyberStriker
| #
Какой вариант предпочесть в финансах? https://planeta-ucheta.ru
1win_vkpi
| #
1win сайт http://www.1win817.ru .
Steel928
| #
Лично мне помог этот ресурс https://urist2020.ru по финансам.
Pingrutty
| #
darknet links https://github.com/aresonioncq0a7/aresonion – darkmarkets
Toliknaf
| #
dark web sites https://github.com/abacuslink4jjku/abacuslink – dark web market
Thunder184
| #
Как избежать https://n1lombard.ru проблем с финансами?
1win_owPi
| #
wan win https://www.dogzz.forum24.ru/?1-10-0-00000155-000-0-0-1742818537 .
mostbet_tzSa
| #
мостбет авиатор https://corgan.borda.ru/?1-0-0-00000265-000-0-0/ .
KevinMaide
| #
Здравствуйте!
Как преодолеть страх перед публичными выступлениями? Подготовьтесь заранее, практикуйте свои речи и не забывайте, что нервозность — это нормальная реакция.
Инвестировать в золото стоит в том случае, если вы хотите сохранить и приумножить свой капитал. Золото считается надежным активом в условиях экономической нестабильности. Стоит ли инвестировать в золото, зависит от ваших финансовых целей и ситуации на рынке. Золото — это не только украшение, но и стабильный актив в портфеле.
Больше информации по ссылке – https://kyocera-mds.ru/
как сделать королевский минет, русский интересный сериал, как сделать скриншот экрана
как сделать файл exe, сериалы топ лучших детективные, фильм смотреть ужасы самые страшные
Удачи!
Red_Code
| #
https://mkkaktual.ru Где учиться финансам?
Byte3899
| #
Типичные https://zoloto-nn.ru промахи в финансах.
Iron275
| #
Изучите этот материал о финансах: https://sempk.ru
Rabyitabe
| #
darkmarket https://github.com/darkwebsitesyhshv/darkwebsites – dark market
SilverBlade
| #
Пошаговая инструкция по финансам: https://citistreet.ru
WilliamIRrutty
| #
dark markets 2025 dark markets
mostbet_xipt
| #
mostbest https://mostbet6003.ru .
DavidUnory
| #
steam desktop authenticator
RobertDaw
| #
Non Gamstop casinos keep it fresh—really cool stuff!
non gamstop casinos no deposit free spins
BlueX3
| #
Практическое применение финансов: https://invest-omsk.ru.
1win_tqpi
| #
1win официальный сайт http://1win817.ru .
Kxyufaita
| #
darkmarkets darknet markets onion address
1win_rqPi
| #
1 вин официальный сайт вход http://dogzz.forum24.ru/?1-10-0-00000155-000-0-0-1742818537 .
Iron_Code
| #
Видеоуроки по финансам: https://matkapsnk.ru.
mostbet_hnSa
| #
mosbet http://corgan.borda.ru/?1-0-0-00000265-000-0-0 .
Thomasquabs
| #
Здравствуйте!
Как развивать уверенность в себе? Постепенно выходите из зоны комфорта, чтобы научиться справляться с новыми ситуациями. Признавайте свои достижения и помните, что ошибки — это часть пути. Занимайтесь тем, что приносит вам удовольствие, и окружайте себя поддерживающими людьми.
Как наладить хорошие отношения с коллегами? Будьте честными и открытыми, проявляйте уважение и проявляйте интерес к мнению других.
Больше информации по ссылке – https://drjahlov.ru/
самые интересные новые фильмы, белками богаты продукты, прикорневой объем как сделать
цветной ник как сделать, интересные факты ледовое побоище, куда можно поехать из россии
Удачи!
DonDonfaita
| #
best darknet markets https://github.com/nexusdarkrtv1u/nexusdark – darknet markets onion
MarkNOlaf
| #
dark web sites tor drug market
mostbet_nlpt
| #
mostbet apk скачать https://www.mostbet6003.ru .
GregoryClile
| #
Единственный знак зодиака, который является магнитом для несчастий и проблем https://x.com/Fariz418740/status/1904737515858174078
PhantomX4
| #
Использование https://nadezhny-lombard.ru на практике финансов.
Eagle_Hunter
| #
Мне самому помог этот https://delta-tomsk.ru ресурс по финансам.
DavidSaili
| #
Digital-агентство реализует качественные решения в регионе по техобслуживанию и комплексному продвижению лендинга командой квалифицированных специалистов с 2011 года. Приоритетное направление нашей — https://www.obsluzhivanie-sajta.ru/ с фиксированной ежемесячной предсказуемой тарифом. Запустим ежедневный мониторинг, аудит безопасности, адаптацию под мобильные устройства, настройку кеширования и круглосуточную доступность веб-ресурса.
Реализуем: PWA-разработку, подключение онлайн-оплаты, оптимизацию SEO, проактивный мониторинг. Поддерживаем надежные CMS для корпоративных сайтов: HostCMS, для интернет-магазинов: PrestaShop, для блогов и лендингов: MODX, для SaaS-платформ: Strapi. Предусмотрено 12+ индивидуальных решений с прозрачными расценками.
Master3890
| #
Что https://lombardlik.ru нельзя делать с финансами?
Eagle_Blade
| #
Финансы для стартапов: https://kamdengi.ru
Nomad9756
| #
Видеоуроки по финансам: https://akfa-y.ru
Pingrutty
| #
tor drug market https://github.com/aresonioncq0a7/aresonion – darknet links
Donaldlaf
| #
dark market url https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet links
who makes orthopedic shoes
| #
Lucky Feet Shoes Anaheim
5761 Ε Sana Ana Canyon Rd ste c,
Anaheim, СA 92807, United States
+17142821000
who makes orthopedic shoes
Storm897
| #
Приветствую. Подскажите, где найти информацию о финансах? https://polyanca.ru
MevexelPlobe
| #
Looking for a top-tier film or video production company in Italy? ORBIS Production is your go-to Italian production company offering full-service video, photo and film production across Milan, Rome, Venice, Florence, Turin, Genoa, Sardinia, Naples, Cortina d’Ampezzo, Verona, Lake Como, Lake Garda, Bari, Sicily, Capri, Forte-dei-Marmi, Siena and all of Italy. Discover our production services, from corporate and commercial films to international service production – https://orbispro.it/en
Phantom636
| #
Эксклюзивное интервью с экспертом по финансам: https://lombard-arena.ru
https://vk.com/topic-5580078_53468063
| #
Онлайн казино: обзор самых популярных платежных систем
Банковские карты продолжают оставаться одним из самых распространенных способов перевода средств. Visa и MasterCard обладают высоким уровнем доверия благодаря своим проверенным практикам обеспечения безопасности и быстрым транзакциям. Однако некоторые площадки могут накладывать ограничения на использование этих карт, что стоит учитывать при выборе.
Еще одним удобным вариантом являются электронные кошельки. Решения, такие как Skrill и Neteller, предлагают высокую скорость обработки запросов и множество дополнительных функций. Эти платформы позволяют не только быстро переводить деньги, но и контролировать финансовые операции, что делает их популярными среди игроков.
Криптовалюты, как Биткойн и Эфириум, также набирают популярность в этой области. Такие средства предлагают анонимность и безопасность, а также убирают необходимость в посредниках. Однако важно понимать, что использование цифровых валют связано с волатильностью курса, что может стать риском для пользователей.
Выбирая подходящий метод для финансовых операций, не забывайте обращать внимание на комиссии, скорость обработки и доступность в вашем регионе. Тщательный анализ приведет к более приятному игровому опыту и поможет избежать неприятных ситуаций.
Методы пополнения счета
Процесс внесения депозитов в игорные платформы стал удобнее благодаря множеству доступных способов. Основные варианты включают банковские карты, электронные кошельки и криптовалюту. Каждое из этих решений обладает своими характеристиками и преимуществами.
Банковские карты, такие как Visa и MasterCard, остаются наиболее распространенным вариантом. Они обеспечивают маскимально простую интеграцию с системами. Обычно процесс пополнения занимает считанные минуты. Некоторые заведения могут попросить верификацию карты, что служит дополнительной защитой для пользователей.
Электронные кошельки, как PayPal, Skrill и Neteller, популярны из-за своей скорости и анонимности. Фонды зачисляются мгновенно, что значительно улучшает пользовательский опыт. Пользователи также могут легко контролировать свои финансы, так как все транзакции фиксируются в кошельке.
Криптовалюта, включая Bitcoin и Ethereum, находит все больше поклонников благодаря своей децентрализованной природе. Транзакции проходят быстро и без посредников, что снижает риски и повышает конфиденциальность. Однако стоит учесть рыночные колебания, влияющие на стоимость активов.
Мобильные платежные системы, такие как Apple Pay и Google Pay, становятся все более востребованными. Множество платформ адаптируются под эти новшества, что упрощает процесс пополнения с помощью смартфона. Это решение идеально подходит для тех, кто ценит скорость и удобство.
При выборе метода пополнения счета важно учитывать комиссии. Некоторые варианты могут предлагать более выгодные условия, что позволяет оптимизировать расходы. Внимательно ознакомьтесь с условиями платформы, чтобы избежать неприятных сюрпризов.
Банковские переводы отличаются высокой степенью безопасности, но могут занять от нескольких дней до недели. Пользователям стоит обратить внимание на комиссии, которые могут существенно варьироваться в зависимости от финансовых учреждений. Рекомендуется заранее изучить условия банка по работе с подобными транзакциями.
https://t.me/otzyvy7kbot
mostbet_bwpt
| #
mostbets https://www.mostbet6003.ru .
GhostWalker
| #
Нестандартный подход к финансам: https://soho-sah.ru
RedSlayer
| #
Авторитетное мнение о https://pk-invest.ru финансах.
Toliknaf
| #
bitcoin dark web https://github.com/abacuslink4jjku/abacuslink – dark web marketplaces
WilliamIRrutty
| #
dark web marketplaces dark market url
Volodyanaf
| #
dark web drug marketplace dark web sites
Kxyufaita
| #
dark web marketplaces dark websites
Curtisjef
| #
В Санкт-Петербурге можно найти знакомства с красивыми девушками, которые обладают не только внешней привлекательностью, но и обаянием, харизмой, что делает каждое общение с ними незабываемым https://t.me/devki_spb1
avtolombardekbsig
| #
деньги под залог авто машин
https://seedvertexnetwork.co.ke/employer/zaim-pod-zalog-pts-11290/
кредит под залог птс
Night856
| #
Столкнулся с проблемой финансами. Решение здесь: https://eskorial-r.ru
Phantom_Striker
| #
Мнение специалистов https://dz-dok.ru о финансах.
Ghost237
| #
Преодоление трудностей при изучении финансов: https://zalognk.ru
gipalBrobe
| #
Арчибальд – академия, которая обучением груминга занимается. Вы можете совершенствовать собственные навыки на живых моделях до бесконечности. Преподаватели отвечают на все вопросы. Дарим методические материалы, учебники, а также видеолекции. Погрузитесь в атмосферу, с которой не захочется расставаться. Ищете курсы груминга в москве и московской области? Grooming-dream.ru – тут реальные отзывы клиентов представлены. Мы выпускаем отменных груминг-специалистов. Предлагаем во время обучения инструменты. Если хотите освоить новую профессию, «Арчибальд» будет самым подходящим для вас местом.
Rabyitabe
| #
darknet links https://github.com/darkwebsitesyhshv/darkwebsites – darknet markets onion
FNDaviditabe
| #
darkmarket darknet site
StormHunter
| #
Пошаговая инструкция https://lombard-ptz.ru по финансам.
DonDonfaita
| #
darknet marketplace https://github.com/nexusdarkrtv1u/nexusdark – darknet market list
Thunder494
| #
Эксклюзивное интервью с экспертом по финансам: https://euroasianlease.ru
ChuckRef
| #
Четыре знака Зодиака ждет переломный момент на этих выходных https://x.com/Fariz418740/status/1905095937828978718
AlphaX3
| #
Памятка для работы с финансами: https://kafe-iris.ru
Pingrutty
| #
dark web market links https://github.com/aresonioncq0a7/aresonion – darkmarket list
Eagle_Voyager
| #
Полное руководство по финансам от А до Я: https://sfera-krasoty.ru
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их происхождение. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы и адаптировать решения под особенности вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
Порошок висмутовый C89836 – UNS Порошок висмутовый C89836 – UNS представляет СЃРѕР±РѕР№ высококачественный химический РїСЂРѕРґСѓРєС‚, идеально подходящий для различных промышленных применений. Ртот порошок отличается отличной чистотой Рё стабильностью, что делает его незаменимым РІ металлообработке, фармацевтике Рё электронике. Если РІС‹ ищете надежный Рё эффективный материал, вам стоит купить Порошок висмутовый C89836 – UNS. РћРЅ обладает уникальными свойствами, такими как высокая тепло- Рё электрическая проводимость. РќРµ упустите возможность использовать этот универсальный порошок для СЃРІРѕРёС… проектов. Выберите лучшее качество СЃ Порошком висмутовым C89836 – UNS.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние возможности.
Наши товары:
Мишень для распыления молибдена
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
Наша продукция:
Латунная квадратная профильная труба 55С…55С…12.5 РјРј ЛС 59-1 РўРЈ 48-21-428-84 Приобретите латунные квадратные трубы РѕС‚ производителя РЅР° Редметсплав.СЂС„. РЁРёСЂРѕРєРёР№ выбор, высокое качество, устойчивость Рє РєРѕСЂСЂРѕР·РёРё Рё простота монтажа – РІСЃРµ это делает наши трубы незаменимым решением для ваших проектов.
Wolf_Hacker
| #
Практическое https://kassacredit.ru применение финансов.
WilliamIRrutty
| #
darknet market dark web markets
Donaldlaf
| #
darkmarkets https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark market list
HaroldVew
| #
Читать далее mega мориарти
Kxyufaita
| #
darknet market dark market list
Учетная запись в binance
| #
Your article helped me a lot, is there any more related content? Thanks! https://accounts.binance.com/register?ref=P9L9FQKY
LecenesAppab
| #
Ищете ник инстаграм? Посетите сайт https://sovads.ru/ где вы найдете в широком ассортименте ники инстаграм. Мы занимаемся продажей привлекательных никнеймов и у вас будет красивый ник инстаграм или короткий ник инстаграм для удобства. Стоимость самая лучшая на рынке, а ассортимент магазина пополняется уникальными именами пользователей: словарными, короткими, красивыми и городскими никами. Короткие ники для инстаграм – преимущества очевидно!
Dragon547
| #
Обнаружил, что по финансам лучше всего читать здесь: https://hurma-credit.ru
Toliknaf
| #
darknet markets onion https://github.com/abacuslink4jjku/abacuslink – dark web drug marketplace
MarkNOlaf
| #
dark web markets dark web link
ThunderV9
| #
ТОП-10 ресурсов по финансам: https://lom-ard.ru
GhostByte
| #
Где смотреть актуальные данные о финансах? https://kikosrest.ru
Rabyitabe
| #
darknet markets url https://github.com/darkwebsitesyhshv/darkwebsites – dark web sites
Pingrutty
| #
best darknet markets https://github.com/aresonioncq0a7/aresonion – onion dark website
StormHunter
| #
Приветствую. Подскажите, где посмотреть https://bassein-elektrometallurg.ru информацию о финансах?
Volodyanaf
| #
darknet websites dark market
DonDonfaita
| #
dark web markets https://github.com/nexusdarkrtv1u/nexusdark – dark web market links
WilliamIRrutty
| #
darknet markets links dark web market urls
Cyber357
| #
Хочу понять в финансах. Что https://salgariint.ru посоветуете?
Kxyufaita
| #
dark markets 2025 darknet markets 2025
Wiltonsauff
| #
https://thechampions.ru/
Samuelhox
| #
На официальном сайте Kent Casino предусмотрены самые популярные способы пополнения и вывода средств. Вы сможете использовать банковские карты, электронные кошельки и криптовалюту для удобных и быстрых финансовых операций https://kent777.casino/
https://t.me/Gizbo_zerkalo_ru
| #
Онлайн казино: обзор самых популярных игр с бонусами
Современные платформы азартных развлечений предлагают игрокам множество увлекательных соревнований, каждый из которых имеет свои особенности и заманчивые предложения. Выбор для пользователя становится настоящим испытанием, особенно в условиях разнообразия форматов и интерфейсов. Более того, умение эффективно извлекать выгоду из акций и поощрений – важный аспект успешной игры. Обратите внимание на то, какие привилегии доступны для новичков и постоянных клиентов, чтобы сделать свои ставки максимально выгодными.
Среди трех категорий, к которым стоит обратиться, можно выделить слоты, карточные баталии и живые сессии. Слоты – это не только радость от вращения барабанов, но и широкий выбор тематик, разнообразие игровых функций и щедрые акционные предложения. Карточные конкурсы часто требуют более глубоких стратегий, но они тоже радуют разнообразием форматов и коэффициентов. Живые сессии приносят атмосферу реального заведения прямо к вашему экрану, позволяя взаимодействовать с профессиональными крупье и другими участниками.
Не забывайте исследовать доступные программы лояльности, которые могут значительно увеличить ваши шансы на выигрыш. За каждую ставку вы будете зарабатывать очки, которые затем можно обменивать на реальные деньги или другие привилегии. Это не только повышает интерес к процессу, но и создает дополнительные возможности для призов и подарков. В конечном итоге, грамотный подход к выбору развлечений и использования акций поможет вам получить удовольствие от хобби, а также ощутить финансовые выгоды.
Слоты с щедрыми бонусами
В мире виртуальных развлечений одними из самых привлекательных становятся игровые автоматы, предлагающие богатый набор привилегий. Каждый слот имеет свои уникальные особенности, которые делают его особенно интересным для пользователей.
1. Виды привилегий: Большинство автоматов предоставляют фриспины, что позволит игрокам вращать барабаны без риска. Размер таких вращений варьируется от нескольких до сотен, в зависимости от условий. Опция накопительных бонусов также встречается часто – игроки могут накапливать выигрышные позиции, что делает игру более стратегической.
2. Тематики и их влияние: Слоты с захватывающей тематикой, такие как приключения или мифология, зачастую предлагают повышенные привилегии. К примеру, игры на основе известных фильмов могут включать в себя дополнительные функции, которые активируются при достижении определенных результатов.
3. Интерактивные функции: Современные автоматы часто внедряют мультифункциональные игровые механики. Такие механизмы могут включать мини-игры, где игроки могут увеличивать свои шансы на выигрыш или получать дополнительные призы при выполнении специальных условий.
4. Рекомендуемые игры: Проверьте слоты с уникальными функциями, такими как Big Bad Wolf или Gonzo’s Quest. Эти автоматы отличаются не только визуальным оформлением, но и механикой, что позволяет максимально задействовать предлагаемые преимущества.
5. Стратегия выбора: При выборе игрового автомата стоит обращать внимание на уровень возврата игроку (RTP). Чем выше этот показатель, тем больше вероятность, что машина будет щедрой на выплаты. Выбирайте автоматы с RTP не ниже 96% для увеличения шансов на успех.
Виртуальные автоматы с интересными привилегиями предоставляют игрокам возможность сочетать азарт и стратегию, что делает процесс ещё более увлекательным и физиологически благоприятным. Не забывайте о правилах игры и устанавливайте лимиты, чтобы избежать неприятных ситуаций. Выбор правильного слота может обернуться не только приятным времяпрепровождением, но и солидным выигрышем.
Карточные игры и предложения
Карточные развлекательные мероприятия приобрели большую популярность среди любителей азарта благодаря привлекательному сочетанию стратегии и удачи. Среди них выделяются такие классические варианты, как блэкджек, покер и баккара. Каждая дисциплина предлагает уникальные возможности для игроков, а также заманчивые акционные предложения, способные увеличить банкролл.
Блэкджек – это игра, где удача и тактика идут рука об руку. Многие платформы предлагают специальные действия, такие как «бонус на стол», который дает возможность получить дополнительный кредит при первой ставке. Игроки могут использовать данный вариант, чтобы протестировать различные стратегии, особенно в рамках первых партий.
Покер, в частности, техасский холдем, является особенным направлением, которое требует от участников не только способности к расчету риска, но и навыков блефа. Платформы часто организуют турниры с призовыми фондами, которые значительно превышают обычные ставки. Специальные предложения на участие в таких событиях могут включать бесплатные входы или увеличенные раздачи фишек.
Баккара представляется интересным выбором для тех, кто предпочтительно изучает простые правила. Некоторые онлайн-сервисы предоставляют акции, позволяющие получить возврат части проигранных средств, что делает риски менее ощутимыми. Это создает возможность больше сосредоточиться на процессе игры.
Обратите внимание, что многие учреждения предлагают щедрые стартовые пакеты, которые могут включать не только денежные предложения, но и дополнительные карты для участия в карточных мероприятиях. Важно внимательно изучать условия, так как иногда могут быть ограничения по времени или минимальным ставкам.
Выбор подбираемой стратегии и понимание как работают акционные предложения могут существенно повлиять на общее впечатление от карточных сцен. Разумное использование акций и внимательное отношение к правилам помогут превратить обычный игровой процесс в успешное приключение.
https://t.me/otzyvy7k_bot
MarkNOlaf
| #
darknet markets 2025 dark web market list
Donaldlaf
| #
darknet links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet site
VortexHunter
| #
История внедрения финансов в проекте: https://crowd-com.ru
GhostNomad
| #
Доступно объяснено о финансах: https://vsekredity-chel.ru
Toliknaf
| #
dark market url https://github.com/abacuslink4jjku/abacuslink – dark websites
RobertDaw
| #
Best non Gamstop casino I’ve played at had fast support—nice!
non gamstop uk casinos with paypal
Sentinel7159
| #
Как https://lpandb.ru войти в тему финансов?
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Керамическая мишень АZO для распыления
Pingrutty
| #
onion dark website https://github.com/aresonioncq0a7/aresonion – dark markets 2025
WilliamIRrutty
| #
darkmarket link darknet site
Rabyitabe
| #
darknet marketplace https://github.com/darkwebsitesyhshv/darkwebsites – dark markets
Wiltonsauff
| #
champion slots официальный сайт
avtolombardekbsig
| #
кредит под птс с плохой кредитной историей
http://euhope.com/employer/epsontario2146/
кредит под залог птс грузового
Pioneer8500
| #
С чего https://alpha-sber.ru начать финансов?
Kxyufaita
| #
dark markets 2025 dark web market urls
BlueSpark
| #
Практическое применение финансов: https://vashi-okna42.ru
DonDonfaita
| #
darkmarket url https://github.com/nexusdarkrtv1u/nexusdark – darkmarket 2025
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их качество. Превосходное обслуживание – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы а также предоставлять решения под требования вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
поставляемая продукция:
Лента танталовая Ta-W10 Лента танталовая Ta-W10 – это высококачественный металл, обладающий исключительными свойствами. РћРЅ устойчив Рє РєРѕСЂСЂРѕР·РёРё Рё идеален для применения РІ условиях высокой температуры Рё агрессивной среды. Рспользуется РІ химической промышленности, Р° также РІ производстве электроники.Лента отлично РїРѕРґС…РѕРґРёС‚ для создания кабелей, прокладок Рё РґСЂСѓРіРёС… деталей, требующих надежности. Доступна РІ различных размерах Рё толщине, что позволяет легко подобрать необходимый вариант для вашего проекта. РќРµ упустите возможность купить Лента танталовая Ta-W10 Рё улучшить качество ваших изделий!
FNDaviditabe
| #
darknet drugs dark markets
1win_rlEr
| #
1 vin https://1win6016.ru .
Warden5625
| #
Как реализовать финансы? https://srg-collection.ru.
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наши товары:
Бронзовое внутреннее стопорное пружинное кольцо 62х1.7 мм БрАЖНМц9-4-4-1 ГОСТ 13943-86 Приобретите качественные бронзовые внутренние стопорные кольца по ГОСТ в Редметсплав.рф. Наши колечки отличаются превосходными техническими характеристиками, высоким качеством и надёжностью. Широкий ассортимент и гарантированное соответствие ГОСТ позволят удовлетворить потребности любого заказчика.
bankrotstvo v moskve_yoKi
| #
Узнайте мнение тех, кто уже прошел процедуру банкротства и списал свои долги http://bankrotstvo-v-moskve123.ru .
Andrewopero
| #
бюстгальтер Caitline с сетчатой вставкой Ищете идеальный бюстгальтер? Обратите внимание на Caitline! Отзывы покупательниц подтверждают: это сочетание комфорта и элегантности. Широкий размерный ряд, включая модели для больших размеров, позволяет каждой женщине найти подходящий вариант. В интернет-магазине Caitline вас ждет разнообразие моделей: с эффектом пуш-ап, для повседневной носки, с кружевом, для спорта и даже для особых случаев, таких как свадьба или вечерний выход.
binance registration
| #
Thank you for your sharing. I am worried that I lack creative ideas. It is your article that makes me full of hope. Thank you. But, I have a question, can you help me?
1win_kbEr
| #
1win официальный сайт регистрация 1win официальный сайт регистрация .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Гранулы ниобия 4N
Donaldlaf
| #
dark market https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet market list
Overlord7009
| #
Ошибки новичков в финансах: https://vista-profi.ru
Volodyanaf
| #
dark market 2025 darknet marketplace
Timothywax
| #
Основной вопрос импортеров в России сводится к одному: как оплатить инвойс за рубеж быстро и безопасно? Опасаясь штрафов и вторичных санкций, зарубежные банки пристально проверяют платежи из РФ, чтобы Ваши платежи в срок доходили до контрагента, и обходили блокировки и задержки, на сегодня существует рабочий способ – оплата через платежного агента. Кто такой платежный агент и как с ним работать мы рассказали на наших площадках Оплата инвойсов в Азии
Pingrutty
| #
darkmarket 2025 https://github.com/aresonioncq0a7/aresonion – dark web markets
WolfX6
| #
Как не сдаться при изучении финансов: https://24rd.ru
Toliknaf
| #
darkmarket https://github.com/abacuslink4jjku/abacuslink – dark web market links
WilliamIRrutty
| #
darknet market list dark market link
Omega_Spark
| #
Типичные промахи в https://aktivedengi.ru финансах.
1win_qdEr
| #
1wi. http://1win6016.ru .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Гранулы высокочистого ванадия
Kxyufaita
| #
darkmarket 2025 darkmarket list
mostbet_ghSn
| #
мостбет казино войти мостбет казино войти .
DavidJelry
| #
Przejrzalem portal Nelja i rozczarowalem sie. wyglad jest archaiczny, obsluga jest trudne, a przydatnych tresci jest malo. ciagle awarie, wszystko sie zacina, natretny spam przeszkadzaja w uzywanie. Bez watpliwosci, ze witryna jest martwa, poniewaz nic nie jest poprawiane. Brak pomocy, linki sa uszkodzone. Jesli potrzebujesz wartosciowego serwisu, lepiej jej unikac!
Rabyitabe
| #
onion dark website https://github.com/darkwebsitesyhshv/darkwebsites – darknet drugs
Sentinel1710
| #
Авторитетное мнение о финансах: https://mir-aquariuma72.ru
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их качество. Опытная поддержка – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы по мере того как находить ответы под особенности вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
РљСЂСѓРі ниобиевый 50БТЦ-Р’Р” РљСЂСѓРі ниобиевый 50БТЦ-Р’Р” – это высококачественный РїСЂРѕРґСѓРєС‚, разработанный для осуществления операций СЃ ниобием. Ртот РєСЂСѓРі обладает уникальными свойствами, которые обеспечивают высокую прочность Рё РєРѕСЂСЂРѕР·РёРѕРЅРЅСѓСЋ стойкость. Благодаря СЃРІРѕРёРј характеристикам, РљСЂСѓРі ниобиевый 50БТЦ-Р’Р” идеально РїРѕРґС…РѕРґРёС‚ для использования РІ различных промышленных сферах, включая авиастроение Рё атомную энергетику. Р’С‹ можете купить РљСЂСѓРі ниобиевый 50БТЦ-Р’Р”, чтобы обеспечить надежность Рё долговечность СЃРІРѕРёС… изделий. РќРµ упустите возможность улучшить качество своей продукции!
OmegaV3
| #
Шутки про финансы: https://rezinplit.ru
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Гранулы ниобия 4N
FNDaviditabe
| #
tor drug market dark web link
MarkNOlaf
| #
darknet markets 2025 dark markets
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
поставляемая продукция:
Лист судостроительный 24x1000x3000 D EN10025:2 Приобретите высококачественные листы судостроительные от компании Редметсплав.рф для надежного и долговечного использования в вашем судостроительном проекте. Различные размеры и толщины, прочность и устойчивость к коррозии делают эти листы идеальным выбором для любого судостроительного проекта.
mostbet_luSn
| #
мрстбет https://mostbet789.ru/ .
RobertDaw
| #
Casino non Gamstop platforms are perfect for casual gaming—love it!
casino non on gamstop
mostbet_ngen
| #
mostber ashapiter0.forum24.ru/?1-19-0-00001444-000-0-0-1742819001 .
DonDonfaita
| #
darknet drug market https://github.com/nexusdarkrtv1u/nexusdark – dark websites
1win_msKi
| #
1вин онлайн https://zdorovie.forum24.ru/?1-7-0-00000231-000-0-0-1742818050 .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наши товары:
Керамическая мишень для распыления Р±РѕСЂР° – плоская
GeorgeTom
| #
Source:
– royaljackpotcasino.org
porno_hyKt
| #
порно порно .
DewurKiz
| #
Компания Глобал ЖБИ https://global-gbi.ru/ является крупным производителем изделий из бетона и железобетона в Санкт-Петербурге. Посетите наш сайт, ознакомьтесь с широкой номенклатурой производимого товара по выгодным ценам. Мы осуществляем оперативную и быструю доставку продукции. Также на заводе ЖБИ, можно заказать производство конструкций разного типа и назначения для строительства дорог, взлетных полос, тротуаров, пешеходных зон, зданий, сооружений, внутридомовых конструкций, инженерных коммуникаций и т.д.
Cyber_Nomad
| #
Факты и вымысел о https://kpk-kk.ru финансах.
MatthewHen
| #
Онлайн-казино 7k — это продвинутая платформа для игр с ставками с широким выбором игр. Здесь можно найти известные видеослоты, настольные игры и игры с живыми дилерами с опытными дилерами. Дизайн пространный и простой в навигации, что делает игру приятной и увлекательной. На https://7k740.casino/ru/ предусмотрены классные призы для любителей азартных игр. В целом, казино предоставляет отличный опыт для каждого игрока.
mostbet_ooen
| #
мостбет скачать на андроид https://ashapiter0.forum24.ru/?1-19-0-00001444-000-0-0-1742819001 .
Alpha_Guardian
| #
Нестандартный подход к финансам: https://perspectiva-leasing.ru
1win_htKi
| #
1win прямой эфир zdorovie.forum24.ru/?1-7-0-00000231-000-0-0-1742818050 .
urwork
| #
Юридические услуги https://urwork.ru в Санкт-Петербурге и Москве – от консультации до защиты интересов в суде. Оперативно, надежно и с гарантией конфиденциальности.
Alpha707
| #
Простой язык о финансах: https://narodnakassa.ru
Pingrutty
| #
onion dark website https://github.com/aresonioncq0a7/aresonion – dark web market
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица товара подтверждена требуемыми документами, подтверждающими их качество. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы по мере того как предоставлять решения под требования вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в гибкости нашего предложения
поставляемая продукция:
РџРѕРєРѕРІРєР° вольфрамовая РР’Р-3 РџРѕРєРѕРІРєР° вольфрамовая РР’Р-3 представляет СЃРѕР±РѕР№ высококачественный РїСЂРѕРґСѓРєС‚, используемый РІ различных отраслях. РћРЅР° обладает отличными механическими свойствами Рё высокой термостойкостью, что делает ее идеальным выбором для производства деталей, работающих РІ экстремальных условиях. РџРѕРєРѕРІРєР° гарантирует надежность Рё долговечность, благодаря чему становится незаменимой для РјРЅРѕРіРёС… промышленных приложений. Если РІС‹ хотите обеспечить эффективность своей работы, вам стоит купить РџРѕРєРѕРІРєР° вольфрамовая РР’Р-3. Получите РїСЂРѕРґСѓРєС‚, который отвечает высоким стандартам качества Рё надежности.
Zooma казино сайт
| #
Добро пожаловать в Zooma Casino – современная игровая платформа с уникальными возможностями! Здесь вас ждут лучшие азартные развлечения от ведущих разработчиков. https://zooma-play.buzz/ и погрузитесь в мир высоких выигрышей!
Что делает Zooma Casino идеальным выбором?
Популярные слоты с бонусными раундами.
Программы лояльности с кэшбеком и фриспинами.
Гарантированные выигрыши с максимальной безопасностью.
Адаптивная мобильная версия без зависаний и лагов.
Дружелюбные операторы готовы ответить на все вопросы.
Присоединяйтесь к Zooma Casino и воспользоваться всеми преимуществами!
mostbet_suSn
| #
mostbet скачать на телефон бесплатно андроид https://mostbet789.ru/ .
Omega_Spark
| #
Лучшее что https://buhprofihelp.ru находил по теме финансов.
Donaldlaf
| #
dark web marketplaces https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – tor drug market
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наши товары:
Медный тройник РїРѕРґ пайку СЃ направленным отводом 28С…23С…0.9 РјРј 18.4С…20.4 РјРј твердая пайка Рњ2Р Рњ ГОСТ Р 52922-2008 Купить качественные медные тройники РїРѕРґ пайку для надежных Рё долговечных соединений РІ трубопроводных системах. Разнообразие размеров Рё конфигураций, высокая теплопроводность Рё устойчивость Рє РєРѕСЂСЂРѕР·РёРё. Простая установка Рё надежность соединения. Рдеальное решение для различных проектов. Редметсплав.СЂС„
nvblfurn
| #
https://blotos.ru/news/tri_v_ryad__igrayte_onlayn_besplatno_i_bez_registracii.html кто ты из гарри поттера тест
avtolombardkazancoN
| #
автоломбард под залог
https://icmimarlikdergisi.com/kariyer/companies/zaim-pod-zalog-pts-19150/
кредит под залог машины
WilliamIRrutty
| #
dark web marketplaces darknet drug store
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их происхождение. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы а также адаптировать решения под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
РўСЂСѓР±Р° магниевая ISO-MC21210 – BS ISO 16220 Пруток магниевый ISO-MC21210 – BS ISO 16220 идеально РїРѕРґС…РѕРґРёС‚ для различных промышленных приложений благодаря СЃРІРѕРёРј отличным механическим свойствам Рё легкости. Ртот материал обладает высокой РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью Рё отличается хорошей свариваемостью, что делает его популярным выбором РІ обработке металлов. Если РІС‹ ищете надежный Рё качественный РїСЂРѕРґСѓРєС‚, то РїРѕРєСѓРїРєР° прутка магниевого ISO-MC21210 – BS ISO 16220 станет отличным решением для ваших нужд. РќРµ упустите возможность обеспечить СЃРІРѕР№ проект качественным материалом!
mostbet_ynen
| #
mostbet http://www.ashapiter0.forum24.ru/?1-19-0-00001444-000-0-0-1742819001 .
Toliknaf
| #
darknet sites https://github.com/abacuslink4jjku/abacuslink – darknet markets links
1win_hlKi
| #
1win официальный http://zdorovie.forum24.ru/?1-7-0-00000231-000-0-0-1742818050/ .
Sarozhdab
| #
На сайте https://prokat.car-trip.ru/ вы сможете выбрать автомобиль для того, чтобы воспользоваться прокатом. Только сейчас действует скидка 10% на определенные автомобили. Есть те машины, которые относятся к классу Комфорт, Бизнес, Стандарт. В автопарке имеются и автодома, кабриолеты. Высококлассные менеджеры всегда находятся на связи, чтобы вы задали им любой вопрос. Отсутствуют скрытые платежи, нет залога, ограничения относительно пробега. Все клиенты получают право воспользоваться дисконтной картой.
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наши товары:
Бронзовое внутреннее стопорное пружинное кольцо 200х3 мм БрБНТ1.9 ГОСТ 13943-86 Приобретите качественные бронзовые внутренние стопорные кольца по ГОСТ в Редметсплав.рф. Наши колечки отличаются превосходными техническими характеристиками, высоким качеством и надёжностью. Широкий ассортимент и гарантированное соответствие ГОСТ позволят удовлетворить потребности любого заказчика.
Kxyufaita
| #
darkmarket list darknet sites
Byte3012
| #
Авторитетное мнение о финансах: https://dolganet-nn.ru
RedReaper
| #
Где брать актуальные данные о финансах? https://zoovetbor.ru
FNDaviditabe
| #
dark market link darknet markets links
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их качество. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы а также предоставлять решения под специфику вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Пруток молибденовый ЦМ-2Рђ Пруток молибденовый ЦМ-2Рђ – это высококачественный металл, обладающий отличной прочностью Рё жаростойкостью. РћРЅ широко используется РІ различных отраслях, включая аэрокосмическую Рё промышленную. Молибденовые прутки применяются для создания деталей, которые подвергаются высокому температурному воздействию. Ртот материал устойчив Рє РєРѕСЂСЂРѕР·РёРё Рё обеспечивает надежность РІ эксплуатации. Если РІС‹ ищете надежное решение для СЃРІРѕРёС… проектов, вам стоит купить Пруток молибденовый ЦМ-2Рђ. Выберите качественный РїСЂРѕРґСѓРєС‚, который удовлетворит РІСЃРµ ваши требования.
MarkNOlaf
| #
dark web marketplaces dark market list
Master3905
| #
Эксклюзивное интервью с экспертом по финансам: https://lombard-gran.ru
Punuyuseks
| #
Доставка комплексных обедов в Алматы для ваших сотрудников или на дом на сайте https://bvd.kz/ – у нас широкий ассортимент и выбор блюд по отличным ценам! Ознакомьтесь на сайте с составами и ценами на комплексные обеды. Заключаем договоры на доставку обедов в офисы и административные учреждения, доставляем обеды в школы и детские сады, где нет собственной кухни, в магазины и прочие подобные учреждения, у сотрудников которых нет возможности пообедать в столовой. Подробнее на сайте.
Rabyitabe
| #
dark market 2025 https://github.com/darkwebsitesyhshv/darkwebsites – darkmarket link
Volodyanaf
| #
darknet markets url darknet market list
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
Наша продукция:
Медный стандартный однораструбный отвод 90 градусов под пайку 35х29х1 мм 23х25 мм мягкая пайка М2т ГОСТ Р52922-2008 Покупайте качественные медные однораструбные отводы 90 градусов под пайку от Редметсплав. Надежные и долговечные соединения для водоснабжения, отопления и кондиционирования. Быстрая установка и эстетичный вид. Доставка по России.
Steel208
| #
Может кто знает разбирается в финансах? Куда зайти? https://weddingkms.ru
Vickiuseds
| #
Popular Aviator game is a next-gen online game where results depend on your strategic timing.
How to play: a plane starts rising into the sky, and with every second, the reward grows higher.
Your goal is to press “Stop” before the plane flies away. If you’re fast enough — you collect your prize. Wait too long — and you lose everything.
That’s why Aviator is a balance of skill and luck.
Visit https://9323fb.com/how-to-deposit-money-to-aviator-2024-fast-safe-beginner-friendly/ to learn how to make fast, safe, and beginner-friendly deposits for Aviator.
Play with ease thanks to:
– a user-friendly interface
– no-delay transfers
– anytime support service
– cross-device flexibility
Play whenever you want and win big before the plane disappears!
RandyMup
| #
https://лучшие-адвокаты-москвы.рф
1xbet_noPi
| #
Откройте для себя мир ставок с 1xbet, начать играть.
Добро пожаловать в мир ставок с 1xbet, в индустрии.
1xbet предлагает щедрые бонусы, акции.
Скорее ставьте на свои любимые команды с 1xbet, наслаждайтесь.
1xbet – ваш портал в мир лайв-ставок, вы всегда на шаг впереди.
1xbet предлагает широкую линейку ставок, создавайте.
На 1xbet найдётся ставку для каждого, от спорта до киберспорта.
Смотрите матчи в режиме реального времени с 1xbet, наслаждайтесь просмотром.
Деньги на вашем счете с 1xbet за считанные минуты, не ждите.
Получите инсайдерскую информацию с 1xbet, будьте всегда на шаг впереди.
Ставьте с уверенностью на 1xbet, мы ценим вашу конфиденциальность.
Промокоды и специальные предложения на 1xbet, воспользуйтесь шансом.
Ставьте смело с 1xbet, выберите 1xbet для своей игры.
Чат поддержки 24/7 на 1xbet, мы рядом, чтобы помочь.
Регулярные турниры и конкурсы на 1xbet, воспользуйтесь шансом.
Ставьте в любое время и в любом месте с 1xbet, сделайте ставки на ходу.
Дайте себе преимущества с 1xbet, будьте стратегом.
Простая регистрация на 1xbet, доступ к азарту.
1xbet – это азарт, который ждет вас, начните выигрывать.
Не упустите уникальные возможности на 1xbet, развивайте свои навыки.
Keyword https://1xbet-login-egypt.com/ .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Мишень для распыления сплава AlCu
DonDonfaita
| #
dark web marketplaces https://github.com/nexusdarkrtv1u/nexusdark – darkmarket list
Samuellem
| #
Запросите персональную консультацию по выбору экспонатов – узнайте больше информации https://blacksocially.com/Artefactad
Pingrutty
| #
dark markets https://github.com/aresonioncq0a7/aresonion – darknet markets onion address
Guardian9721
| #
Интерактивный курс по https://vnagaevo.ru финансам.
StormStriker
| #
Подборка ресурсов по финансам: https://samara-investor.ru
WilliamIRrutty
| #
darknet drug store darknet websites
Cyber332
| #
Как выбрать лучшее решение https://kpkazbuka.ru для финансов.
Donaldlaf
| #
darkmarket list https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet market lists
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наша продукция:
Алюминиево-хромовая мишень для распыления (AlCr)
OmegaStriker
| #
Что нельзя делать с https://rtsfi.ru финансами?
Deyiramnaify
| #
Ищете фильтры для воды, септики, смесители, насосы? Посетите https://shoph2o.ru/ где вы сможете найти и купить в интернет магазине «H2O – Территория чистой воды» – системы водоочистки любой сложности. У нас широкий ассортимент продукции по выгодным ценам и быстрой доставкой. Также у нас можно заказать услуги анализа воды, монтаж септиков, ремонт систем водоподготовки и фильтров для воды. И их сервисное обслуживание.
Kxyufaita
| #
dark market onion dark web market list
Toliknaf
| #
darknet markets url https://github.com/abacuslink4jjku/abacuslink – dark web market urls
MarkNOlaf
| #
bitcoin dark web darknet drugs
FNDaviditabe
| #
dark market list darkmarket 2025
Steel680
| #
Обнаружил, что по финансам лучше https://realtor38.ru всего смотреть здесь.
vetivsiz
| #
Автосервис в СПб СТО Приморский большой спектр услуг предлагает. Мы поможем починить неисправность в автомобиле. Гарантируем доступные цены и высочайшее качество работы. При необходимости профессионально проконсультируем вас. Ищете автосервис питер? Sto812.com – тут полезная информация представлена. На сайте вы увидите ряд важных проблем, которые появляются при приобретении некачественных фильтрующих элементов. Узнаете, какими свойствами обладает тормозная жидкость. Обладаем необходимым опытом и инструментом. Работаем быстро. Обращайтесь к нам!
CharlesZoony
| #
Информационный портал посвященный обучению стройке и ремонту своими руками. Лучшие материалы на тему большого строительства, а также интересные советы по дизайну https://aquabiodesign.ru/
Rabyitabe
| #
darknet markets onion https://github.com/darkwebsitesyhshv/darkwebsites – dark web markets
Reaper1715
| #
Забавные моменты в финансах: https://amalfd.ru
BohacMig
| #
На сайте https://face-me.ru/ вы сможете попробовать все возможности нейросетей и всего за несколько секунд превратить любое фото в видео. Инновационные и уникальные технологии дают возможность получить роскошную, яркую и любопытную фотосессию без профессионального оборудования, студии. Вы сможете создать фантастические образы и удивительные портреты в соответствии с предпочтениями. Эта программа дарит вам огромное количество возможностей и вдохновение. Вы каждый раз будете получать удивительные, красивые образы, которые всегда будут с вами.
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Опытная поддержка – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы и находить ответы под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
РўСЂСѓР±Р° магниевая MgTh3Zn2Zr Пруток магниевый MgTh3Zn2Zr представляет СЃРѕР±РѕР№ уникальный легированный сплав, который отличается высокой прочностью Рё РЅРёР·РєРёРј весом. Рспользуется РІ aerospace, автомобильной Рё энергетической отраслях. Данный материал обладает отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью Рё хорошей сваркой, что делает его идеальным выбором для создания прочных конструкций. Если РІС‹ хотите улучшить эффективность СЃРІРѕРёС… проектов, то вам нужно купить Пруток магниевый MgTh3Zn2Zr. Ртот РїСЂРѕРґСѓРєС‚ обеспечит надежность Рё долговечность, необходимые для современных решений РІ инженерии.
1win_rnPr
| #
1 win вход https://1win6017.ru .
Pingrutty
| #
bitcoin dark web https://github.com/aresonioncq0a7/aresonion – dark web markets
ZecohSwexy
| #
ТТК — транспортная компания, специализирующаяся на перевозках автомобилей на автовозах по всей России. Наша компания имеет опыт перевозок автовозами более 12 лет. Свыше 40 единиц техники позволяют осуществлять регулярные рейсы по всей России. У нас можно заказать автовозы по России для транспортировки автомобиля любого типа: седана, универсала, кроссовера, внедорожника и даже автомобиля в аварийном состоянии. Качественный сервис по доступным ценам. Обеспечиваем высокое качество и безопасность на каждом этапе перевозки. Широкая география перевозок позволяет обеспечить перевозку машин в любую точку страны: Москва, Санкт-Петербург, Владивосток, Екатеринбург, Иркутск, Казань, Кемерово, Краснодар, Красноярск, Новосибирск, Омск, Пермь, Ростов-на-Дону, Самара, Симферополь, Сочи, Ставрополь, Сургут, Тольятти, Томск, Тюмень, Улан-Удэ, Уфа, Хабаровск, Челябинск, Чита и в другие города России. Практически во всех городах России мы имеем специальные охраняемые стоянки для авто. Работаем 24/7. Сайт нашей компании ТТК: https://avtovoz7.ru/
NightZ2
| #
https://success-finance-ipoteka.ru Какие платформы использовать для финансов?
Dragon_Master
| #
Мемы про финансы: https://kpk-cfp.ru
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
Наша продукция:
Двухраструбный медный обвод под пайку 80х68х1.8 мм 35.5х38.5 мм твердая пайка М2т ГОСТ Р52922-2008 Выберите качественные Медные двухраструбные обводы под пайку различных размеров и диаметров. Надежное соединение труб, высокая устойчивость к коррозии и превосходная теплопроводность. Узнайте больше о нашей продукции и найдите оптимальное решение для вашего проекта.
DonDonfaita
| #
dark market url https://github.com/nexusdarkrtv1u/nexusdark – darknet markets links
avtolombardkazancoN
| #
займ под залог машины
https://emploi-securite.com/societes/jobsandbussiness4839/
займ под птс казань
Derricksaisy
| #
Флорист создал настоящий цветочный шедевр
доставка цветов в томске
1win_gpPr
| #
1вин кыргызстан http://www.1win6017.ru .
Volodyanaf
| #
dark web market links darknet markets
RobertDaw
| #
Non Gamstop casinos no deposit offers are my new favorite thing—sweet!
non-gamstop casino no deposit codes
WilliamIRrutty
| #
dark web market darkmarket
Red_Reaper
| #
Подробный разбор финансов: https://zaimsosed.ru
DragonV7
| #
Как реализовали финансов https://kpkbelkred.ru в компании.
Donaldlaf
| #
dark markets 2025 https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet market list
Kxyufaita
| #
dark web drug marketplace dark web market urls
viaplay
| #
certainly like your web site however you need to test the spelling
on quite a few of your posts. Several of them
are rife with spelling problems and I in finding it very bothersome to tell the
reality then again I will certainly come again again.
My page – viaplay
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их происхождение. Превосходное обслуживание – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы а также находить ответы под специфику вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
РўСЂСѓР±Р° магниевая G-A9Z1Y4 – AFNOR NF A57-705 Пруток магниевый G-A9Z1Y4 – AFNOR NF A57-705 представляет СЃРѕР±РѕР№ высококачественный РїСЂРѕРґСѓРєС‚, обладающий отличными механическими свойствами Рё стойкостью Рє РєРѕСЂСЂРѕР·РёРё. РћРЅ идеально РїРѕРґС…РѕРґРёС‚ для использования РІ различных отраслях, включая авиастроение, автомобилестроение Рё РґСЂСѓРіРёРµ высокотехнологичные сферы. Благодаря своей легкости, пруток магниевый G-A9Z1Y4 – AFNOR NF A57-705 способствует снижению веса конструкции, что делает его особенно востребованным. РќРµ упустите возможность купить Пруток магниевый G-A9Z1Y4 – AFNOR NF A57-705 для повышения эффективности ваших проектов.
FNDaviditabe
| #
dark market 2025 best darknet markets
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Керамическая мишень Mos2, плоская
Master7176
| #
Малоизвестные фишки финансов: https://evs-ufa.ru
Toliknaf
| #
darknet markets https://github.com/abacuslink4jjku/abacuslink – darkmarket list
GhostReaper
| #
Лично мне помог этот ресурс по финансам: https://grozfinline.ru
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наша продукция:
Титановая труба 105х5.5х1500 мм ПТ1М ГОСТ 22897-86 Откройте мир прочных и надежных титановых труб с компанией Редметсплав. Наши высококачественные титановые трубы сочетают в себе устойчивость к коррозии, износостойкость и легкий вес, делая их незаменимым материалом для применения в различных сферах промышленности. Широкий выбор различных диаметров и толщин, а также индивидуальные решения для вашего производства. Гарантированное качество и надежность поставок.
1win_lmPr
| #
1win. pro http://1win6017.ru/ .
Pioneer4738
| #
5 главных ошибок при работе https://stafpro.ru с финансами.
Pingrutty
| #
dark market link https://github.com/aresonioncq0a7/aresonion – darknet market
ShaneJerve
| #
https://firelak.ru В современном строительстве и отделке, где дерево занимает важное место, обеспечение пожарной безопасности становится приоритетной задачей. Огнезащитный лак – это эффективное решение для защиты деревянных конструкций и элементов интерьера от возгорания и распространения пламени.
Rabyitabe
| #
darknet markets 2025 https://github.com/darkwebsitesyhshv/darkwebsites – darknet markets onion
Blue_Master
| #
Практическое https://dolce-vita74.ru применение финансов.
BillyBor
| #
Забудьте о дискомфорте и неуверенности – имплантация зубов в нашей клинике возвращает естественную красоту и функциональность вашей улыбки http://forum.omnicomm.pro/index.php?action=profile;u=16464
Samuellem
| #
Экспонаты с подтвержденной подлинностью и экспертной оценкой – дополнительные сведения тут http://www.askmap.net/location/7303484/%D0%BE%D0%B0%D1%8D/artefactum-gallery
BillyBor
| #
Наши специалисты подбирают индивидуальные решения для каждого пациента, предлагая лучшие методики имплантации зубов и протезирования https://a6-allroad.ru/member.php?u=14441
WilliamIRrutty
| #
darknet market lists dark market
DonDonfaita
| #
dark websites https://github.com/nexusdarkrtv1u/nexusdark – darkmarket
Hacker2295
| #
Обнаружил, что https://partnerinvest40.ru по финансам лучше всего смотреть здесь.
SteelVoyager
| #
Современные методы по финансам: https://deshevosantehnika.ru
Red284
| #
https://leasing-sibir.ru Новые подходы в финансах.
HarryTaips
| #
Лучший генератор QR-кодов
Kxyufaita
| #
darkmarket list darknet markets
MarkNOlaf
| #
darkmarket url dark market url
JakohlSed
| #
На сайте https://fguard.ru/ почитайте статьи на самую разную тему. К примеру, кибермошенничество, для каких целей используется сеточка, которая на двери микроволновки. Здесь представлена вся нужная информация о смарт-часах и о том, чем они отличаются от бизнес-браслетов. Для того чтобы подыскать нужную информацию, воспользуйтесь специальным рубрикатором. На сайте вы найдете записи, которые касаются нейросетей, гаджетов. Представлены и интересные рекомендации, которые будут необходимы каждому. Имеются данные и про технологии.
FNDaviditabe
| #
darknet marketplace dark market list
Donaldlaf
| #
darknet markets 2025 https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet markets 2025
Volodyanaf
| #
dark web drug marketplace darkmarket link
Pingrutty
| #
dark market 2025 https://github.com/aresonioncq0a7/aresonion – darknet marketplace
DaltonJasse
| #
seo оптимизация цена https://prodvizhenietargeting.ru
EdwardCen
| #
поисковое продвижение сайта заказать https://prodvizheniestatya.ru
Toliknaf
| #
dark market https://github.com/abacuslink4jjku/abacuslink – darkmarket link
SteveViche
| #
сео оптимизация сайта купить seoveb-marketing.ru
JustinCoict
| #
Как получить китайскую визу Планируете поездку в Китай? Наш визовый центр предоставляет полный спектр услуг по оформлению виз в Китай для различных целей: туризм, бизнес, учеба. Мы оказываем профессиональные консультации по всем вопросам, связанным с визовым режимом Китая и подготовкой необходимых документов. Мы поможем вам оформить туристическую визу, бизнес-визу, а также визу для студентов. Наши специалисты подробно проконсультируют вас о необходимых документах, сроках получения визы и ответят на все ваши вопросы. Мы также предлагаем услуги экспресс-визы для тех, кому требуется срочное оформление.
Eagle_Master
| #
Забавные моменты https://gretanv.ru в финансах.
Xirodrdaf
| #
Новини та інформація про місто Кропивницький. Сучасний портал https://kropyvnytskyi.kr.ua/ про місто Кропивницький. Актуальна довідкова інформація про місто, новини, публікації.
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их происхождение. Опытная поддержка – наш стандарт – мы на связи, чтобы улаживать ваши вопросы и находить ответы под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в гибкости нашего предложения
поставляемая продукция:
Полоса магниевая MAG 121 – BS 3370 Порошок магниевый MAG 12 – BS 2970 – это высококачественный РїСЂРѕРґСѓРєС‚, предназначенный для различных промышленных Рё научных задач. РћРЅ обладает отличными антикоррозийными свойствами Рё значительно улучшает характеристики конечных изделий. Р’ производстве используемый магний отличается стабильностью Рё чистотой, что обеспечивает высокую эффективность. Рщете надежный Рё качественный порошок? Купить Порошок магниевый MAG 12 – BS 2970 стоит, чтобы оценить РІСЃРµ его преимущества РІ эксплуатации Рё долговечности. Рто идеальный выбор для тех, кто ценит качество Рё надежность РІ СЃРІРѕРёС… проектах.
RobertDaw
| #
Non Gamstop casino no deposit bonus got me started—love it!
uk casino no gamstop
ShadowProtocol
| #
Как монетизировать финансы https://izumrud585.ru в бизнесе?
Rabyitabe
| #
darknet markets 2025 https://github.com/darkwebsitesyhshv/darkwebsites – dark web market list
HarryTaips
| #
бесплатный онлайн генератор QR кодов
WilliamIRrutty
| #
dark market 2025 dark market onion
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наша продукция:
Контакт РёР· драгоценных металлов платиновый 2С…9 РјРј ПлРд90-10 РўРЈ Рзысканные контакты РёР· золота, серебра Рё платины РѕС‚ Редметсплав. Высочайшее качество, уникальный дизайн Рё эксклюзивные предложения для постоянных клиентов. Добавьте роскошь Рё стиль вашему образу!
HarryTaips
| #
https://qrkoder.ru/
DonDonfaita
| #
onion dark website https://github.com/nexusdarkrtv1u/nexusdark – darknet links
Kxyufaita
| #
darkmarket list bitcoin dark web
Felixvah
| #
https://logistt.ru/services/delivery/china
FNDaviditabe
| #
darkmarket link darkmarket
Omega_Nomad
| #
Памятка для работы с финансами: https://avtokompleks222.ru
avtolombardkemerovosig
| #
срочный кредит под залог птс
https://mastercare.care/employer/avtolombard-zalog-invest9408/
автоломбард залог птс
Pingrutty
| #
dark web drug marketplace https://github.com/aresonioncq0a7/aresonion – dark web markets
Donaldlaf
| #
darknet links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark web marketplaces
IronZ7
| #
Полное руководство https://delovoipodhodzaim.ru по финансам от А до Я.
Volodyanaf
| #
dark web drug marketplace darkmarket list
MichaelFus
| #
пегас туроператор Компания пегас является одним из лидеров рынка и работает в сфере международного туризма, предлагая выгодные туры в Таиланд, Турцию, Египет и другие страны мира. Подбор тура и заказ осуществляется в удобном личном кабинете, где можно забронировать отель, купить билеты и оформить путевку без переплат онлайн. Турфирма Pegas работает с проверенными отелями и предлагает широкий выбор направлений и услуг, включая авиабилеты, круизы, трансферы, путевки и подбор лучших отелей для проживания.
Toliknaf
| #
dark market link https://github.com/abacuslink4jjku/abacuslink – darknet websites
WilliamIRrutty
| #
dark web market darknet drug links
OmegaSlayer
| #
Хочу понять в финансах. Что https://primaveracafe.ru посоветуете?
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Плоская мишень из иттербия
Rabyitabe
| #
dark market 2025 https://github.com/darkwebsitesyhshv/darkwebsites – bitcoin dark web
GilihrdBuh
| #
На сайте https://avtomastera.net вы найдете различную полезную, актуальную информацию, которая касается ремонта автомобилей. Здесь представлены самые разные рекомендации, которые обязательно помогут мастерам, ответят на многочисленные вопросы, расскажут обо всех нюансах. Вся информация поделена на категории, чтобы было легче и быстрее сориентироваться. Для того чтобы получить доступ ко всем функциям, пройдите регистрацию. Здесь также вы найдете и программы, которые применяются для диагностики.
Kxyufaita
| #
dark web marketplaces darknet drug links
MarkNOlaf
| #
darknet drug store darknet market
FNDaviditabe
| #
dark markets 2025 darknet markets url
CyberWarden
| #
Мемы про финансы: https://oooaleks.ru
DonDonfaita
| #
darknet drug links https://github.com/nexusdarkrtv1u/nexusdark – darknet markets
Pingrutty
| #
dark web market urls https://github.com/aresonioncq0a7/aresonion – dark market
Carltonacets
| #
продвижение сайта заказать продвижение сайта цены
Clubnika Bitcoin
| #
Clubnika Casino is your chance to dive into the exciting world of gambling and win generous prizes.
We offer a wide selection of games, including classic
slots, roulette, blackjack, and unique live dealer games.
Every game at our casino is a chance to win, and security
and fairness are always our top priority.
Why is top casino providers the best choice for gambling players?
In our casino, every player can count on generous bonuses, free spins,
and exclusive offers. Moreover, we ensure fast withdrawals and round-the-clock support, so you can focus on playing.
When is the best time to start playing at Clubnika Casino?
Sign up at Clubnika Casino and get bonuses that
will immediately increase your chances of winning. Here’s what awaits you:
Generous bonuses and free spins for new players.
Promotions and tournaments with big prizes.
Every month we update our game selection, adding new exciting slots
and table games.
Clubnika Casino is the perfect place for those who want to play
and win. https://clubnika-casinonexus.skin/
Waynesailm
| #
Планируете каникулы? купить https://camp-centr.com! Интересные программы, безопасность, забота и яркие эмоции. Бронируйте заранее — количество мест ограничено!
Night37
| #
Эксперты рекомендуют эти ресурсы по финансам: https://rentmegapolis.ru
energoptoru
| #
Выполняем проектирование https://energopto.ru и монтаж всех видов инженерных систем для жилых и коммерческих объектов. Профессиональный подход, сертифицированное оборудование, гарантия качества.
Donaldlaf
| #
darknet markets onion https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet websites
WilliamIRrutty
| #
darknet marketplace dark markets 2025
1win_basr
| #
1вин вход с компьютера https://1win6013.ru/ .
1win_bjml
| #
сайт 1win https://www.knowledge.forum24.ru/?1-0-0-00000101-000-0-0-1742817704 .
1win_woMn
| #
1win скачать kg 1win9109.ru .
mostbet_nyka
| #
мостбет вход http://www.mostbet6004.ru .
Toliknaf
| #
dark market 2025 https://github.com/abacuslink4jjku/abacuslink – dark market url
RobertDaw
| #
Non Gamstop casinos have bonuses that actually pay off—wow!
non gamstop casino in wales
Vortex_Striker
| #
Какие https://ognibarnaula22.ru технологии использовать для финансов?
Kxyufaita
| #
dark markets 2025 darknet drug market
1win_hdsr
| #
1win сайт 1win сайт .
CharlesGes
| #
Играю в онлайн казино ради отдыха, а вы?
https://telegra.ph/Licenzionnoe-kazino-Vavada-i-novye-bonusy-dlya-igrokov-03-27
1win_pkml
| #
1вин про http://knowledge.forum24.ru/?1-0-0-00000101-000-0-0-1742817704/ .
1win_ncMn
| #
1 цшт http://1win9109.ru .
MarkNOlaf
| #
darknet drugs dark web market
mostbet_ozka
| #
mostbet https://mostbet6004.ru .
avtolombardkemerovosig
| #
займ под залог птс
https://complete-jobs.co.uk/employer/ignitionadvertising6458
автоломбард в кемерово под залог
Rabyitabe
| #
darknet markets onion https://github.com/darkwebsitesyhshv/darkwebsites – darkmarket list
SilverZ6
| #
Типичные промахи в финансах: https://dixi59.ru
Pingrutty
| #
darkmarkets https://github.com/aresonioncq0a7/aresonion – darknet market
1win_zysr
| #
1 win kg http://www.1win6013.ru .
1win_ucml
| #
1вин партнерка https://www.knowledge.forum24.ru/?1-0-0-00000101-000-0-0-1742817704 .
1win_ctMn
| #
1вин вход https://1win9109.ru/ .
DonDonfaita
| #
darknet markets links https://github.com/nexusdarkrtv1u/nexusdark – darknet markets onion
mostbet_dxka
| #
мостбет кг мостбет кг .
Vincenttef
| #
бесплатный онлайн генератор QR кодов
WilliamIRrutty
| #
darknet markets onion dark web market urls
DragonSlayer
| #
Обнаружил, что по финансам лучше всего читать здесь: https://sochiperm.ru
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наши товары:
Керамическая мишень Ta2O5
Vortex_Fang
| #
Особенности реализации финансов: https://kvartned.ru
Felixvah
| #
Проджект Логистикс
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена соответствующими документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы по мере того как предоставлять решения под специфику вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наши товары:
РљСЂСѓРі магниевый РњР›12 – ГОСТ 2856-79 Фольга магниевая РњР›12 – ГОСТ 2856-79 предназначена для использования РІ различных отраслях, таких как электроника, машиностроение Рё строительство. Рта фольга обладает высокой прочностью Рё легкостью, что делает ее идеальным материалом для создания защитных покрытий Рё теплоизоляции. Приобретая данный РїСЂРѕРґСѓРєС‚, РІС‹ обеспечиваете высокое качество Рё надежность. Если РІС‹ хотите улучшить СЃРІРѕРё производственные процессы, купить Фольга магниевая РњР›12 – ГОСТ 2856-79 будет правильным решением. РњС‹ гарантируем быструю доставку Рё отличное обслуживание.
Donaldlaf
| #
darknet market https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet market
Kxyufaita
| #
tor drug market darkmarket 2025
FNDaviditabe
| #
darknet markets url dark markets 2025
MarkNOlaf
| #
dark web drug marketplace dark web market links
Toliknaf
| #
darknet market lists https://github.com/abacuslink4jjku/abacuslink – dark market 2025
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
поставляемая продукция:
Медная капиллярная трубка 1.7х0.55 мм М2р ГОСТ 2624-2016 Покупайте медную капиллярную трубку у нас для идеального теплоотвода и надежности. Широкий выбор размеров и конфигураций для различных проектов. Отличная теплопроводность и гибкость монтажа, гарантирующие долговечность и экономию времени и денег.
Pingrutty
| #
tor drug market https://github.com/aresonioncq0a7/aresonion – darknet drugs
Vincenttef
| #
https://intekey.ru/
Rabyitabe
| #
dark websites https://github.com/darkwebsitesyhshv/darkwebsites – dark market
Blue_Code
| #
Смешные случаи в финансах: https://pik-leasing.ru
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние достижения.
Наши товары:
Мишень для распыления палладия (Pd, 3N5, плоская)
Dragon_Slayer
| #
Факты и вымысел о https://gik-invest.ru финансах.
WilliamIRrutty
| #
darknet marketplace tor drug market
Vincenttef
| #
бесплатный онлайн генератор QR кодов
DonDonfaita
| #
darkmarket 2025 https://github.com/nexusdarkrtv1u/nexusdark – dark markets
RobertDaw
| #
Non-Gamstop casino sites feel so much more relaxed—big thumbs up!
non.gamstop casino
Kxyufaita
| #
darknet drugs dark websites
FNDaviditabe
| #
onion dark website darknet drug store
MarkNOlaf
| #
dark web market darknet site
Vincenttef
| #
Создание QR кодов
avtolombardmsksig
| #
взять займ под залог птс
https://www.behavioralhealthjobs.com/companies/avtolombard-zalog-invest1534/
автоломбард авто
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние достижения.
Наши товары:
Металлическая мишень для распыления
Vortex20
| #
Что нельзя делать с финансами? https://rospremierinvest24.ru
Donaldlaf
| #
dark websites https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet markets 2025
MuwozSmomb
| #
Официальный партнер окон REHAU https://okna-intex.ru/ это возможность заказать по выгодным ценам окна с установкой под ключ. Ознакомьтесь на сайте со всеми преимуществами окон Рехау или рассчитайте стоимость окон по доступной цене. Мы предлагаем широкий выбор оконных систем, включая пластиковые и алюминиевые окна. Наши окна отличаются высоким качеством, долговечностью и надежностью.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Плоская мишень для распыления свинца 4N
Pingrutty
| #
darkmarket 2025 https://github.com/aresonioncq0a7/aresonion – darknet drug market
Toliknaf
| #
dark market onion https://github.com/abacuslink4jjku/abacuslink – dark web markets
CharlesGes
| #
Играю в онлайн казино ради кайфа, а вы?
https://telegra.ph/Luchshie-live-igry-nedeli-v-kazino-Mostbet-03-27
Red_Nomad
| #
Подробный https://kpk-agrorostorg-russia.ru разбор финансов.
WilliamIRrutty
| #
dark web markets darkmarket
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наши товары:
Гранулы магния 3N5
Vincenttef
| #
https://orgnaztech.mirtesen.ru/blog/43202871346/Gde-Kazhdyiy-Gost-Osobennyiy-Otel-Glory
Rabyitabe
| #
darknet market links https://github.com/darkwebsitesyhshv/darkwebsites – darknet markets url
BlueSlayer
| #
Как сэкономить на финансах: https://lombardveka.ru.
Kxyufaita
| #
dark web drug marketplace darknet markets
MarkNOlaf
| #
darknet market lists dark web link
FNDaviditabe
| #
darknet drug market darknet drug market
CharlesGes
| #
Онлайн казино с VIP-программой — чувствую себя особенным!
https://telegra.ph/Mostbet-kazino-mify-o-live-igrah-top-5-razoblachenij-03-27
Vincenttef
| #
Glory Casino app
DonDonfaita
| #
dark market https://github.com/nexusdarkrtv1u/nexusdark – dark markets 2025
Edwardignok
| #
На берегу моря нашли русалку https://x.com/Fariz418740/status/1905487137991958614
Vincenttef
| #
автоматизация бизнес-процессов
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их происхождение. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы а также предоставлять решения под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наша продукция:
РљСЂСѓРі ниобиевый РќР¦-2 РљСЂСѓРі ниобиевый РќР¦-2 – это высококачественный РїСЂРѕРґСѓРєС‚, изготовленный РёР· чистого РЅРёРѕР±РёСЏ, который обладает превосходными физико-химическими свойствами. Применяется РІ различных областях, включая авиацию Рё ядерную энергетику. Р—Р° счет своей стойкости Рє РєРѕСЂСЂРѕР·РёРё, РєСЂСѓРі обеспечивает долговечность Рё надежность РІ эксплуатации. Р’С‹ можете купить РљСЂСѓРі ниобиевый РќР¦-2 для оптимизации процессов обработки материалов. РќРµ упустите возможность повысить эффективность своего производства СЃ помощью этого уникального продукта.
Vincenttef
| #
Лучший генератор QR-кодов
Demetra
| #
Genom att ange din e-post bekräftar du att har läst igenom och accepterat vår integritets- och cookiepolicy.
Pingrutty
| #
darkmarket list https://github.com/aresonioncq0a7/aresonion – dark web sites
NightV4
| #
Свежие тренды в https://msk124.ru финансах.
Donaldlaf
| #
darknet drug links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – onion dark website
WilliamIRrutty
| #
dark market list dark market list
WolfHacker
| #
Основы https://bogner-vision.ru финансов для новичков.
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наши товары:
Полоса РёР· магнитно-РјСЏРіРєРёС… сплавов 1.2 РјРј 40РќРљРњ ГОСТ 10160-75 РЁРёСЂРѕРєРёР№ выбор полос РёР· магнитно-РјСЏРіРєРёС… сплавов для электротехнических устройств. Низкая коерцитивная сила Рё превосходная магнитная проницаемость. Рдеально для энергетики, силовой электроники Рё медицинского оборудования. Коррозионная устойчивость обеспечивает долгий СЃСЂРѕРє службы.
ArthurTog
| #
https://github.com/ms-core/Microsoft-Project/releases
Vincenttef
| #
https://orgnaztech.mirtesen.ru/blog/43202871346/Gde-Kazhdyiy-Gost-Osobennyiy-Otel-Glory
Gefevpek
| #
Visit https://bestcasinoideal.com/ and discover expert reviews, top strategies and essential tips. Explore online slots, table games, live dealer experiences. We have compiled a complete collection of the best online casinos and gambling sites for 2025, tested and rated by experts.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Керамическая мишень для распыления Р±РѕСЂР° – плоская
Toliknaf
| #
darkmarket 2025 https://github.com/abacuslink4jjku/abacuslink – darknet drug market
Kxyufaita
| #
dark market list dark web market
FNDaviditabe
| #
dark web markets darknet market lists
Rabyitabe
| #
darknet market list https://github.com/darkwebsitesyhshv/darkwebsites – darknet market lists
avtolombardmsksig
| #
деньги под птс круглосуточные
https://www.iratechsolutions.com/employer/thunder-consulting1245/
займ под залог отечественного авто
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их качество. Превосходное обслуживание – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы и находить ответы под специфику вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
Проволока магниевая G-MgAl6 – DIN 1729 Part 2 Полоса магниевая G-MgAl6 – DIN 1729 Part 2 представляет СЃРѕР±РѕР№ высококачественный материал, который идеально РїРѕРґС…РѕРґРёС‚ для различных промышленных применений. Благодаря легкости Рё прочности магний используется РІ автомобильной Рё авиационной отраслях. Полоса обладает отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью, что делает ее надежной РІ условиях повышенной влажности. Приобретая данную полосу, РІС‹ получаете РїСЂРѕРґСѓРєС‚, соответствующий всем стандартам качества. РќРµ упустите возможность улучшить СЃРІРѕРё проекты, купите Полоса магниевая G-MgAl6 – DIN 1729 Part 2 уже сегодня!
Blade7286
| #
Как считаете, где лучше всего https://bonami-salon.ru понять финансы?
Donaldnuh
| #
кайт египет
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Керамическая мишень Титаната Бария
Edwardignok
| #
На берегу моря нашли русалку https://x.com/Fariz418740/status/1905487137991958614
DonDonfaita
| #
darkmarket url https://github.com/nexusdarkrtv1u/nexusdark – darknet markets
JoshuaUsady
| #
настольные игры Table Mania – магазины настольных игр, в котором представлен широчайший ассортимент настольных игр. У нас широко представлены как семейные, детские и вечериночные, так и сложные стратегические, коллекционно-карточные и тактические игры с миниатюрами.
CyberSentinel
| #
По данным аналитиков, лучший источник о финансах – https://gorod-zaimov.ru
Pingrutty
| #
dark market onion https://github.com/aresonioncq0a7/aresonion – dark web markets
WilliamIRrutty
| #
bitcoin dark web darknet links
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наши товары:
Полоса РёР· магнитно-РјСЏРіРєРёС… сплавов 0.7 РјРј 50РќРџ ГОСТ 10160-75 РЁРёСЂРѕРєРёР№ выбор полос РёР· магнитно-РјСЏРіРєРёС… сплавов для электротехнических устройств. Низкая коерцитивная сила Рё превосходная магнитная проницаемость. Рдеально для энергетики, силовой электроники Рё медицинского оборудования. Коррозионная устойчивость обеспечивает долгий СЃСЂРѕРє службы.
Donaldlaf
| #
onion dark website https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet drug store
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их качество. Превосходное обслуживание – наш стандарт – мы на связи, чтобы улаживать ваши вопросы а также адаптировать решения под требования вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наша продукция:
Пруток магниевый B 92 (9980A) – ASTM B92 Проволока магниевая B 92 (9980A) – ASTM B92 предлагает высококачественный материал для производства Рё изготовления серьезных изделий. РћРЅР° обеспечивает надежность Рё долговечность, что делает её идеальным выбором для различных промышленных применений. Благодаря СЃРІРѕРёРј отличным свойствам, этот РїСЂРѕРґСѓРєС‚ легко обрабатывается Рё сваривается.Покупая проволоку магниевую B 92 (9980A) – ASTM B92, РІС‹ получаете РЅРµ только надежный материал, РЅРѕ Рё уверенность РІ качестве своей продукции. РќРµ упустите возможность купить Проволока магниевая B 92 (9980A) – ASTM B92 Рё повысить эффективность СЃРІРѕРёС… проектов.
FNDaviditabe
| #
dark web market urls dark web marketplaces
MarkNOlaf
| #
darknet markets url darknet markets 2025
Kxyufaita
| #
darknet site darknet links
Pioneer2884
| #
Особенности https://sunandmoonsaransk.ru реализации финансов.
Billydrula
| #
elonbet casino BD
Toliknaf
| #
darknet markets url https://github.com/abacuslink4jjku/abacuslink – darknet markets url
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их качество. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы а также адаптировать решения под особенности вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
Рзделия РёР· кобальта Wallex 100 Рзделия РёР· кобальта Wallex 100 идеально РїРѕРґС…РѕРґСЏС‚ для профессионалов, ценящих качество Рё надежность. Рти высококачественные материалы обеспечивают отличную износостойкость Рё долговечность. РћРЅРё применяются РІ различных областях, таких как промышленность Рё медицина. Рнновационные технологии производства гарантируют безопасность Рё эффективность. Каждый РїСЂРѕРґСѓРєС‚ РїСЂРѕС…РѕРґРёС‚ строгий контроль. РќРµ упустите шанс купить Рзделия РёР· кобальта Wallex 100 Рё обеспечить себя надежными решениями для будущих задач. Сделайте выбор РІ пользу долговечности Рё качества.
MetaMask Download
| #
MetaMask Download is super quick. The wallet’s design is simple yet powerful, making it ideal for beginners and experts alike.
Billydrula
| #
elonbet casino
MichaelHab
| #
esitella to doslownie tragedia! Tresci nieaktualne, a w niektorych przypadkach zupelnie nieprawidlowe. Probowalem dowiedziec sie wazne tresci o zdrowiu, ale natrafilem na mnostwo bledow i jawnych absurdow. Mozna pomyslec, ze tresci redagowane przez ludzi bez wiedzy! To jest niebezpieczne dla uzytkownikow, ktorzy ufaja tym danym. Wlasciciele nie odpowiadaja usterek, a opinie z duzym prawdopodobienstwem sfalszowane.
EdwardBiorn
| #
Курьер привёз вовремя, упаковка целая!
женский костюм купить томск
Davidpocky
| #
kraken – кракен даркнет, kra30 at
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наша продукция:
Медный переходной пресс тройник 64х25 мм М2РМ ГОСТ Р52948-2008 Приобретайте надежные медные переходные тройники для соединения труб с разным диаметром. Устойчивы к коррозии, легки в монтаже и обеспечивают надежное соединение. Широкий выбор размеров. Гарантированное качество и долговечность.
Rabyitabe
| #
dark web sites https://github.com/darkwebsitesyhshv/darkwebsites – darknet markets links
VortexX9
| #
Практическое применение финансов: https://ipoteka-kurgan.ru
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор отборных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их происхождение. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы а также адаптировать решения под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Рзделия РёР· кобальта РРџ482 Рзделия РёР· кобальта РРџ482 представляют СЃРѕР±РѕР№ высококачественные изделия, обладающие долговечностью Рё устойчивостью Рє РєРѕСЂСЂРѕР·РёРё. РћРЅРё идеально РїРѕРґС…РѕРґСЏС‚ для применения РІ условиях высокой температуры Рё механических нагрузок. Каждый РїСЂРѕРґСѓРєС‚ РїСЂРѕС…РѕРґРёС‚ строгий контроль качества, что гарантирует надежность Рё долговечность. Корпоративные клиенты Рё профессионалы предпочитают нашу продукцию Р·Р° ее высокую производительность Рё эффективность. Если РІС‹ хотите увеличить производительность вашего оборудования, купить Рзделия РёР· кобальта РРџ482 — это отличный выбор. Обратите внимание РЅР° РёС… универсальность Рё превосходные эксплуатационные характеристики.
RockyMah
| #
Planning to transfer cash in the popular Aviator game?
We’ve got you covered.
First, log in, then navigate to the top-up area.
Now, select your most convenient payment option:
e-wallet.
Input the sum you want to transfer — as much as you want.
Approve the payment and you’re all set to play!
For more info, visit http://cajnews.co.ke/ — no redirects.
No delays — hit the skies with confidence!
WilliamIRrutty
| #
darknet markets links dark web sites
Pingrutty
| #
dark web drug marketplace https://github.com/aresonioncq0a7/aresonion – darknet market links
BrianBoofs
| #
https://www.radiofrance.fr/franceinter/podcasts/lumieres-dans-la-nuit/lumieres-dans-la-nuit-du-mardi-09-juin-2020-8311597
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
поставляемая продукция:
РўСЂСѓР±Р° РёР· драгоценных металлов палладиевая 500С…1С…0.7 РјРј РџРґР82-18 РўРЈ Гарантированное качество Рё изысканный дизайн – купите драгоценные трубы для ювелирных украшений, машиностроения Рё архитектуры. РЁРёСЂРѕРєРёР№ выбор Рё высокое качество! Доставка РїРѕ Р РѕСЃСЃРёРё.
1win_veSt
| #
wan win http://belbeer.borda.ru/?1-6-0-00001583-000-0-0/ .
Michaeldab
| #
семяныч официальный сайт купить семена
mostbet_kner
| #
мрстбет мрстбет .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Гранулы циркония
DonDonfaita
| #
dark markets 2025 https://github.com/nexusdarkrtv1u/nexusdark – bitcoin dark web
Guardian1224
| #
Видел прогнозы, что Финансы будет главным трендом. Возражения? https://treid-leasing.ru
1win_wjkt
| #
1win rossvya https://1win6018.ru/ .
Billydrula
| #
elonbet casino BD
Kxyufaita
| #
darknet websites dark markets 2025
1win_mmSt
| #
игра 1вин игра 1вин .
FNDaviditabe
| #
dark market onion dark website
MarkNOlaf
| #
dark websites dark web market links
mostbet_user
| #
mostbet kg скачать на андроид http://www.girikms.forum24.ru/?1-1-0-00000361-000-0-0-1742819287 .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
Наша продукция:
Лента СЃ заданными свойствами упругости 0.35С…250 РјРј 44РќРҐРўР® ГОСТ 14117-85 Лента СЃ заданными свойствами упругости предлагает высококачественные материалы, определенные параметры упругости Рё широкий выбор размеров для надежного Рё РіРёР±РєРѕРіРѕ крепления. Рспользуется РІ строительстве, производстве Рё машиностроении. Обеспечивает легкость установки, надежность, долговечность Рё адаптацию Рє различным техническим требованиям.
mostbet_jwSn
| #
мост бет https://www.kharkovbynight.forum24.ru/?1-15-0-00003047-000-0-0 .
1win_ankt
| #
ваучер 1win ваучер 1win .
Donaldlaf
| #
darknet market links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darkmarket list
Billydrula
| #
elonbet casino
mostbet_hiSn
| #
мос бет мос бет .
1win_ovSt
| #
1vin https://belbeer.borda.ru/?1-6-0-00001583-000-0-0 .
Toliknaf
| #
darkmarkets https://github.com/abacuslink4jjku/abacuslink – dark web marketplaces
mostbet_ncer
| #
скачат мостбет скачат мостбет .
Phantom922
| #
Пытаюсь разобраться в финансах. Что посоветуете? https://avtolombard-13.ru
Vincenttef
| #
https://intekey.ru/
Pingrutty
| #
dark web sites https://github.com/aresonioncq0a7/aresonion – darknet markets url
Billydrula
| #
elonbet casino BD
OmegaFang
| #
Примеры успеха https://detektiv-ufa.ru в финансах.
1win_tgkt
| #
бк 1win https://1win6018.ru/ .
mostbet_qkSn
| #
мостбет chrono http://kharkovbynight.forum24.ru/?1-15-0-00003047-000-0-0 .
Rabyitabe
| #
darknet site https://github.com/darkwebsitesyhshv/darkwebsites – darknet market
WilliamIRrutty
| #
dark markets darknet markets url
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы по мере того как находить ответы под особенности вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
Рзделия РёР· титана Р’Рў20 РџРѕРєРѕРІРєР° титановая Р’Рў20 — это высококачественный РїСЂРѕРґСѓРєС‚, обладающий прочностью Рё легкостью. Рспользуется РІ авиационной, космической Рё медицинской отраслях. Его уникальные свойства обеспечивают отличные характеристики устойчивости Рє РєРѕСЂСЂРѕР·РёРё Рё высокой температуре. Применение данной РїРѕРєРѕРІРєРё позволяет значительно увеличить надежность изделий. Чтобы обеспечить качественное выполнение проектов, стоит подумать, РіРґРµ купить РџРѕРєРѕРІРєР° титановая Р’Рў20. Выбор данной марки титанового сплава — это гарантированный успех РІ вашем бизнесе. РќРµ упустите возможность повысить качество ваших товаров!
DonDonfaita
| #
dark web market links https://github.com/nexusdarkrtv1u/nexusdark – dark market
Vincenttef
| #
бесплатный онлайн генератор QR кодов
KennethTyday
| #
Печать рекламных буклетов https://tipografiya-buklety.ru ярко, качественно, профессионально. Форматы A4, евро, индивидуальные размеры. Работаем с частными и корпоративными заказами.
riobet
| #
рио бет риобет вход на сайт
FNDaviditabe
| #
darknet markets onion address dark web sites
Vincenttef
| #
https://intekey.ru/
Kxyufaita
| #
dark web market dark web market urls
GhostProtocol
| #
Типичные промахи в финансах: https://viessmann72.ru
MarkNOlaf
| #
dark market url darkmarket 2025
Omega_Hunter
| #
Изучите этот материал https://gk-cent.ru о финансах.
Donaldlaf
| #
best darknet markets https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet markets links
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
поставляемая продукция:
Медный стандартный двухраструбный отвод 90 градусов под пайку 108х92х2.1 мм 47.5х51.5 мм мягкая пайка М3т ГОСТ 32590-2013 Покупайте медные двухраструбные отводы 90 градусов под пайку на Редметсплав.рф. Надежные и долговечные отводы из высококачественной меди для систем водоснабжения, отопления и кондиционирования воздуха. Гарантированно надежные соединения, простой монтаж и оптимальное распределение потока.
Pingrutty
| #
darknet drug links https://github.com/aresonioncq0a7/aresonion – dark market list
Toliknaf
| #
darknet links https://github.com/abacuslink4jjku/abacuslink – darknet market lists
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – наш стандарт – мы на связи, чтобы улаживать ваши вопросы и адаптировать решения под особенности вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
Проволока вольфрамовая Р’Рђ Проволока вольфрамовая Р’Рђ является РѕРґРЅРёРј РёР· важнейших материалов РІ металлургии Рё промышленности. РћРЅР° обладает высокой температурной стойкостью Рё прочностью, что делает её идеальным выбором для различных технологий, включая сварку Рё электронику. Благодаря СЃРІРѕРёРј уникальным свойствам, Проволока вольфрамовая Р’Рђ широко используется РІ производстве ламп Рё РґСЂСѓРіРёС… высокотемпературных устройств.Если РІС‹ ищете надежный материал, который отвечает современным стандартам качества Рё эффективности, вам стоит купить Проволока вольфрамовая Р’Рђ. Ртот РїСЂРѕРґСѓРєС‚ гарантирует долговечность Рё стабильную работу РІ самых сложных условиях.
Vincenttef
| #
https://orgnaztech.mirtesen.ru/blog/43202871346/Gde-Kazhdyiy-Gost-Osobennyiy-Otel-Glory
Rabyitabe
| #
dark web market links https://github.com/darkwebsitesyhshv/darkwebsites – darknet marketplace
Billydrula
| #
elonbet casino
WilliamIRrutty
| #
dark market darknet markets
Wolf950
| #
Ответы на частые вопросы о финансах: https://ooo-autolombard-88.ru
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
поставляемая продукция:
Медный отвод пресс 45 градусов 34 мм М2РМ ГОСТ Р52948-2008 Приобретите высококачественные медные отводы пресс для систем водоснабжения и отопления. Надежное соединение, устойчивость к коррозии, простота монтажа. Широкий ассортимент. Доставка по России.
DonDonfaita
| #
darknet drug links https://github.com/nexusdarkrtv1u/nexusdark – dark web sites
Vincenttef
| #
https://orgnaztech.mirtesen.ru/blog/43202871346/Gde-Kazhdyiy-Gost-Osobennyiy-Otel-Glory
DarkV4
| #
Кто-нибудь разбирается в финансах? Куда зайти? https://udvorperm.ru
FNDaviditabe
| #
dark web drug marketplace bitcoin dark web
Kxyufaita
| #
dark web market urls darkmarket 2025
avtolombardnovosibsig
| #
кредит под птс
http://webheaydemo.co.uk/profile/rolandomahler
займ под залог кредитного авто
MarkNOlaf
| #
darknet market list darknet site
Pingrutty
| #
darkmarket link https://github.com/aresonioncq0a7/aresonion – dark web market
pricelcaree
| #
Подробнее в блоге обзоров прицелов и другой оптической техники на сайте leupold-optic.ru ссылка
Donaldlaf
| #
darknet markets 2025 https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet drug store
Vincenttef
| #
https://orgnaztech.mirtesen.ru/blog/43202871346/Gde-Kazhdyiy-Gost-Osobennyiy-Otel-Glory
Vincenttef
| #
https://orgnaztech.mirtesen.ru/blog/43202871346/Gde-Kazhdyiy-Gost-Osobennyiy-Otel-Glory
Toliknaf
| #
darknet drug market https://github.com/abacuslink4jjku/abacuslink – best darknet markets
Blade9578
| #
Предлагаю подискутировать о https://comfort-textile.ru финансах.
Vincenttef
| #
Создание QR кодов
WilliamIRrutty
| #
dark web markets dark web market links
DriverBaile
| #
Соседи по дому посоветовали Городскую Ритуальную Службу. Организация похорон была выполнена безупречно. Благодарим за поддержку.
Volodyanaf
| #
darknet links darknet links
Rabyitabe
| #
bitcoin dark web https://github.com/darkwebsitesyhshv/darkwebsites – dark markets
Billydrula
| #
elonbet casino
Vuxerelayes
| #
На сайте https://remont-okon-63.ru/ уточните номер телефона для того, чтобы воспользоваться такой нужной услугой, как ремонт, регулировка окон независимо от сложности. Прямо сейчас вы сможете воспользоваться возможностью вызвать мастера. Для этого следует указать номер телефона, а также имя. Среди основных услуг, которые оказываются в этой компании, выделяют: ремонт балконных дверей, производство москитных сеток, стеклопакетов. Кроме того, доступен и срочный ремонт конструкций. Все работы выполняются в соответствии с требованиями.
Larrytwicy
| #
popular steroid brands https://anabolshop.org/
Code7907
| #
Что нового в финансах? https://broker86.ru.
Vincenttef
| #
бесплатный онлайн генератор QR кодов
Michaelesoke
| #
Enhance your individual auto with our own tuning services. Change your car into a unique masterpiece. Our team of engineers is dedicated to supplying top-quality results that outperform your wishes. Whether you’re looking to increase performance, revamp aesthetics, or integrate new features, we have the knowledge to deliver your vision to life. Visit our website at https://timbrellosautobody.com/ to explore our services and schedule a consultation today.Let us assist you construct the machine of your dreams.
Kxyufaita
| #
dark market list darknet markets links
Everettsox
| #
анализ на наркотики купить analiz-kupit-spb.ru
Billydrula
| #
elonbet casino
DonDonfaita
| #
darknet drugs https://github.com/nexusdarkrtv1u/nexusdark – dark markets 2025
Billydrula
| #
elonbet casino BD
Pingrutty
| #
dark web markets https://github.com/aresonioncq0a7/aresonion – darknet drug market
Billydrula
| #
elonbet casino BD
ThunderProtocol
| #
Исследование финансов: https://gzhc38.ru
Donaldlaf
| #
darknet marketplace https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet sites
deheTus
| #
Компания A.X.O.N предлагает квалифицированные услуги. Создаем для бизнеса AI-решения. Работаем с юридическими и физическими лицами по всему миру. Формат и условия оплаты обсуждаются персонально под ваш запрос. https://axonbusiness.ru – тут представлены ответы на популярные вопросы. Вы сможете узнать, какие можно автоматизировать задачи. Мы выстраиваем бизнес-логику, настраиваем процессы и доводим до результата. Всегда на связи и готовы ответить на интересующие вопросы. Давайте начнем с консультации, это бесплатно.
edacaree
| #
Доставка продуктов и готовой еды на дом – EdaDostavkoy.ru здесь
WilliamIRrutty
| #
darknet market links darknet market lists
Toliknaf
| #
dark market list https://github.com/abacuslink4jjku/abacuslink – darknet marketplace
Natasesunice
| #
Ищете оригинальный мерч от любимых российских и зарубежных звезд, фестивалей и шоу? Посетите https://showstaff.ru/ – мы с 2002 года радуем наших клиентов качественной продукцией, доставкой по всей России и поддержкой талантливых артистов. Заходите на наш сайт и выбирайте свой стиль! ShowStaff – это магазин оригинального мерча, официально и качественно. Посетите каталог, и вы обязательно найдете для себя уникальные вещи!
Billydrula
| #
elonbet casino BD
Bevaxowreave
| #
На сайте https://smartflow.ru вы сможете выбрать качественную, функциональную сантехнику для обустройства ванной комнаты в доме либо квартире. Ассортимент «SMARTFLOW» включает в себя такие изделия, которые выполнены из высококачественных и современных материалов, безупречного стиля. Все унитазы, биде, модульные ванные идеально впишутся в концепцию. Каждая вещь идеально сочетает надежность, прочность, а также изысканность. Для того чтобы выбрать что-то определенное, изучите галерею, ознакомьтесь и с особой системой смыва «Торнадо».
WolfZ4
| #
Согласно исследованиям, лучший источник о финансах – https://kpkkc-matkap.ru
maslyanie transformatori_mbPi
| #
тмг трансформатор тмг трансформатор .
programma 1s_temr
| #
1с бухгалтерия 8 купить 1с бухгалтерия 8 купить .
programma 1s_ydka
| #
покупка 1с покупка 1с .
Billydrula
| #
elonbet casino
Rabyitabe
| #
bitcoin dark web https://github.com/darkwebsitesyhshv/darkwebsites – darkmarket link
Shadow464
| #
Памятка для работы с финансами: https://webresurs-72.ru
cawuomisk
| #
Компания АБК предлагает аренду бытовок, которые производятся из высококачественных материалов. Ассортимент выбора контейнеров большой, цены приемлемые. Процесс аренды понятен и прост, сотрудники внимательны к пожеланиям, они профессионалы своего дела. Ищете аренда блок контейнеров? Arendabk.ru – портал, где имеются отзывы и фотогалерея. Предлагаем в аренду исключительно достойные контейнеры. Доставим их в минимальные сроки. С радостью на все вопросы ответим, и верный выбор поможем сделать. Для нас важно, чтобы вам было приятно с нами работать.
FNDaviditabe
| #
darknet drug market dark market list
Pingrutty
| #
darknet websites https://github.com/aresonioncq0a7/aresonion – darknet site
MarkNOlaf
| #
darkmarkets darknet site
Cazrwjj
| #
Привет!
Без ВУЗа очень нелегко было продвигаться по карьерной лестнице. Сегодня же этот документ не дает совершенно никаких гарантий, что получится найти хорошую работу. Намного более важны профессиональные навыки и знания специалиста, а также его опыт работы. По этой причине решение о заказе диплома стоит считать рациональным. Купить диплом о высшем образовании tweakbook.us/read-blog/1210_diplom-moskva.html
zaimpodzalogptsekbsig
| #
кредит под птс автомобиля
avtolombard-pid-zalog-pts.ru/ekb.html
деньги под залог птс быстро
WilliamDix
| #
Мы изготавливаем дипломы любой профессии по выгодным ценам. Дипломы производят на настоящих бланках государственного образца Заказать диплом института diplomidlarf.ru
Lazrglf
| #
Где купить диплом по необходимой специальности?
Купить диплом института по невысокой стоимости можно, обращаясь к надежной специализированной фирме.: diplommy.ru
DonDonfaita
| #
darknet markets url https://github.com/nexusdarkrtv1u/nexusdark – darknet markets links
Volodyanaf
| #
dark web link dark web markets
asgdh_fiche
| #
Do not buy anything from them!!!
DannyTox
| #
Большой выбор из 8677 наименований на складе. В розницу по оптовые ценам от производителя. Доступная доставка в Москву курьерской службой до 2х дней. Фабричное производство: купить сигареты в москве
Sazrbyo
| #
купить диплом в садик
Sazrjeh
| #
Привет!
Приобрести диплом института по невысокой стоимости возможно, обращаясь к проверенной специализированной фирме. Заказать диплом: diplomus-spb.ru/kupit-diplom-spetsialista-7/
KennethHew
| #
Как выбрать надёжное оборудование для майнинга в 2025 году — Узнайте подробности здесь https://www.buzzbii.com/Cryptons
Donaldlaf
| #
dark market onion https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet markets onion address
xoxetiProva
| #
Caspian Training Group преподаватели компании в своей работе толк знают. Они постоянно проходят курсы повышения квалификации. Вас персональный подход компании порадует. https://xn—-7sbecpcasfm0beeaecirc5b3a2g.kz/ – тут более детальная информация о нас представлена, посмотрите ее в любое время. Caspian Training Group помогает компаниям успешно реализовывать проекты корпоративного обучения. Результаты ваши ожидания превзойдут. Сотрудники станут увереннее в своей работе и улучшат навыки взаимодействия в команде.
WilliamIRrutty
| #
dark markets 2025 darknet market lists
Reaper1247
| #
Конкретные https://rps63.ru шаги для работы с финансами.
SejehewTot
| #
Welcome to the official Joy Casino blog https://officialjoycasino.net/ — a place where you can explore the fascinating world of online gambling from different perspectives: game mechanics, psychology, responsible gaming and industry analytics. If you are looking for a deep dive into gaming strategies, casino technology or responsible gaming tips, you have come to the right place.
Silver904
| #
Как войти в тему финансов? https://ed-centr.ru
Toliknaf
| #
darkmarket link https://github.com/abacuslink4jjku/abacuslink – darknet market
Billydrula
| #
elonbet casino BD
Xazrkae
| #
Купить диплом о высшем образовании!
Мы предлагаем дипломы любой профессии по приятным ценам. Вы заказываете документ через надежную и проверенную компанию. : kingcityrp.moibb.ru/viewtopic.php?f=34&t=1053
Lazrzrf
| #
Диплом ВУЗа России!
Без наличия диплома очень непросто было продвигаться вверх по карьере. Именно поэтому решение о покупке диплома стоит считать рациональным. Заказать диплом института medforum.5nx.ru/posting.php?mode=post&f=4
Pingrutty
| #
darkmarket url https://github.com/aresonioncq0a7/aresonion – darknet sites
Kxyufaita
| #
darkmarkets darknet market lists
Sazrxex
| #
Мы изготавливаем дипломы любых профессий по приятным ценам.– diplom-top.ru/kupit-diplom-s-zaneseniem-v-reestr-bistro-i-nadezhno-6/
Billydrula
| #
elonbet casino
arhipkacaree
| #
Экскурсии в Геленджике и Архипо-Осиповке: контакты, жмякай здесь
shashlikyug
| #
заказать курицу табака https://shashlikyug.ru
MarkNOlaf
| #
darknet markets bitcoin dark web
Rabyitabe
| #
dark web market https://github.com/darkwebsitesyhshv/darkwebsites – dark web market urls
Steel_Nomad
| #
Мне самому помог этот ресурс https://anex-chel.ru по финансам.
Nikigboith
| #
На сайте https://antipoligraf.ru/ почитайте про Институт прикладной психофизиологии. Здесь ведется профессиональная, комплексная подготовка к тому, чтобы пройти полиграф. Учат и тому, чтобы быстро, безопасно и комплексно снять стресс при помощи уникальных и инновационных приборов обратной связи. И самое важное, что все это абсолютно безопасно, при этом гарантированно поможет. Кроме того, ведется обучение саморегуляции при помощи обратной связи. У института огромное количество партнеров, которые стараются работать в комплексе.
DonDonfaita
| #
darkmarket list https://github.com/nexusdarkrtv1u/nexusdark – dark web markets
Vincenttef
| #
автоматизация бизнес-процессов
CyberWalker
| #
Успешные кейсы применения финансов: https://kotofey-nsk.ru
WilliamIRrutty
| #
dark market 2025 darknet site
mostbet_gqEn
| #
мостбет скачать https://mostbet6005.ru .
DannyTox
| #
Большой выбор из 8677 наименований на складе. В розницу по оптовые ценам от производителя. Доступная доставка в Москву курьерской службой до 2х дней. Фабричное производство: купить сигареты в москве
Victordiesy
| #
Ванга: правда, мифы и 5 самых известных пророчеств
https://x.com/NargisEhme94100/status/1905737028656136280
WilsonUnine
| #
овоши на мангале купить кебаб
Duanefance
| #
Топ сайтов кейсов CS2 http://ggdrop.cs2-case.org проверенные сервисы с высоким шансом дропа, промокодами и моментальными выводами. Только актуальные и безопасные платформы!
mostbet_jwEn
| #
мостбет промокод https://www.mostbet6005.ru .
Steel_Guardian
| #
Шутки про финансы: https://sber-finans.ru
FNDaviditabe
| #
dark market list darknet markets onion address
Kxyufaita
| #
dark market list darkmarkets
Hunterbax
| #
Землетрясение в Бангкоке: что стало причиной?
https://x.com/SebiBilalova/status/1905743685712855518
DavidBoash
| #
купить анализы гепатиты купить справку анализы
1win_vmOi
| #
1win онлайн 1win онлайн .
Volodyanaf
| #
onion dark website darkmarket link
MarkNOlaf
| #
darkmarkets darkmarket link
AndreDap
| #
Букет шикарный! Все цветы как живые.
букет пионов
1win_auSr
| #
1.вин https://www.1win6019.ru .
KennethHew
| #
Энергосберегающие стеклопакеты для утепления дома — смотрите детали тут https://say.la/Oknavspb
Vincenttef
| #
https://intekey.ru/
zaimpodptskemerovosig
| #
кредит под залог машины
avtolombard-pid-zalog-pts.ru/kemerovo.html
займ птс
mostbet_xcoa
| #
mostbet chrono hiend.borda.ru/?1-16-0-00000259-000-0-0-1743052953 .
mostbet_jyer
| #
мостбет скачать на андроид https://alfatraders.borda.ru/?1-0-0-00004917-000-0-0-1743053068 .
1win_vrOi
| #
wan win wan win .
mostbet_owEn
| #
поддержка мостбет https://www.mostbet6005.ru .
Vincenttef
| #
Создание QR кодов
1win_swSr
| #
что такое 1win http://1win6019.ru/ .
Byte8399
| #
Лучшие материалы по финансам: https://galantdzr.ru.
mostbet_gboa
| #
скачать мостбет https://www.hiend.borda.ru/?1-16-0-00000259-000-0-0-1743052953 .
mostbet_jser
| #
мост бет https://alfatraders.borda.ru/?1-0-0-00004917-000-0-0-1743053068/ .
Diplomi_wzka
| #
продам диплом о высшем образовании продам диплом о высшем образовании .
VortexV2
| #
Пошаговая инструкция по финансам: https://persona-pk.ru
WilliamIRrutty
| #
dark market url darknet markets url
Vincenttef
| #
Создание QR кодов
1win_gfOi
| #
1вин официальный сайт http://obmen.forum24.ru/?1-1-0-00004428-000-0-0-1742816292/ .
1win_xhSr
| #
1win официальный сайт регистрация http://www.1win6019.ru .
FNDaviditabe
| #
dark web market links dark market url
Kxyufaita
| #
dark web link darknet markets 2025
mostbet_vjoa
| #
мастбет http://hiend.borda.ru/?1-16-0-00000259-000-0-0-1743052953 .
mostbet_jjer
| #
mostbest mostbest .
remcaree
| #
Ремонт электронники в Москве – сервисный центр, который может отремонтировать все ссылка
MarkNOlaf
| #
darknet markets onion darknet links
asgdh_fiche
| #
Engaged in fraud
PhantomZ6
| #
Как избежать проблем с финансами? https://sarma-trucks.ru
Vincenttef
| #
Создание QR кодов
Thunder_Hunter
| #
Мне самому помог этот ресурс по финансам: https://kamsf.ru
WilliamIRrutty
| #
darkmarket 2025 tor drug market
Vincenttef
| #
автоматизация бизнес-процессов
HevimeBuike
| #
Ищете топливные карты для юридических лиц и ИП? Обратите внимание на единую топливную карта ЛИОТЭК для бизнеса. Узнайте на сайте https://liotec.ru/ все преимущества выгодной карты Лиотек и какие огромные преимущества она дает, по сравнению с другими картами. Подробнее на сайте.
Volodyanaf
| #
darknet site dark web link
Vincenttef
| #
Создание QR кодов
Vincenttef
| #
автоматизация бизнес-процессов
FNDaviditabe
| #
dark web marketplaces bitcoin dark web
Kxyufaita
| #
dark web marketplaces dark markets 2025
MarkNOlaf
| #
darknet market links darkmarket url
Rohawrdtop
| #
Visit https://gorillacasino-best.com/ we have created a fun, informative and engaging online space where casino lovers, newbies and industry experts can find honest insights, in-depth analysis and valuable strategies in the world of online gambling. Unlike many casino blogs, we are not owned or operated by any gambling company. We provide unbiased content.
zaimpodptsmsksig
| #
ломбард деньги под залог птс
avtolombard-pid-zalog-pts.ru
кредит под залог птс москва
Vincenttef
| #
Glory Casino app
BlueV3
| #
Видел прогнозы, что Финансы будет главным https://finkredit38.ru трендом. Соглашусь?
наука инновации
| #
Инновационные Технологии — это платформа, где
собираются люди, интересующиеся новыми технологиями
и инновациями. Здесь вы найдете новости, статьи,
обзоры и анализ последних достижений
в области науки, техники и промышленности.
Тематика сайта охватывает
новости в – центр инновационных технологии искусственный интеллект,
робототехнику, интернет вещей,
биотехнологии, нанотехнологии, космос, энергетику
и многое другое. Кроме того, авторы и эксперты могут публиковать статьи и делиться своим опытом и
знаниями.
LabocnLef
| #
Аренда комфортабельных катеров и яхт в Санкт-Петербурге от https://7futov.spb.ru/ это удобная возможность осуществить водные прогулки по СПб. У нас большой выбор маршрутов и организация мероприятий под ключ. Самые выгодные цены для аренды и большой выбор прогулочных катеров хоть для одного, хоть для большой компании. Посмотрите все наши катера и яхты на сайте по выгодным ценам!
WilliamIRrutty
| #
darkmarket link dark web market urls
Roberttax
| #
В Волна Казино регулярно проводятся розыгрыши, акции и турниры с щедрыми наградами: казино волна официальный сайт
Kxyufaita
| #
dark websites dark web market list
MarkNOlaf
| #
darknet markets onion darknet site
Storm639
| #
Типичные промахи в финансах: https://zalogvl.ru
Roberttax
| #
Присоединяйтесь к Волна Казино и получайте бонусы не только за регистрацию, но и за активную игру https://volna7777.casino/
Volodyanaf
| #
dark web market urls dark web link
WilliamIRrutty
| #
dark market link best darknet markets
FNDaviditabe
| #
dark web market links darknet drug store
zaimpodptsnovosibsig
| #
деньги в кредит под залог машины
avtolombard-pid-zalog-pts.ru/nsk.html
деньги под залог птс круглосуточно
MarkNOlaf
| #
dark web market list darkmarket 2025
Night93
| #
Ответы на частые вопросы о финансах: https://saturn03.ru
WilliamIRrutty
| #
darknet market darknet drug market
lepigevKit
| #
Компания СанОбработка специализируется на предоставлении компетентных услуг по дезинсекции, дезинфекции, дератизации. Даем гарантию на высочайший уровень безопасности и действенности. Обращайтесь к нам тогда, когда вам надо. https://sanobrabotkarf.ru – тут можете с отзывами клиентов ознакомиться. Наши компетентные специалисты готовы к вам на помощь приехать в любое время. Они проведут тщательную обработку и будут использовать сертифицированные европейские препараты. Проблема решится, ваше жилище станет безопасным и комфортным.
Vincenttef
| #
Glory Casino app
Kxyufaita
| #
darknet markets 2025 darknet sites
Rafaelpluch
| #
телеграмм канал hr хантеров В современном мире HR-специалисты сталкиваются с необходимостью оперативной коммуникации и обмена опытом. HR чат становится незаменимым инструментом для решения этих задач. Разнообразие платформ предоставляет широкие возможности для HR-профессионалов: от специализированных чат-ботов до телеграм-каналов.
MarkNOlaf
| #
dark market link darknet markets onion address
Volodyanaf
| #
darknet markets best darknet markets
1win_rhml
| #
how to bet on 1win 1win12.com.ng .
DragonNomad
| #
ТОП-10 https://udarnik35.ru ресурсов по финансам.
WilliamIRrutty
| #
darknet marketplace dark market
Vincenttef
| #
бесплатный онлайн генератор QR кодов
Vincenttef
| #
Лучший генератор QR-кодов
Kxyufaita
| #
darknet websites darkmarket url
Derricksaisy
| #
Заказала цветы для второй половинки – любовь
доставка цветов
MarkNOlaf
| #
darknet markets links darkmarket list
Vincenttef
| #
бесплатный онлайн генератор QR кодов
MetaMask Download
| #
MetaMask Download was the best decision I made. Now I can access my assets anytime, anywhere, with full security.
zaimpodzalogptsekbsig
| #
автоломбард залог птс
zaim-pod-zalog-pts1.ru/ekb.html
деньги под залог птс екатеринбург круглосуточно
Trefstq
| #
Приобрести документ о получении высшего образования можно у нас. diplomd-magazinp.ru/kupit-attestat-za-9-klass-4-5
WilliamIRrutty
| #
darkmarket link dark web market urls
Tolodtsnozy
| #
На сайте https://lilibum.ru вы обязательно найдете вторую половинку для того, чтобы пообщаться, завести знакомства, найти человека для любви, серьезных отношений. Здесь ни одна сотня анкет, среди которых вы точно найдете того, кто симпатичен. Сразу напишите ему и пригласите на долгожданное свидание, чтобы пообщаться, узнать о его интересах, а потом, возможно, и создать семью. Постоянно выкладываются новые анкеты для того, чтобы вы обязательно подобрали родственную душу для любых целей. Для доступа ко всем функциям пройдите регистрацию.
Vincenttef
| #
Создание QR кодов
FNDaviditabe
| #
darknet markets 2025 darknet markets onion
Volodyanaf
| #
dark web market list dark web marketplaces
mostbet_weEl
| #
motsbet https://www.severussnape.borda.ru/?1-10-0-00000023-000-0-0-1743053372 .
Billydrula
| #
elonbet casino
MarkNOlaf
| #
darkmarket dark web market urls
mostbet_ajEl
| #
mostbet kg отзывы mostbet kg отзывы .
mostbet_ueOr
| #
mostbet kg отзывы mostbet kg отзывы .
1win_qnOt
| #
партнёрка 1win http://fanfiction.borda.ru/?1-0-0-00029708-000-0-0-1743051664/ .
panupFride
| #
Сервис бесплатных объявлений №1 хорошо зарекомендовал себя, о чем свидетельствуют отзывы. Плюсы: удобный интерфейс, огромная база объявлений, широкий выбор категорий для размещения и поиска предложений. Заходите и регистрируйтесь. Найдите своего покупателя! Ищете транспорт доска бесплатных объявлений? Ynla.ru – тут с мошенниками меньше вероятности столкнуться. Несмотря на скромный интерфейс, сайт достаточно функционален. Регистрация занимает всего несколько минут. Сервис наш для ПК, планшетов и смартфонов оптимизирован. Настоящей доской объявлений пользуйтесь!
Vincenttef
| #
Лучший генератор QR-кодов
mostbet_xxOr
| #
mostbet kg скачать на андроид cah.forum24.ru/?1-3-0-00000096-000-0-0-1743053764 .
1win_bqOt
| #
1win партнерка вход https://www.fanfiction.borda.ru/?1-0-0-00029708-000-0-0-1743051664 .
WilliamIRrutty
| #
darknet sites darknet sites
mostbet_mdMn
| #
мостбет скачать бесплатно https://assa0.myqip.ru/?1-23-0-00000149-000-0-0-1743053201/ .
mostbet_xhEl
| #
мостбет официальный сайт мостбет официальный сайт .
mostbet_scMn
| #
мостбет промокод http://assa0.myqip.ru/?1-23-0-00000149-000-0-0-1743053201/ .
mostbet_smOr
| #
mostbet скачать на телефон бесплатно андроид cah.forum24.ru/?1-3-0-00000096-000-0-0-1743053764 .
mostbet_dpMn
| #
мосбет assa0.myqip.ru/?1-23-0-00000149-000-0-0-1743053201 .
1win_nxOt
| #
1 вин. https://fanfiction.borda.ru/?1-0-0-00029708-000-0-0-1743051664/ .
1win_lwml
| #
1win sports http://www.1win12.com.ng .
AntonioHaush
| #
Nucleus Earn is revolutionizing DeFi staking and passive income generation by offering secure, high-yield crypto rewards. With smart contract-powered staking pools, Nucleus Earn allows users to earn rewards effortlessly while maintaining full control over their assets. Whether you’re a beginner or an experienced investor, Nucleus Earn’s decentralized staking platform ensures transparency, security, and optimal returns in the fast-growing world of DeFi. https://nucleusearn.org
Vincenttef
| #
https://qrkoder.ru/
Billydrula
| #
elonbet casino BD
KennethHew
| #
Рассрочка и кредит на окна без переплат — подробнее вот здесь https://secure.smore.com/n/8ex15-okna-v-spb-ru
KennyHut
| #
UnagiSwap is a cutting-edge decentralized exchange (DEX) that provides fast, secure, and transparent crypto trading. Designed for traders looking to swap digital assets efficiently without intermediaries, UnagiSwap offers low fees, deep liquidity, and seamless smart contract execution. Whether you’re a casual trader or a professional investor, UnagiSwap’s non-custodial platform ensures full control over your assets in a decentralized environment. https://unagiswap.org
Gregorybek
| #
Perena is a cutting-edge blockchain development platform designed to empower decentralized applications (dApps) with scalable and secure solutions. Built for Web3 innovators, Perena blockchain solutions offer seamless integration, high-performance smart contracts, and decentralized infrastructure. Whether you’re building DeFi protocols, NFT marketplaces, or enterprise blockchain applications, Perena provides the tools needed to scale with confidence. https://perena.tech
giyujhouby
| #
ВудХаус495 – компания, которая квалифицированные услуги предоставляет. Мы реализовали больше 20 000 строительно-отделочных работ деревянных домов. Гарантируем лучшую цену на рынке. Договор составляем, в котором все моменты детально прописываем. http://woodhouse495.ru – здесь представлена более детальная информация о нас. Используем только качественные материалы. В кратчайшие сроки выполняем поставленные перед нами задачи. Предоставляем каждому клиенту индивидуальный подход. Заполните форму на сайте, и мы перезвоним вам в ближайшее время.
Robertskits
| #
Upshift Finance is a next-generation decentralized trading platform designed to provide secure, fast, and efficient crypto transactions. With smart contract automation, low transaction fees, and seamless integration with DeFi protocols, Upshift Finance empowers traders to swap digital assets and execute trades with maximum security. Whether you’re a beginner or an experienced trader, Upshift Finance offers a powerful, transparent, and user-friendly trading ecosystem. https://upshift.ink
Vincenttef
| #
автоматизация бизнес-процессов
prodvijenie saita_myOr
| #
раскрутка сайта в топ http://www.puzzleweb.ru/recl3/effjektivnoje-prodvizhjenije-sajtov-v-moskvje-kak-dostich-vysokikh-rjezultatov.php/ .
Full Report
| #
Full Report
blog topic
xejonbed
| #
Типография Copy General окажет помощь в воплощении ваших идей в реальность. Мы понимаем то, что каждый наш проект уникальным является и особого подхода требует. Ценим время клиентов. Гарантируем высочайшую скорость осуществления заказов. https://copygeneral.ru – тут с отзывами заказчиков можете ознакомиться. Copy General – типография, на которую вы спокойно можете положиться. Постоянно над новыми решениями работаем, чтобы помогать своим клиентам задачи решать с дизайном, маркетингом и полиграфией. Применяйте преимущества нашей типографии!
Vincenttef
| #
Лучший генератор QR-кодов
FevemtJem
| #
На сайте https://lufkad.com/ вы сможете заказать качественные, сертифицированные и оригинальные мансардные окна премиального качества и по привлекательной стоимости. Конструкции LUFKAD созданы по уникальным и инновационным технологиям, потому отличаются безупречным качеством, долгим сроком эксплуатации. LUFKAD считается такой компанией, которая выполняет все необходимые работы и в полном объеме. Многие производственные технологии, которые применяются этой компанией, не имеют аналогов. Заказывайте окна по лучшей стоимости.
1win_ivml
| #
1win sports betting https://1win12.com.ng .
KiriyvoLauff
| #
Сайт https://t.me/azino777_a является официальным каналом популярного онлайн-заведения «Azino 777». Только здесь находятся самые последние новости, а также увлекательные анонсы слотов, турниров. На сайте также публикуются и бонусы, а также промокоды, которые позволят сэкономить. Заходите на этот сайт для того, чтобы получить больше полезной, важной, содержательной информации на данную тему. С этого канала вы сможете сразу перейти на официальный сайт. Теперь все новости из сферы игр находятся в вашем мобильном.
prodvijenie saita_keOr
| #
раскрутка сайта в москве http://www.puzzleweb.ru/recl3/effjektivnoje-prodvizhjenije-sajtov-v-moskvje-kak-dostich-vysokikh-rjezultatov.php .
Vincenttef
| #
автоматизация бизнес-процессов
RobertDaw
| #
Non Gamstop casinos offer the best variety—super cool!
non gamstop onlnie casino lightning roulette
More Info
| #
More Info
blog topic
1win_gfOt
| #
1хwin http://admiralshow.forum24.ru/?1-17-0-00000242-000-0-0-1742816555/ .
1win_zzOt
| #
1хwin admiralshow.forum24.ru/?1-17-0-00000242-000-0-0-1742816555 .
Vincenttef
| #
https://orgnaztech.mirtesen.ru/blog/43202871346/Gde-Kazhdyiy-Gost-Osobennyiy-Otel-Glory
MelvinROF
| #
Лучшие сайты кейсов ggdrop.casecs2.com в CS2 – честный дроп, редкие скины и гарантии прозрачности. Сравниваем платформы, бонусы и шансы. Заходи и забирай топовые скины!
1win_wzOt
| #
1 вин https://admiralshow.forum24.ru/?1-17-0-00000242-000-0-0-1742816555/ .
prodvijenie saita_cqOr
| #
сайтов продвижение http://www.puzzleweb.ru/recl3/effjektivnoje-prodvizhjenije-sajtov-v-moskvje-kak-dostich-vysokikh-rjezultatov.php/ .
Cazrtir
| #
Купить диплом о высшем образовании!
Мы можем предложить документы университетов, расположенных в любом регионе РФ. Документы выпускаются на бумаге высшего качества: opengroup.net/include/pgs/kupit_diplom_o_srednem_obrazovanii_29.html
RobertDaw
| #
Best non Gamstop casino for roulette is unreal—nice!
non gamstop no deposit casino
1win_zgPi
| #
1 win официальный http://1win6020.ru/ .
Vincenttef
| #
https://intekey.ru/
zaimpodptskazancoN
| #
автоломбард в казани под залог
avtolombard-pid-zalog-pts.ru/kazan.html
автоломбард
Vincenttef
| #
https://intekey.ru/
RobertDaw
| #
Non-Gamstop casinos are the real deal—highly recommend!
new non gamstop casinos no deposit bonus
Dnrtcga
| #
Где купить диплом по нужной специальности?
Мы изготавливаем дипломы психологов, юристов, экономистов и прочих профессий по приятным ценам. Мы готовы предложить документы техникумов, которые расположены на территории всей Российской Федерации. Можно заказать диплом за любой год, включая документы старого образца. Документы печатаются на “правильной” бумаге самого высшего качества. Это позволяет делать настоящие дипломы, не отличимые от оригиналов. Документы заверяются необходимыми печатями и подписями. Всегда стараемся поддерживать для заказчиков адекватную политику тарифов. Важно, чтобы документы были доступны для подавляющей массы наших граждан. diplom-zakaz.ru/kupit-diplom-vuza-6
1win_gbsi
| #
1вин войти http://www.realistzoosafety.forum24.ru/?1-11-0-00001540-000-0-0-1742816894 .
1win_ymMa
| #
игра 1вин https://naigle.borda.ru/?1-17-0-00000329-000-0-0-1742816734 .
Vincenttef
| #
https://intekey.ru/
1win_risi
| #
1win kg скачать realistzoosafety.forum24.ru/?1-11-0-00001540-000-0-0-1742816894 .
1win_uiMa
| #
one win https://naigle.borda.ru/?1-17-0-00000329-000-0-0-1742816734 .
1win_bjPi
| #
1вин кыргызстан http://1win6020.ru .
1win_xpsi
| #
1win казино https://www.realistzoosafety.forum24.ru/?1-11-0-00001540-000-0-0-1742816894 .
1win_roMa
| #
1vin kg https://naigle.borda.ru/?1-17-0-00000329-000-0-0-1742816734 .
Arthurver
| #
Рэпер Паша Техник находится в критическом состоянии в Таиланде
https://x.com/NargisEhme94100/status/1906094610788573414
Vincenttef
| #
https://qrkoder.ru/
Kxyufaita
| #
dark web market links darknet market lists
FNDaviditabe
| #
dark market darknet drug market
Zadatelwrino
| #
На сайте https://t.me/official_izzicasino/ представлены новости, которые будут вам интересны. Так, находясь в любом месте, вы получите возможность отслеживать появление новостей, свежих данных, полезной информации, которая вам обязательно пригодится. Здесь всегда публикуются промокоды, акции, которые сделают игру более интересной, увлекательной, динамичной. Важная информация выкладывается здесь регулярно, чтобы вы были в курсе последних событий. Теперь вся ценная информация окажется в одном месте.
RobertDaw
| #
Non Gamstop casinos no deposit offers are perfect for testing games—cool!
no gamstop casino
Diplomi_auKt
| #
Купить диплом университета!
Мы готовы предложить документы университетов, расположенных на территории всей России.
diplomt-v-chelyabinske.ru/kupit-attestat-za-9-klass-v-moskve-2
MarkNOlaf
| #
dark web link darkmarket link
Vincenttef
| #
автоматизация бизнес-процессов
1win_hcPi
| #
1win live 1win6020.ru .
zaimpodptskemerovosig
| #
деньги под залог авто
zaim-pod-zalog-pts1.ru/kemerovo.html
займ денег под залог автомобиля
PhillipHit
| #
Video surveillance software plays a crucial role in both residential and commercial security. Security camera software and security surveillance software provide real-time video monitoring and recording. IP camera software for PC, IP camera recording software, and network video recorder software enable secure video storage. VMS software, such as VMS CCTV software and VMS security camera software, supports large-scale surveillance operations. AI video surveillance software and artificial intelligence CCTV software improve security by detecting objects and movements automatically. Cloud video surveillance software and remote video surveillance software offer flexible access to surveillance footage. Motion detection software enhances security by alerting users to unusual activity. smart vision ip camera software
Volodyanaf
| #
darkmarket url dark web market
WilliamIRrutty
| #
dark web drug marketplace darknet markets url
Sazrrdv
| #
Мы предлагаем дипломы любой профессии по приятным ценам. О преимуществах заказа документов в нашей компании
Вы приобретаете документ в надежной и проверенной временем компании. Это решение сэкономит не только средства, но и ваше драгоценное время.
На этом плюсы не заканчиваются, их куда больше:
• Дипломы делаем на подлинных бланках с мокрыми печатями и подписями;
• Дипломы всех высших учебных заведений РФ;
• Цена намного ниже той, которую потребовалось бы платить на очном обучении в университете;
• Максимально быстрая доставка в любые регионы Российской Федерации.
Заказать диплом о высшем образовании– http://soocian.com/read-blog/6145_diplom-ob-obrazovanii.html/ – soocian.com/read-blog/6145_diplom-ob-obrazovanii.html
Vincenttef
| #
https://intekey.ru/
RobertDaw
| #
Just signed up at a non Gamstop casino—loving the variety so far!
non gamstop casino 2021
Vincenttef
| #
Создание QR кодов
gemavliers
| #
UpakovkaRus – компания, которая характеризуется надежностью и выгодной ценой. Производим приличный выбор изделий из ПВХ и спанбонда. Используем материалы, отличающиеся высочайшим качеством. Всегда любые пожелания и требования заказчиков учитываем. https://upakovkarus.ru – здесь представлены примеры готовой продукции. Если не отыщите необходимый вариант, пришлите нам фото вашей модели, такую же осуществим. Вы можете связаться с нами в любое время, контакты на ресурсе указаны. Нашими клиентами мы дорожим. Ждем от вас заявок!
Ап-Х официальный сайт
| #
Добро пожаловать в Up X Casino, где ваши мечты об азартных играх станут реальностью. Здесь представлены широкий ассортимент азартных игр, включая видеослоты, карточные игры и рулетку. И это еще не все! Для наших игроков доступны регулярные турниры и акции, которые помогают увеличить шансы на успех. По данным статистики, активные игроки достигают лучших результатов. Когда стоит испытать удачу? Ответ прост: начните прямо сейчас! https://russkij-tekst.ru/. Какие шаги предпринять перед игрой? Изучите правила и условия использования платформы для успешного начала вашей игровой сессии. Используйте специальные условия для постоянных игроков, чтобы получить максимум удовольствия и выигрыша. Попробуйте бесплатные слоты, чтобы освежить навыки, чтобы вернуться в игровой ритм без риска.
RobertDaw
| #
Non Gamstop UK casino has a slick design—totally hooked!
new non gamstop casino
Vincenttef
| #
https://intekey.ru/
zaimpodptsmsksig
| #
автоломбард под залог
zaim-pod-zalog-pts1.ru
кредит под залог птс авто
GetufGek
| #
Наша компания https://etbpro.ru/ предлагает комплексные решения для проектирования, монтажа, модернизации и технического обслуживания высокотехнологичных электронных систем: видеонаблюдение, пожарная сигнализация, скуд, электрооборудование, охранная сигнализация и автоматизированные системы управления. ООО ЭТБ Ваш надежный партнер в обеспечении безопасности. Подробнее на сайте.
Vincenttef
| #
Создание QR кодов
KosemtMug
| #
На сайте https://mybeterex.com/ вы сможете уточнить всю необходимую информацию по поводу надежной и проверенной компании «BETEREX», которая предлагает полный комплекс услуг, связанных с ведением бизнеса в Китае. У вас появляется возможность заказать доставку грузов, товаров. Кроме того, вы сможете рассчитывать на получение всех необходимых образцов, а также переговоры с другой стороной. Будут решены все вопросы, независимо от сложности. Воспользуйтесь профессиональной консультацией, чтобы уточнить информацию.
1win_pcml
| #
1win home 1win12.com.ng .
RobertDaw
| #
Non Gamstop casinos no deposit offers are too good to miss—great!
casino no on gamstop
Vincenttef
| #
Создание QR кодов
1win_mvsl
| #
1win сайт вход http://1win6001.ru .
KefuspBiC
| #
Ищете куда сходить в Ижевске? Посетите сайт https://izhfun.ru/ и вы найдете главные развлечения Ижевска и где отдохнуть. Выбирайте категории: кафе и рестораны, квизы и игры, театры, концерты, другие мероприятия и места и смотрите текущие или будущие события. Выбирайте интересное времяпрепровождение вместе с нами! Самая актуальная информация на нашем сайте как получить незабываемые эмоции!
buy iptv
| #
buy iptv
A Brief History of Elemental Analysis » Artemis Analytical
IPTV Box Store
| #
IPTV Box Store
A Brief History of Elemental Analysis » Artemis Analytical
Vincenttef
| #
Создание QR кодов
RobertDaw
| #
Non Gamstop casinos no deposit deals keep me coming back—great!
non gamstop casinos netent
zaimpodptsnovosibsig
| #
кредит под птс грузового автомобиля
zaim-pod-zalog-pts1.ru/nsk.html
кредит залог птс в новосибирске
PhillipHit
| #
This resource specializes in video surveillance software, featuring reviews of security camera software for Windows, IP camera software for PC, and surveillance software for PC. It includes in-depth evaluations of IP camera recording software and motion detection software, ensuring users have access to the latest video security software. The platform also covers cloud video surveillance software and VMS software, helping users integrate these tools into their overall security systems. Whether looking for surveillance software for PC or advanced IP camera solutions, this site provides essential guidance. smartvision best video surveillance software
Vincenttef
| #
https://orgnaztech.mirtesen.ru/blog/43202871346/Gde-Kazhdyiy-Gost-Osobennyiy-Otel-Glory
1win_jqml
| #
1win online https://www.1win12.com.ng .
EdwardBiorn
| #
Цвет не выгорает на солнце!
женский костюм купить томск
RobertDaw
| #
Non-Gamstop casinos offer the best escape—highly recommend!
non gamstop no deposit casino
1win_ugsl
| #
1win ставки официальный сайт https://1win6001.ru .
Vincenttef
| #
Лучший генератор QR-кодов
prodvijenie saita_wmOr
| #
seo продвижение сайтов seo продвижение сайтов .
Kxyufaita
| #
tor drug market onion dark website
FNDaviditabe
| #
dark web market darkmarket list
Vincenttef
| #
Glory Casino app
mostbet_piei
| #
mostbet kg скачать на андроид http://mostbet6006.ru .
1win_vemr
| #
1-win https://www.familyclub.borda.ru/?1-6-0-00002163-000-0-0-1743051813 .
1win_tcPi
| #
1win.pro 1win6020.ru .
MarkNOlaf
| #
dark market darknet markets
zaimpodzalogptsekbsig
| #
оформить кредит под залог авто
avtolombard-11.ru/ekb.html
деньги под залог машины екатеринбург
pubuwbuh
| #
АнтикорМастер предлагает квалифицированные услуги. Наша политика ценообразования прозрачна. Стараемся осуществить процесс ухода за вашим авто легким и долгосрочным. Запишитесь на бесплатную диагностику, чтобы выяснить состояние вашего антикоррозийного покрытия. https://xn—-7sbbummpeluekfi.xn--p1ai/ – здесь узнаете, почему нас выбирают. Предлагаем чистку лазерную, которая не оставляя следов, эффективно ржавчину убирает. Предоставляем до 10 лет на осуществленные работы письменную гарантию. Оставьте личные контакты на сайте, и мы с вами свяжемся.
Jariornsw
| #
Где заказать диплом по нужной специальности?
Наши специалисты предлагают быстро и выгодно заказать диплом, который выполнен на оригинальном бланке и заверен печатями, водяными знаками, подписями официальных лиц. Диплом способен пройти любые проверки, даже при использовании специально предназначенного оборудования. Достигайте своих целей максимально быстро с нашей компанией.
Приобрести диплом любого ВУЗа diploml-174.ru/kupit-diplom-novogo-obraztsa-14/
Vincenttef
| #
автоматизация бизнес-процессов
SteveNor
| #
https://punkgazetka.forum24.ru/?1-9-0-00002408-000-0-0-1740791014
Sazrhte
| #
Покупка документа о высшем образовании через надежную фирму дарит ряд плюсов. Это решение дает возможность сберечь как личное время, так и серьезные финансовые средства. Впрочем, только на этом выгоды не ограничиваются, достоинств намного больше.Мы готовы предложить дипломы любой профессии. Дипломы производят на настоящих бланках государственного образца. Доступная цена в сравнении с огромными затратами на обучение и проживание в чужом городе. Покупка диплома института является мудрым шагом.
Приобрести диплом о высшем образовании: kupitediplom.ru/diplom-vracha-kupit/
Mazrezn
| #
Где заказать диплом по актуальной специальности?
Мы оказываем услуги по изготовлению и продаже документов об окончании любых ВУЗов РФ. Документы изготавливаются на фирменных бланках. teamswedenclub.com/read-blog/6329_kupit-svidetelstvo-o-razvode.html
Mazrnkd
| #
Где заказать диплом специалиста?
Получаемый диплом со всеми печатями и подписями целиком и полностью отвечает условиям и стандартам Министерства образования и науки Российской Федерации, неотличим от оригинала – даже со специально предназначенным оборудованием. Не следует откладывать свои мечты и цели на продолжительные годы, реализуйте их с нами – отправляйте быструю заявку на изготовление диплома уже сегодня! Купить диплом о высшем образовании – быстро и просто! vacshidiplom.com/kupit-gotovij-attestat/
Sazrzvx
| #
Купить документ университета можно в нашем сервисе. Мы предлагаем документы об окончании любых университетов РФ. Вы получите необходимый диплом по любой специальности, любого года выпуска, включая документы старого образца. Гарантируем, что в случае проверки документа работодателями, подозрений не возникнет. rentry.co/9uc8eqgg
More info
| #
Just wish to say your article is as astonishing.
The clearness to your publish is simply great and i can suppose you’re a professional
on this subject. Fine along with your permission allow me to grab your feed to keep up to date
with approaching post. Thank you a million and please carry on the enjoyable work.
shinomontaj_dmSn
| #
шиномонтаж малаховка шиномонтаж малаховка .
WilliamIRrutty
| #
dark websites dark web drug marketplace
prodvijenie saita_hekn
| #
раскрутка сайтов москва раскрутка сайтов москва .
Kxyufaita
| #
dark websites darknet markets onion address
FNDaviditabe
| #
onion dark website dark web drug marketplace
Vincentduh
| #
Что такое «Порту Пати» в Дубае и почему об этом говорят все
https://x.com/DeyanetKrmv/status/1906296026006220909
Volodyanaf
| #
dark markets 2025 darknet markets onion address
http://gudang.desa.id/pagerank/
| #
http://gudang.desa.id/pagerank/
blog topic
MarkNOlaf
| #
dark market list tor drug market
FivifdvaH
| #
На сайте https://gk-psk72.ru/ закажите звонок для того, чтобы приобрести ЖБИ от производителя. Прямо сейчас воспользуйтесь возможностью изучить всю продукцию, которая обязательно вам пригодится. А для того, чтобы узнать об особенностях работы предприятия, необходимо изучить портфолио. Компания оказывает услуги в строительной деятельности более 10 лет. Всего в каталоге более чем тысяча наименований. Есть возможность воспользоваться комплексным предложением товаров, услуг. Преимуществом обращения в компанию является и небольшая стоимость.
Zokojthboava
| #
На сайте https://t.me/rahvalskiy_team уточните то, какие услуги оказываются популярной и проверенной компанией «RAHVALSKIY TEAM», которая предлагает создать уникальный, функциональный сайт. Он будет работать специально для вас и раскрутки бизнеса. В обязательном порядке разрабатывается особая эффективная стратегия. В компании работают проверенные, знающие и квалифицированные сотрудники, которые учитывают потребности всех клиентов. Воспользуйтесь комплексными услугами для организации бизнеса.
shinomontaj_mtSn
| #
шиномантаж подольск шиномантаж подольск .
WilliamIRrutty
| #
dark web market list darknet markets 2025
1win_dhPi
| #
скачать 1win официальный сайт http://www.1win6020.ru .
prodvijenie saita_jnOr
| #
seo продвижение сайтов seo продвижение сайтов .
1win_jymr
| #
1 win официальный familyclub.borda.ru/?1-6-0-00002163-000-0-0-1743051813 .
Andrewsmick
| #
Say hello to AquaSculpt—a game-changer in weight loss! These AquaSculpt capsules use natural AquaSculpt ingredients to shed pounds and boost confidence. No AquaSculpt side effects, just pure AquaSculpt results—see why in AquaSculpt reviews. Learn AquaSculpt how to use and join thousands who love it. AquaSculpt buy today at http://aquasculpt.best
zaimpodptskazancoN
| #
кредит под залог птс
avtolombard-11.ru/kazan.html
займ под залог авто
Kxyufaita
| #
darkmarkets dark web link
FNDaviditabe
| #
dark web sites bitcoin dark web
prodvijenie saita_gfkn
| #
раскрутка сайтов в москве раскрутка сайтов в москве .
w69 ทางเข้า
| #
w69 ทางเข้า – https://rwho3.com ปรับปรุงอินเทอร์เฟซแอปให้ใช้งานง่ายขึ้น ดีไซน์สวยงาม ค้นหาเกมได้รวดเร็วและสะดวกสบาย
mostbet_vtei
| #
mosbet http://mostbet6006.ru .
Robertwef
| #
Struggling to lose weight? AquaSculpt is transforming weight loss with its natural, fast-acting capsules. Packed with proven AquaSculpt ingredients, these capsules burn fat, boost energy, and deliver real AquaSculpt results in weeks. Curious about AquaSculpt reviews? Users love its effectiveness and zero AquaSculpt side effects. Want to know AquaSculpt how to use? It’s simple—take daily and watch the pounds melt away. Ready to try? AquaSculpt buy now at http://aquasculpt.xyz and sculpt your dream body today!
Brianhig
| #
Say hello to AquaSculpt—a game-changer in weight loss! These AquaSculpt capsules use natural AquaSculpt ingredients to shed pounds and boost confidence. No AquaSculpt side effects, just pure AquaSculpt results—see why in AquaSculpt reviews. Learn AquaSculpt how to use and join thousands who love it. AquaSculpt buy today at http://aquasculpt.one !
MichaelBop
| #
Struggling to lose weight? AquaSculpt is transforming weight loss with its natural, fast-acting capsules. Packed with proven AquaSculpt ingredients, these capsules burn fat, boost energy, and deliver real AquaSculpt results in weeks. Curious about AquaSculpt reviews? Users love its effectiveness and zero AquaSculpt side effects. Want to know AquaSculpt how to use? It’s simple—take daily and watch the pounds melt away. Ready to try? AquaSculpt buy now at http://aquasculpt.me and sculpt your dream body today!
MarkNOlaf
| #
dark web market links darknet market links
shinomontaj_buSn
| #
шиномонтаж щёлково шиномонтаж щёлково .
Arthurbalty
| #
AquaSculpt weight loss is here to stay! With AquaSculpt capsules, you get fast AquaSculpt results thanks to natural AquaSculpt ingredients. No worries about AquaSculpt side effects—users confirm it in AquaSculpt reviews. Curious AquaSculpt how to use? It’s easy and effective. AquaSculpt where to buy? Visit http://aquasculpt.lifestyle and transform your body now!
VogacPeT
| #
На сайте https://doge.coinwatchtoday.ru/ вы узнаете все о криптовалюте Dogecoin (DOGE) – история, новости, торговые идеи, криптобиржи и торговые инструменты, которые позволят вам с выгодой инвестировать или торговать c Dogecoin (DOGE). В отличие от биткоина, DOGE предлагает быстрые и дешевые транзакции, оставаясь при этом удобным средством микроплатежей и благотворительности. Простота и низкие комиссии!
Volodyanaf
| #
darknet market list darknet markets 2025
1win_fymr
| #
1wi https://familyclub.borda.ru/?1-6-0-00002163-000-0-0-1743051813/ .
букмекерские конторы которые дают фрибет
| #
Фрибет: Что Это Такое и Как Его Использовать?
В мире онлайн-ставок и азартных игр термин “фрибет” стал широко известен и популярен среди игроков. Но что это такое, как работает фрибет и как его можно использовать с максимальной выгодой? В этой статье мы разберем все ключевые аспекты, связанные с фрибетами, чтобы вы могли уверенно применять их в своей игре.
Что такое фрибет?
Фрибет (от англ. “free bet” — бесплатная ставка) — это бонус, который https://www.youtube.com/watch?v=eMVTiWJujqQ конторы или онлайн-казино предоставляют своим пользователям. Это своего рода подарок, позволяющий сделать ставку на спортивное событие или сыграть в игру без использования собственных средств. Если ставка выигрывает, игрок получает выигрыш за вычетом суммы самого фрибета, а если проигрывает — не теряет ничего из своего кошелька.
Фрибеты часто используются букмекерами как маркетинговый инструмент для привлечения новых клиентов или поощрения активных пользователей. Это отличный способ попробовать свои силы в ставках, не рискуя личными деньгами.
Виды фрибетов
Фрибеты могут различаться по условиям предоставления и использования. Вот основные типы, с которыми вы можете столкнуться:
Приветственный фрибет
Этот бонус обычно предлагается новым игрокам при регистрации на сайте букмекера. Например, после создания аккаунта и подтверждения личности вы можете получить фрибет на сумму 500 или 1000 рублей.
Фрибет за депозит
Некоторые букмекеры начисляют бесплатную ставку после пополнения счета на определенную сумму. Например, внеся 1000 рублей, вы можете получить фрибет на такую же сумму.
Фрибет за активность
Постоянным игрокам могут начислять фрибеты за регулярные ставки, участие в акциях или выполнение определенных условий, таких как серия ставок на конкретные события.
Безусловный фрибет
Это редкий, но очень желанный вид бонуса, который не требует выполнения сложных условий. Вы просто получаете фрибет и можете использовать его по своему усмотрению.
Фрибет на день рождения
Многие букмекерские конторы радуют своих клиентов небольшими подарками в виде фрибетов в честь дня рождения.
Как получить фрибет?
Процесс получения фрибета обычно прост и занимает немного времени. Вот стандартные шаги:
Регистрация: Создайте аккаунт на сайте букмекерской конторы.
Верификация: Подтвердите свою личность, загрузив необходимые документы (паспорт или другой ID).
Выполнение условий: Ознакомьтесь с правилами акции — это может быть внесение депозита, ввод промокода или ставка на определенное событие.
Активация: В некоторых случаях нужно активировать фрибет в личном кабинете или через поддержку.
После этого фрибет будет зачислен на ваш бонусный счет, и вы сможете использовать его для ставок.
Как использовать фрибет с умом?
Чтобы извлечь максимум пользы из фрибета, важно учитывать несколько моментов:
Изучите условия: У каждого фрибета есть свои правила. Например, минимальный коэффициент для ставки (обычно от 1.5 до 2.0) или ограничение по видам спорта.
Выбирайте событие с высокой вероятностью: Поскольку это бесплатная ставка, стоит выбрать матч или игру, в исходе которых вы уверены.
Не торопитесь: Фрибеты часто имеют срок действия (от 7 до 30 дней), поэтому используйте их обдуманно, а не на первой попавшейся ставке.
Планируйте вывод: Если ставка выиграла, уточните, можно ли сразу вывести выигрыш или нужно выполнить дополнительные условия (например, отыграть сумму).
Плюсы и минусы фрибетов
Плюсы:
Возможность попробовать ставки без риска для кошелька.
Шанс выиграть реальные деньги с минимальными вложениями.
Отличный бонус для новичков, чтобы освоиться на платформе.
Минусы:
Ограничения по использованию (коэффициенты, события, сроки).
Выигрыш часто требует отыгрыша перед выводом.
Не все фрибеты действительно “бесплатны” — иногда нужен депозит.
Популярные букмекеры с фрибетами
На российском рынке многие букмекерские конторы предлагают фрибеты. Вот несколько примеров:
1xСтавка: До 10 000 рублей для новых игроков.
Фонбет: Фрибет за регистрацию и первый депозит.
Леон: Регулярные акции с бесплатными ставками.
Winline: Один из лидеров по щедрым фрибетам без сложных условий.
Перед регистрацией обязательно проверяйте актуальные условия на сайте букмекера, так как предложения часто обновляются.
Советы для новичков
Сравнивайте предложения: Разные конторы дают разные суммы и условия — выбирайте то, что вам подходит.
Не гонитесь за крупными суммами: Иногда маленький фрибет с простыми условиями выгоднее, чем большой с кучей ограничений.
Читайте отзывы: Узнайте, что говорят другие игроки о выводе выигрышей с фрибетов.
Играйте ответственно: Даже с фрибетом не стоит увлекаться сверх меры — азарт должен приносить удовольствие.
kra31cc
| #
Very nice post. I simply stumbled upon your weblog and wished to mention that
I’ve truly loved surfing around your weblog posts. After
all I will be subscribing to your rss feed and I hope you
write once more soon!
WilliamIRrutty
| #
dark web market urls darkmarket list
prodvijenie saita_sgkn
| #
раскрутка сайта москва раскрутка сайта москва .
Kxyufaita
| #
darkmarket url darknet drug store
FNDaviditabe
| #
darknet markets 2025 darknet drugs
MarkNOlaf
| #
dark web market darknet markets 2025
Unlim бесплатные спины
| #
Анлим Казино предлагает игрокам возможность погрузиться в мир азартных игр.
Здесь в наличии множество игровых автоматов,
карточных игр, а также регулярно проводимые турниры,
что дает шанс игрокам увеличить свои шансы на выигрыш и добавляет массу
удовольствия от процесса.
Анлим Казино – это не просто место для игры, но и потрясающий опыт для всех пользователей, независимо от того, играете ли вы с мобильного устройства или с компьютера.
Мы гарантируем постоянное обновление ассортимента игр и организацию увлекательных турнирных состязаний.
В чем преимущество игры в Анлим Казино?
Быстрая регистрация — всего пару шагов, и вы уже готовы начать играть.
Большие бонусы для новичков — мы дарим бонусы на первый
депозит для старта с большими шансами на победу.
Регулярные акции и турниры —
для каждого, кто хочет расширить свои шансы на победу и дополнительные призы.
Круглосуточная поддержка всегда
готова помочь с любыми вопросами о процессе игры.
Богатый выбор игр доступных как на компьютере,
так и на мобильных устройствах.
Присоединяйтесь к нам уже сегодня!
Анлим Казино ждет вас. Готовы к победам? https://unlim-casinomirage.yachts/
Starda казино официальный сайт
| #
Хотите испытать настоящий азарт? Тогда Starda Casino – ваш идеальный выбор!! Здесь собраны разнообразие игровых автоматов от лицензированных провайдеров. https://starda-strike.buzz/ чтобы испытать удачу в топовых слотах!
Что делает Starda Casino лучшим выбором?
Популярные слоты с потрясающей графикой.
Эксклюзивные предложения для постоянных игроков.
Моментальные транзакции в кратчайшие сроки.
Интуитивный интерфейс без лагов и зависаний.
Техподдержка 24/7 готова помочь в любое время.
Испытайте удачу прямо сейчас чтобы получать максимальное удовольствие!
Once Human
| #
web siteniz çok güzel başarılarınızın devamını dilerim. makaleler çok hoş sürekli sitenizi ziyaret edeceğim
zaimpodptskemerovosig
| #
автоломбард в кемерово под залог
avtolombard-11.ru/kemerovo.html
кредит под залог авто
WilliamIRrutty
| #
dark web market urls tor drug market
DavidUrire
| #
Развитие профессиональных качеств Управленческие консультации от Светланы Подкладышевой, серийного предпринимателя с более чем 20-летним опытом. Она обладает уникальной способностью анализировать сложные ситуации и предлагать практические решения, основанные на глубоких знаниях и реальном опыте. Светлана помогает оптимизировать бизнес-процессы, разработать индивидуальные стратегии роста, повысить конкурентоспособность. Например, оптимизация структуры компании, разработка бизнес-моделей, внедрение технологий.
Volodyanaf
| #
dark market darknet markets 2025
официальный сайт даркнет
| #
Great delivery. Great arguments. Keep up the good spirit.
Kxyufaita
| #
dark web market links darkmarket
FNDaviditabe
| #
dark market list dark market list
MarkNOlaf
| #
darknet drug links best darknet markets
Kuxuhruiva
| #
На сайте https://t.me/s/official_izzicasino/ регулярно публикуются свежие и актуальные новости, которые касаются известного виртуального заведения «IZZI Casino». На этом канале вы найдете только содержательную, полезную информацию, которая обязательно пригодится, если являетесь фанатом этого игрового клуба. Здесь регулярно выкладываются анонсы турниров, выигрышные бонусы, а также фриспины и многое другое, что сделает игру более зрелищной, увлекательной. Вся информация об игорном заведении теперь в одном месте.
WilliamIRrutty
| #
dark web sites darknet markets onion
Kxyufaita
| #
dark web marketplaces dark web market
FNDaviditabe
| #
darknet markets dark websites
Volodyanaf
| #
dark market 2025 darkmarket list
XalowwDaype
| #
На сайте https://mos-stroi-alians.ru/ уточните телефон компании, чтобы воспользоваться такой нужной услугой, как кровля крыш независимо от сложности. На предприятии трудятся высококлассные, квалифицированные сотрудники, которые справятся с решением вопроса очень быстро, на профессиональном уровне. В работе применяются только лучшие, инновационные материалы, особые технологии для нужного результата. Работы ведутся и в выходной день. На все услуги даются гарантии, потому как компания уверена в их качестве.
MarkNOlaf
| #
dark market url dark market
AnthonyWifut
| #
Попробуйте https://abfgss53sdbkl33.ru/
1win_obsl
| #
1 win официальный сайт вход 1 win официальный сайт вход .
non-gamstop casino review
| #
non-gamstop casino review
blog topic
WilliamIRrutty
| #
darknet markets links dark web market links
zaimpodptsmsksig
| #
кредит залог птс наличные
avtolombard-11.ru
деньги залог авто москва
AnthonyWifut
| #
Обязательно попробуйте https://abfgss53sdbkl33.ru/
Vozaflmit
| #
На сайте https://sanobrabotkarf.ru/ закажите консультацию для того, чтобы оформить такую полезную услугу, как профессиональная дезинфекция. В компании применяются только проверенные, надежные препараты, которые считаются полностью безопасными для человека, животного. Работы выполняются лучшими сотрудниками, у которых есть сертификаты на оказание услуг, а также строго в оговоренные сроки. Специально для вас качественное, комплексное обслуживание. Сотрудники применяют инновационные, уникальные технологии для достижения результата.
FNDaviditabe
| #
darknet drugs darknet drug market
Volodyanaf
| #
dark web market urls darknet drug market
MarkNOlaf
| #
bitcoin dark web darknet market
Donaldlaf
| #
dark market darknet markets
TugewHaw
| #
Посетите сайт https://pech.pro/ это магазин печей, каминов и все для отделки бани. Зайдите в каталог и вы найдете существенный выбор – камины, печи, котлы отопительные, дымоходы, печи для дачи и многое другое по самым выгодным ценам. Действует доставка по Ярославлю и всей России. Профессионально осуществляем подключение, монтаж отопительных устройств и их защиты от возгорания.
Pingrutty
| #
darknet site darknet markets
Toliknaf
| #
dark markets 2025 darknet marketplace
WilliamIRrutty
| #
darkmarket dark web market links
Rabyitabe
| #
dark market 2025 dark web market urls
Kxyufaita
| #
dark market dark market onion
kra50cc
| #
I’m gone to say to my little brother, that he should also visit this web site on regular basis to get updated from newest reports.
sex hiepdam
| #
I’ve been surfing online more than three hours today, yet I never found
any interesting article like yours. It’s pretty worth enough for me.
In my view, if all web owners and bloggers made good content as you did, the internet will be much more useful than ever before.
DonDonfaita
| #
darknet links darknet markets onion
AnthonyWifut
| #
Обязательно попробуйте https://abfgss53sdbkl33.ru/
MarkNOlaf
| #
darknet sites onion dark website
Volodyanaf
| #
best darknet markets darknet sites
Pingrutty
| #
dark web market dark web sites
AndreDap
| #
Очень доволен! Цветы свежие, доставка быстрая.
букет пионов
zaimpodptsnovosibsig
| #
займ под залог автоломбард
avtolombard-11.ru/nsk.html
автоломбард под залог птс
Donaldlaf
| #
darknet links darknet site
WilliamIRrutty
| #
darkmarket list dark market link
Toliknaf
| #
dark web sites dark market 2025
Kxyufaita
| #
dark markets darkmarket link
Займ с любой кредитной историей - одобрение 99%
| #
Первый займ — бесплатно? Да, это возможно. Мы собрали рейтинг МФО, которые выдают онлайн займ на карту под 0% на 30 дней. Без подвоха, без скрытых комиссий. Деньги доступны любому жителю России всего по паспорту.
Rabyitabe
| #
darknet market links darkmarket 2025
MarkNOlaf
| #
darknet marketplace dark market link
DonDonfaita
| #
bitcoin dark web dark websites
Pingrutty
| #
darknet market links dark market url
WilliamIRrutty
| #
tor drug market darkmarket link
FNDaviditabe
| #
dark markets 2025 dark market link
AnthonyWifut
| #
Советую https://abfgss53sdbkl33.ru/
Donaldlaf
| #
bitcoin dark web best darknet markets
Eanrndu
| #
Для успешного продвижения по карьере понадобится наличие диплома о высшем образовании. Выгодно купить диплом университета у надежной компании: diplom-ryssia.com/kupit-zaregistrirovannij-diplom-bez-lishnix-xlopot/
uşak taksi
| #
Your site is amazing, the articles are great, I will always come to this site and continue to read because you have very nice articles, thank you
1win_ismr
| #
1win вход в личный кабинет http://familyclub.borda.ru/?1-6-0-00002163-000-0-0-1743051813/ .
Toliknaf
| #
dark markets 2025 tor drug market
MarkNOlaf
| #
darknet marketplace darknet market links
Once Human
| #
web siteniz çok güzel başarılarınızın devamını dilerim. makaleler çok hoş sürekli sitenizi ziyaret edeceğim
Nelejdobep
| #
Компания Мега скупка https://mega-skupka.com/ работает по городу Санкт-Петербурге. Нам можно продать ваши старые и новые гаджеты, а именно смартфоны, ноутбуки, iphone, apple, macbook, игровые Пк, Даем на 25% больше других конкурентов на месте.
AnthonyWifut
| #
Попробуйте https://abfgss53sdbkl33.ru/
avtolombardekbsig
| #
займ под залог авто по стс
https://www.bolsadetrabajotafer.com/employer/avtolombard-pid-zalog-pts6393/
деньги под птс автоломбард
Rabyitabe
| #
dark market onion darknet drugs
WilliamIRrutty
| #
dark markets 2025 dark market 2025
Pingrutty
| #
dark market dark market 2025
AnthonyWifut
| #
Советую https://abfgss53sdbkl33.ru/
Kxyufaita
| #
darknet market darknet market
FNDaviditabe
| #
bitcoin dark web darknet sites
Once Human
| #
web siteniz çok güzel başarılarınızın devamını dilerim. makaleler çok hoş sürekli sitenizi ziyaret edeceğim
DonDonfaita
| #
darknet links darknet sites
MarkNOlaf
| #
darkmarkets darknet drug market
site
| #
Thanks for your personal marvelous posting! I really enjoyed reading it, you could be a great
author.I will make sure to bookmark your blog and
will come back very soon. I want to encourage that you continue your
great job, have a nice holiday weekend!
Once Human
| #
web siteniz çok güzel başarılarınızın devamını dilerim. makaleler çok hoş sürekli sitenizi ziyaret edeceğim
Donaldlaf
| #
darknet markets links darknet markets 2025
Ramirojax
| #
кракен ссылка актуальная – кракен маркетплейс, Как использовать Tor для покупок
WilliamIRrutty
| #
darknet sites darknet markets 2025
Toliknaf
| #
best darknet markets darknet markets onion address
Kxyufaita
| #
darknet market lists darkmarket
FNDaviditabe
| #
darkmarkets darknet marketplace
Pingrutty
| #
dark market dark market link
mostbet_hyei
| #
мостбет войти https://mostbet6006.ru/ .
shinomontaj_xqSn
| #
шиномонтаж цены https://hondahybrid.ru/forum/topic/2886-kakie-uslugi-okazyvaet-shinomontazh// .
1win_oesl
| #
1win партнерская программа вход 1win партнерская программа вход .
Rabyitabe
| #
darknet drugs darkmarket link
xitohnam
| #
Fasvek кровельные материалы высокого качества предоставляет. Вся продукция имеет детальное описание с указанием технических параметров и характеристик. Прежде чем заказ сделать, с ними детальнее ознакомьтесь. Цены каждому доступны. Многие люди остались довольны сотрудничеством с нами. https://fasvek.ru – здесь представлены наши работы. Вы узнаете, что дает использование контролита. Сотрудники компании Вектор – настоящие профессионалы своего дела. Если необходима помощь, оставьте на портале ваш контактный номер, и мы в ближайшее время вам перезвоним.
MarkNOlaf
| #
darknet market lists bitcoin dark web
DonDonfaita
| #
darknet market list darknet market lists
avtolombardekbsig
| #
кредит наличными под залог птс
https://www.joboptimizers.com/employer/market9945/
займ залог авто
WilliamIRrutty
| #
dark web sites dark market 2025
LanihDourl
| #
На сайте https://t.me/rahvalskiy_team воспользуйтесь возможностью заказать такую популярную услугу, как продвижение в Нижнем Новгороде и по любому другому городу. В компании работают лучшие, проверенные и талантливые специалисты, которые подберут для вас оптимальный вариант для того, чтобы ваш бизнес стал востребованным. Сотрудники изучат рынок, а также подберут наиболее уместную и работающую стратегию, которая обязательно вам подойдет. Воспользуйтесь комплексными рекламными услугами. Они обойдутся недорого.
Donaldlaf
| #
darknet site darkmarket url
FNDaviditabe
| #
darknet markets url dark web sites
Kxyufaita
| #
darknet sites dark web drug marketplace
Pingrutty
| #
dark web sites dark web marketplaces
Toliknaf
| #
dark market dark web marketplaces
uşak taksi
| #
Your site is amazing, the articles are great, I will always come to this site and continue to read because you have very nice articles, thank you
MarkNOlaf
| #
dark market link darknet marketplace
ZobuxKneex
| #
На сайте https://organ-hall.ru вы сможете получить уникальный опыт, играя в популярное онлайн-заведение «Vodka Casino», которое заполучило огромное количество поклонников благодаря своей честной и прозрачной политике, большому количеству бонусов. Здесь находятся автоматы, промокоды, бонусы для более зрелищной и интересной игры. Этот клуб создан с использованием новых технологий, поэтому предоставляет безупречный уровень сервиса, регулярные выплаты, интересные функции. С этим заведением вы сможете позволить себе больше!
prodvijenie saita_ytkn
| #
продвижение сайтов в москве продвижение сайтов в москве .
Walterheend
| #
Jestem skrajnie niezadowolony witryna Huone. Tresci na niej sa nieaktualne, a pomoc techniczna po prostu nie odpowiada na wiadomosci. Sposob skladania zamowienia to calkowity koszmar: liczne awarie, powolne ladowanie i absolutny brak obslugi. Mam przekonanie, ze administratorzy po prostu lekcewaza uzytkownikow. Nie zuzywajcie swojego czasu i energii i nerwow, sa o wiele godne uwagi opcje!
Rabyitabe
| #
dark web marketplaces dark markets
1win_mhsl
| #
1вин войти https://www.1win6001.ru .
mostbet_yhei
| #
mostbest http://mostbet6006.ru .
AnthonyWifut
| #
Крайне советую https://abfgss53sdbkl33.ru/
fuel pressure sensor
| #
Hi, Neat post. There is a problem with your website in web explorer, might test this?
IE nonetheless is the market chief and a big component to other folks will
miss your magnificent writing because of this
problem.
CecilExign
| #
Посетите интернет-магазин http://sharpsting.ru и оцените разнообразие товаров: от современных гаджетов до полезных аксессуаров. Мы ценим каждого клиента и стремимся предоставить лучший сервис. Присоединяйтесь к числу наших довольных покупателей уже сегодня
WilliamIRrutty
| #
darknet markets links darknet market list
Pingrutty
| #
dark market list darknet market lists
Donaldlaf
| #
dark markets 2025 bitcoin dark web
JosephNum
| #
For depositing in the Aviator betting game(https://ascenddata.co.ke/how-to-deposit-money-in-the-aviator-game-a-step-by-step-guide/), it’s necessary to complete these instructions.
At the beginning, sign into your profile on the website that has Aviator.
Then, open the Wallet tab.
Click on your supported transaction type, such as e-wallet (Neteller, etc.).
Enter the deposit value, make sure all is correct, and finalize the payment.
Most of the time, your gaming funds will be topped up within seconds, allowing you to enjoy the crash game right away!
Keep in mind: fees may vary on the site.
Kxyufaita
| #
bitcoin dark web darknet markets 2025
FNDaviditabe
| #
dark market dark web link
Sazrchd
| #
купить диплом о дополнительном профессиональном образовании
JimmyLothe
| #
http://fabnews.ru/forum/showthread.php?p=92469#post92469
Toliknaf
| #
onion dark website best darknet markets
shinomontaj_vgSn
| #
шиномантаж сергиев посад hondahybrid.ru/forum/topic/2886-kakie-uslugi-okazyvaet-shinomontazh/ .
Rabyitabe
| #
best darknet markets darknet market list
MarkNOlaf
| #
darknet markets links dark web drug marketplace
Volodyanaf
| #
dark web sites darkmarket list
DonDonfaita
| #
darkmarket darknet markets
Pingrutty
| #
darkmarket 2025 darknet site
Derricksaisy
| #
Розы в яркой летней упаковке – страсть и солнце
купить цветы в томске
JimmyLothe
| #
http://gazeta.ekafe.ru/viewtopic.php?f=110&t=38467
WilliamIRrutty
| #
darknet drug store dark web drug marketplace
mostbet_jwei
| #
скачать мостбет http://www.mostbet6006.ru .
Jamesdusty
| #
Цветы свежие, сочные, будто впитали в себя лето!
доставка цветов
GeraldBaf
| #
регистрация дачного дома
Alfonsomuh
| #
Привет, любитель поиграть в контру и получить за это скинов от Гейба. У тебя есть полны и нвентарь и ты ищешь продать скины с выподом на карту . Читай подборку сайтов которые тебе помогут сделать это мгновенно и с удобным способом вывода для тебя.
avtolombardkazancoN
| #
займ под залог авто казань
https://melocasting.com/employer/zaim-pod-zalog-pts-18232/
залог под птс авто
Donaldlaf
| #
dark web market dark web market urls
Kxyufaita
| #
darknet market links onion dark website
FNDaviditabe
| #
darkmarket 2025 darknet market
Ronaldbut
| #
МикоФарм Фунги
Toliknaf
| #
darknet markets links darknet marketplace
taksi uşak
| #
Your site is amazing, the articles are great, I will always come to this site and continue to read because you have very nice articles, thank you
prodvijenie saita_mfkn
| #
продвижение сайтов в москве продвижение сайтов в москве .
MarkNOlaf
| #
dark web sites dark market
Rabyitabe
| #
dark web drug marketplace darkmarket url
Pingrutty
| #
darkmarket url darknet market lists
1win_msmr
| #
1winn https://familyclub.borda.ru/?1-6-0-00002163-000-0-0-1743051813/ .
Once Human
| #
web siteniz çok güzel başarılarınızın devamını dilerim. makaleler çok hoş sürekli sitenizi ziyaret edeceğim
DonDonfaita
| #
darkmarket list darknet market links
Volodyanaf
| #
dark market url dark web drug marketplace
WilliamIRrutty
| #
bitcoin dark web dark market link
Once Human
| #
web siteniz çok güzel başarılarınızın devamını dilerim. makaleler çok hoş sürekli sitenizi ziyaret edeceğim
Donaldlaf
| #
darkmarket darkmarket 2025
Kxyufaita
| #
darknet markets url dark web market list
FNDaviditabe
| #
dark markets 2025 bitcoin dark web
Ronaldbut
| #
https://mycofarm.ru/
Fuyazquini
| #
На сайте https://koch-market.ru/ закажите звонок для того, чтобы воспользоваться услугами детейлинг центра. Есть возможность воспользоваться такими услугами, как оклейка кузова автомобиля, винилография на мотоцикл, тюнинг фар, оклейка антигравийной пленкой. Для того чтобы сориентироваться в выборе, необходимо изучить примеры работ – те проекты, которые реализованы. Также на сайте представлены и содержательные, любопытные статьи на данную тему. Все специалисты, работающие в компании, отличаются большим опытом.
PosoxdBum
| #
На сайте https://t.me/azino777_a вы найдете огромное количество свежих, актуальных и содержательных новостей, которые представлены на тему популярного онлайн-заведения «Azino 777» . Его по достоинству оценило огромное количество посетителей, ведь площадка работает максимально честно, уважает своих посетителей, ценит их время. Именно здесь, в первую очередь, появляются интересные, увлекательные новости. Вы узнаете о них первым. На этом канале публикуются промокоды, бонусы для более зрелищной игры.
kra37at
| #
Undeniably believe that which you said. Your favorite reason seemed
to be on the net the easiest thing to be aware of.
I say to you, I definitely get irked while people consider worries that they
just don’t know about. You managed to hit the nail upon the
top as well as defined out the whole thing without having side effect , people can take a signal.
Will probably be back to get more. Thanks
Toliknaf
| #
dark market onion darknet market lists
AnthonyWifut
| #
Советую https://abfgss53sdbkl33.ru/
Вулкан Платинум слоты с джекпотом
| #
Vulkan Platinum — это не просто казино, а настоящая игровая платформа для тех, кто ценит качество и
надежность. Здесь можно наслаждаться не
только популярными слотами, но и уникальными игровыми предложениями,
которые могут существенно увеличить ваш доход.
Игровой процесс в Vulkan Platinum всегда увлекательный и непредсказуемый.
Что делает Vulkan Platinum платежные методы отличным выбором для игроков?
Мы предлагаем безопасную игровую среду, где каждый момент проходит с максимальным комфортом и без забот.
С нами выигрывать не только приятно, но и выгодно, благодаря нашим бонусам и акциям.
Когда самое время начать игру в Vulkan Platinum?
Чем раньше, тем лучше! Процесс регистрации занимает всего несколько минут, и вы сразу сможете наслаждаться всеми преимуществами казино.
Вот что вас ждет в Vulkan Platinum:
Мы гарантируем безопасные и быстрые выплаты,
а также надежную защиту ваших
данных.
Независимо от того, предпочитаете
ли вы слоты или настольные игры, у нас есть всё
для вашего комфорта.
Для лояльных клиентов Vulkan Platinum всегда подготовит специальные
предложения и бонусы.
Vulkan Platinum — это ваше место
для больших выигрышей и незабываемых моментов. https://vulkan-rush.top/
Pingrutty
| #
darknet sites darknet markets links
Once Human
| #
web siteniz çok güzel başarılarınızın devamını dilerim. makaleler çok hoş sürekli sitenizi ziyaret edeceğim
MarkNOlaf
| #
darknet marketplace dark market onion
Rabyitabe
| #
dark web sites darknet market list
avtolombardkazancoN
| #
автоломбард в казани круглосуточно
http://zenithgrs.com/employer/jabenefits8178/
займ под залог авто
Nivugtelese
| #
Viziteaza site-ul https://betwave.ro/ ?i vei gasi o lista cu toate cazinourile online din Romania, precum ?i recenzii ale acestora. Va prezentam cele mai bune condi?ii de la cazinou cu retrageri rapide. Va spunem ce bonusuri exista pentru reaprovizionare sau inregistrare. Cum sa alegi cel mai bun cazinou online pentru tine – cite?te pe site!
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
DonDonfaita
| #
darknet drug market best darknet markets
WilliamIRrutty
| #
darknet links dark web marketplaces
alex-machine
| #
отзывы ремонт посудомоечных ремонт посудомоечных машин в москве
Ronaldbut
| #
МикоФарм Фунги
uşak taksi
| #
Your site is amazing, the articles are great, I will always come to this site and continue to read because you have very nice articles, thank you
Diplomi_poKt
| #
Купить диплом университета!
Мы можем предложить документы институтов, расположенных в любом регионе Российской Федерации.
diplomk-vo-vladivostoke.ru/kupit-attestat-texnikuma-7
Volodyanaf
| #
bitcoin dark web darknet drugs
Kxyufaita
| #
darkmarket link dark market url
FNDaviditabe
| #
darknet markets onion address dark markets 2025
Donaldlaf
| #
darknet markets 2025 dark market url
Pingrutty
| #
darknet market dark web market
Toliknaf
| #
darknet site darknet drug market
MarkNOlaf
| #
dark web market links darkmarket link
RobertDaw
| #
Casino non Gamstop platforms are perfect for casual gaming—love it!
no gamstop casino
Rabyitabe
| #
dark market url dark markets 2025
1win_wcot
| #
1win партнерка вход http://alfatraders.borda.ru/?1-0-0-00004932-000-0-0-1743258210 .
drenajnie raboti spb_tlst
| #
сделать дренаж на участке цена в спб сделать дренаж на участке цена в спб .
DonDonfaita
| #
dark websites dark market 2025
sofisimo_c_zzen
| #
Откройте для себя новые горизонты на sofisimo.com, увлекательные материалы.
Погрузитесь в уникальный контент sofisimo.com, изучая.
Сайт sofisimo.com – ваша отправная точка, ищите.
sofisimo.com – остров знаний, вас ждут.
sofisimo.com – ваш помощник в обучении, развивая.
Станьте частью сообщества на sofisimo.com, делиться опытом.
sofisimo.com – это источник креативности, необычные решения.
Посетите sofisimo.com для открытия новых возможностей, мир знаний.
sofisimo.com для любителей знаний, где каждый.
sofisimo.com – это больше, чем просто сайт, научатся.
Проведите время с пользой на sofisimo.com, вдохновение.
Каждый день с sofisimo.com – это новая возможность, учиться.
Свяжитесь с сообществом на sofisimo.com, ценятся.
sofisimo.com – ключ к вашему успеху, жаждет.
Узнайте секреты успеха на sofisimo.com, который.
sofisimo.com – это ваша платформа, но это для всех.
sofisimo.com ждет вас, ваши мечты становятся реальностью.
Ищите новую информацию на sofisimo.com, вдохновение не заканчивается.
sofisimo.com: место, где встречаются идеи, где.
comprar muebles https://sofisimo.com/ .
1win_nfMr
| #
вин 1 http://www.1win6002.ru .
drenajnie raboti spb_pdel
| #
дренажно распределительная система дренажно распределительная система .
WilliamIRrutty
| #
dark web drug marketplace dark markets
Ronaldbut
| #
https://mycofarm.ru/
Lazruzg
| #
Приобрести диплом любого ВУЗа!
Мы изготавливаем дипломы любой профессии по приятным тарифам— diplomass.com/ekb-kupit-diplom-4/
mostbet_ldOt
| #
мостюет http://www.mostbet6007.ru .
Jarioryzv
| #
Где приобрести диплом специалиста?
Наши специалисты предлагают быстро заказать диплом, который выполняется на бланке ГОЗНАКа и заверен печатями, водяными знаками, подписями. Данный диплом пройдет любые проверки, даже с применением профессиональных приборов. Решайте свои задачи быстро с нашим сервисом. Заказать диплом о высшем образовании! linux.listbb.ru/viewtopic.php?f=2&t=1301
Mazrgqq
| #
Мы предлагаем дипломы любой профессии по приятным ценам. Стараемся поддерживать для покупателей адекватную ценовую политику. Для нас важно, чтобы дипломы были доступными для большого количества наших граждан.
Покупка диплома, который подтверждает обучение в университете, – это грамотное решение. Заказать диплом университета: diplomoz-197.com/diplom-kupit-ofitsialno-2/
Pingrutty
| #
darknet markets links dark market url
Cazrwwi
| #
Здравствуйте!
Мы готовы предложить дипломы психологов, юристов, экономистов и других профессий по приятным ценам. Стоимость может зависеть от выбранной специальности, года получения и университета: diplomans.com/
Volodyanaf
| #
darknet links darknet markets
Kxyufaita
| #
onion dark website dark market url
FNDaviditabe
| #
dark market best darknet markets
Donaldlaf
| #
dark web sites darknet drug links
DavidUrire
| #
Оптимизация процессов управления Управленческие консультации от Светланы Подкладышевой, серийного предпринимателя с более чем 20-летним опытом. Она обладает уникальной способностью анализировать сложные ситуации и предлагать практические решения, основанные на глубоких знаниях и реальном опыте. Светлана помогает оптимизировать бизнес-процессы, разработать индивидуальные стратегии роста, повысить конкурентоспособность. Например, оптимизация структуры компании, разработка бизнес-моделей, внедрение технологий.
Toliknaf
| #
darknet markets onion address darknet market
avtolombardkemerovosig
| #
автоломбард залог машины
https://jobs.web4y.online/employer/avtolombard-111946/
деньги под залог автомобиля
degiwmup
| #
https://boilers24.com/ and you will find spare parts for boilers of many brands. Here you can buy boxes and control boards, actuators, gas valves, oil nozzles, ignition electrodes, flame sensors and much more. Check out the catalog with the entire range at competitive prices.
Trefkyq
| #
Заказать документ о получении высшего образования вы можете у нас в столице. diplomass.com/kupit-diplom-sssr-2
MarkNOlaf
| #
darknet markets onion dark web sites
https://vodka-fun.top/
| #
Dreaming of big victories? Then welcome to Vodka Casino – the perfect place to play! Here, we offer thousands of slot machines from top studios. https://vodka-fun.top/ and claim generous bonuses!
What exclusive features does our casino offer?
Popular casino games featuring high RTP.
Lucrative promotions for new and existing players.
Guaranteed winnings without delays or hidden fees.
Optimized platform on smartphones and tablets.
Professional service always ready to assist.
Test your luck to enjoy gambling to the fullest!
RobertDaw
| #
Non-Gamstop casino platforms are so intuitive—really nice!
non gamstop casino 2021
Clubnika best live casino games
| #
Clubnika Casino is the place for true gambling enthusiasts, where everyone finds their spot.
Here, you’ll find the best games, profitable offers, and
unique opportunities to win. We guarantee the safety, fairness, and transparency of all gaming processes.
Why is Clubnika live dealer games your ideal choice?
By joining Clubnika Casino, you gain access to generous
bonuses, free spins, and regular promotions that
will significantly enhance your chances of success.
At Clubnika Casino, you can be confident that any questions will
be resolved promptly thanks to our round-the-clock support.
When is the best time to join Clubnika Casino? Don’t waste
any time, start right now and get bonuses that will speed up your
journey to victory. Here’s what awaits you at Clubnika Casino:
Generous bonuses for newcomers and regular free spins.
Tournaments with big prizes and incredible winning opportunities.
Game updates every month so you’ll never get bored.
Join Clubnika Casino and become part of the thrilling world of winnings!. https://clubnika-casinoeuphoria.website/
Rabyitabe
| #
darknet marketplace darkmarket link
source
| #
I am not sure where you are getting your info, but good topic.
I needs to spend some time learning more or understanding more.
Thanks for fantastic info I was looking for this information for my mission.
drenajnie raboti spb_eust
| #
смета на устройство дренажа https://drenazh-uchastka-krasnodar.ru/ .
drenajnie raboti spb_vbel
| #
дренаж под ключ в ленинградской области дренаж под ключ в ленинградской области .
WilliamIRrutty
| #
darknet drug market darknet market
Pingrutty
| #
dark market dark markets
DonDonfaita
| #
dark market url dark markets 2025
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
Ronaldbut
| #
МикоФарм Фунги
1win_diMr
| #
один вин http://www.1win6002.ru .
Starda Casino
| #
Welcome to Starda Casino – the best place for gambling entertainment! Here, you’ll find top-tier entertainment from renowned studios. https://starda-rush.top/ and get a chance to hit the jackpot!
What makes our casino special?
Popular games with high RTP.
Exclusive offers with free spins and cashback.
Instant transactions without hidden fees.
Modern design for a seamless experience.
Round-the-clock assistance answering within minutes.
Start playing now and experience the ultimate excitement!
Kxyufaita
| #
darknet markets darkmarket list
FNDaviditabe
| #
dark web market list darkmarket link
mostbet_qdOt
| #
мостюет mostbet6007.ru .
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
Monitoring transporta_iiOi
| #
установка мониторинга транспорта http://kontrol-avto.ru .
Donaldlaf
| #
dark markets 2025 dark market 2025
drenajnie raboti spb_wqst
| #
дренаж санкт петербург http://drenazh-uchastka-krasnodar.ru .
MarkNOlaf
| #
dark web market darknet drugs
Toliknaf
| #
darknet links darknet drug market
drenajnie raboti spb_bqel
| #
дренажные системы спб дренажные системы спб .
RobertDaw
| #
Non-Gamstop casino sites feel so much more relaxed—big thumbs up!
non.gamstop casinos
1win_hdot
| #
1wi. http://www.alfatraders.borda.ru/?1-0-0-00004932-000-0-0-1743258210 .
Rabyitabe
| #
bitcoin dark web dark web sites
WilliamIRrutty
| #
darkmarket link darknet markets onion address
Pingrutty
| #
darkmarket link onion dark website
CalowlaW
| #
На сайте https://boostclicks.ru/ вы сможете ознакомиться с рекомендациями, ценными и важными советами про API интеграцию, скрипты, а также любопытные сервисы из мира трафика. Для того чтобы вам было удобней ориентироваться, все материалы поделены на разделы, что позволит отыскать то, что действительно актуально и интересно в данный момент. Регулярно на портале появляется новый, интересный контент на данную тему. Имеется и список тех статей, которые пользуются особой популярностью. Все они созданы лучшими и высококлассными экспертами с огромным опытом.
1win_wlMr
| #
1win ru https://1win6002.ru/ .
Kxyufaita
| #
dark markets darknet drug market
1win_ioMl
| #
1 win.com 1win6049.ru .
mostbet_ypOt
| #
мостбет скачать на андроид https://mostbet6007.ru/ .
DonDonfaita
| #
dark market onion dark website
Ronaldbut
| #
МикоФарм Фунги
1win_dxOi
| #
1 вин. balashiha.myqip.ru/?1-12-0-00000437-000-0-0-1743258848 .
avtolombardkemerovosig
| #
автоломбард под птс в кемерово
https://es-africa.com/employer/newrba6811/
кредит под залог птс
Monitoring transporta_svOi
| #
купить gps глонасс мониторинг транспорта купить gps глонасс мониторинг транспорта .
MarkNOlaf
| #
darknet markets darkmarket 2025
Donaldlaf
| #
darknet site dark market link
WilliamIRrutty
| #
darkmarket url dark websites
Toliknaf
| #
dark market list dark web market links
RobertDaw
| #
Non-Gamstop casinos make gaming feel unrestricted—highly recommend!
non gamstop no deposit casino
Pingrutty
| #
dark web marketplaces darknet drug links
Kxyufaita
| #
darknet links dark markets 2025
aksjdf_lit
| #
Witamy w SlottyWay Kasyno – wiodącej, w pełni licencjonowanej platformie gier online, łączącej najnowocześniejsze rozwiązania technologiczne z doskonałą i przyjazną obsługą klienta. Nasza biblioteka gier jest niezwykle bogata i zróżnicowana – od klasycznych automatów z klimatem tradycyjnych kasyn, przez najnowsze sloty wideo, aż po nowoczesne gry stołowe na żywo z krupierami, które zapewnią Ci wrażenia rodem z prestiżowych salonów gier: Slottyway Vladimir dolboeb. Stawiamy na pełne bezpieczeństwo i transparentność. Nasze zaawansowane systemy ochrony, w połączeniu z szyfrowaniem SSL, gwarantują ochronę Twoich danych osobowych i finansowych. Współpracujemy wyłącznie z uznanymi dostawcami oprogramowania i stosujemy rygorystyczne standardy bezpieczeństwa.
Jamessok
| #
לאהוב. אתם תזיינו אותי? – כן, מותק, אנחנו הולכים לזיין אותך כמו שצריך. – אנחנו הולכים לקחת אותך בדרך הבוגרת. רק את צריכה בדירה לולדימיר בדירה דיסקרטית. מה דעתך? התחת היפה של אשתך! ולדימיר ברוסלן בתחינה, והוא לא איחרה לחכות. לאחר שראה את המבט הזה כהזמנה, read content
Rabyitabe
| #
dark market 2025 dark web market list
Monitoring transporta_szOi
| #
контроль транспорта цена контроль транспорта цена .
MarkNOlaf
| #
darknet drug links darknet marketplace
Ronaldbut
| #
https://mycofarm.ru/
DonDonfaita
| #
dark web market links darknet sites
WilliamIRrutty
| #
best darknet markets darknet markets
Donaldlaf
| #
darkmarket darknet markets onion
FNDaviditabe
| #
darknet markets links darknet marketplace
Pingrutty
| #
darknet drugs darknet markets 2025
RobertDaw
| #
Non Gamstop casino UK support is top-notch—really impressed!
non gamstop casinos uk 10 deposit
Toliknaf
| #
darkmarket url onion dark website
MegexBox
| #
На сайте https://stakecasino.tech изучите подробную, актуальную информацию, которая касается популярного онлайн-казино «Stake Casino». Это место для любителей азарта, ведь здесь есть еще и букмекерская контора. Каждому игроку предлагается огромное количество интересных возможностей для того, чтобы получить море приятных, положительных эмоций. А заодно вы сможете заработать много денег на все, что пожелаете. Вас порадует понятный, простой интерфейс, а также огромное количество бонусов, безупречная защита.
nevedCar
| #
Adguard-faq.com предоставляет информацию. Разъясним, что значит, подписка на Адгуард VPN и как управлять ею. Расскажем, где на AdGuard можно взять промокоды. Работа с Adguard имеет много положительных сторон. На сайте рассмотрим самые очевидные. Ознакомьтесь уже сейчас с ответами на часто задаваемые вопросы. https://adguard-faq.com – здесь расскажем, зачем нужны официальные сайты-зеркала продуктов AdGuard. Узнаете, как купить продукты экосистемы приватности Адгуард. Ищите отменный блокировщик рекламы? Adguard – отличный выбор для вас!
avtolombardmsksig
| #
автоломбард авто
https://analyticsjobs.in/jobs/companies/avtolombard-118791/
взять кредит под птс
MarkNOlaf
| #
darknet sites darknet markets onion address
Rabyitabe
| #
darkmarket link darknet websites
1win_uqMl
| #
1win официальный сайт 1win официальный сайт .
ZavamlReK
| #
Продать технику легко, обратитесть в скупку78. Компания https://spbskupka78.ru/ занимает лидирующие позиции по выкупу устройств и техники у населения. В данный момент компания ввела оценку через мессенджеры whats app и телеграмм. Наша компания добавила в прайс оценку таких устройства, как: айфоны от 7 до 16 модели, iPad от 2014 года до 2024 (процессор M4), макбуки от 2012 года до 2024 на процессоре М4, ноутбуки бизнес сегмента и игровые, аудиосистемы, микрофоны, ресиверы и усилители звука, телевизоры и мониторы.
Ronaldbut
| #
https://mycofarm.ru/
Jamessok
| #
הכנת דו ” ח? הבחור נופף בידו וצחק. – אתה מדבר על זה… לא, כמובן. כשעלה שני נשפים התמונות בעצמי, אז החלטתי לזרוק למקס עוד כמה את זה ככה, בלי להסיט את מבטו. בהתחלה הוא היה ביישן…. אבל אז העז…. תפס אותי בחלק האחורי של הראש ויירט את היוזמה. פטרובנה היא נערות ליווי ברחובות
Lazrbrd
| #
Где заказать диплом по нужной специальности?
Приобрести диплом ВУЗа по невысокой стоимости возможно, обращаясь к проверенной специализированной фирме.: prodiplome.com
WilliamIRrutty
| #
dark markets dark market
DonDonfaita
| #
darknet markets darknet markets onion
WilliamDix
| #
Мы изготавливаем дипломы любых профессий по доступным ценам. Дипломы производятся на настоящих бланках государственного образца Заказать диплом ВУЗа diplom-onlinex.com
Kxyufaita
| #
darknet market links dark websites
Pingrutty
| #
darknet markets onion address darknet drugs
1win_gtOi
| #
1win зайти https://www.balashiha.myqip.ru/?1-12-0-00000437-000-0-0-1743258848 .
Donaldlaf
| #
dark websites darknet market list
GohifeFer
| #
На сайте https://xn—-7sbecpcasfm0beeaecirc5b3a2g.kz/ получите расчет на то, во сколько вам обойдется корпоративное обучение. «CASPIAN TRAINING GROUP» более 20 лет оказывает помощь в том, чтобы вы прошли успешное обучение. Для того чтобы получить консультацию, коммерческое предложение, необходимо ввести в специальную форму свои личные данные, телефон, электронную почту. Компания оказывает такие услуги, как: бизнес-тренинги, обучение персонала языкам, бизнес-симуляции. В этой компании ни один десяток предприятий проходит обучение за год.
1win_blMl
| #
1win официальный сайт скачать http://1win6049.ru/ .
MarkNOlaf
| #
darkmarket link dark market url
RobertDaw
| #
Non Gamstop casinos no deposit offers keep me playing—awesome!
non gamstop casinos uk betz.casino
Toliknaf
| #
darknet drug store darknet drug store
Rabyitabe
| #
dark market onion tor drug market
WilliamIRrutty
| #
darkmarket 2025 darknet markets
Kxyufaita
| #
dark web market list dark markets 2025
FNDaviditabe
| #
darkmarket url onion dark website
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
Ronaldbut
| #
МикоФарм Фунги
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
Pingrutty
| #
onion dark website onion dark website
Josephkaw
| #
последние спортивные новости
DonDonfaita
| #
darknet markets links dark market 2025
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
avtolombardmsksig
| #
займ под залог автоломбард
https://cdltruckdrivingcareers.com/employer/ishayoga7336/
кредит под птс авто
MarkNOlaf
| #
darknet drugs darknet site
w69aias
| #
ติดตั้งแอป w69aias รับประสบการณ์เดิมพันที่เหนือกว่า พร้อมฟีเจอร์ใหม่ช่วยให้เล่นเกมได้ง่ายขึ้น
w69 slot asia login
| #
ดาวน์โหลดแอป w69 slot asia login
แล้วสนุกกับสล็อต แจ็คพอตแตกง่าย และอัตราจ่ายที่ดีที่สุด
Donaldlaf
| #
dark web link darknet markets links
เกมส์ สล็อต w69 slot
| #
ดาวน์โหลดแอป เกมส์ สล็อต w69 slot – https://rxq58.com ได้ง่ายๆ
เพียงไม่กี่ขั้นตอน พร้อมรับโบนัสต้อนรับ 100$ สนุกกับการเดิมพันที่ดีที่สุดบนมือถือ
Bevaxowreave
| #
На сайте https://smartflow.ru вы сможете выбрать качественную, функциональную сантехнику для обустройства ванной комнаты в доме либо квартире. Ассортимент «SMARTFLOW» включает в себя такие изделия, которые выполнены из высококачественных и современных материалов, безупречного стиля. Все унитазы, биде, модульные ванные идеально впишутся в концепцию. Каждая вещь идеально сочетает надежность, прочность, а также изысканность. Для того чтобы выбрать что-то определенное, изучите галерею, ознакомьтесь и с особой системой смыва «Торнадо».
สล็อต ออนไลน์ w69
| #
ติดตั้งแอป สล็อต ออนไลน์ w69 แล้วรับการแจ้งเตือนแมตช์สำคัญ โปรโมชั่นพิเศษ
และข่าวสารก่อนใคร
RobertDaw
| #
Non Gamstop UK casino has a great feel—big win!
non gamstop casinos uk no deposit bonus
w69ทางเข้าหลัก
| #
แอป w69ทางเข้าหลัก รองรับระบบฝาก-ถอนที่รวดเร็ว ให้คุณทำธุรกรรมได้ง่ายดายเพียงปลายนิ้ว
WilliamIRrutty
| #
dark web sites darknet markets 2025
Toliknaf
| #
tor drug market darknet markets
w69con
| #
w69con ปรับแต่งอินเทอร์เฟซแอปใหม่
ดีไซน์สวยงาม ค้นหาง่าย และใช้งานได้สะดวกกว่าเดิม
JerryDab
| #
In Azerbaijan, the 12-day holiday vacation has ended https://pin.it/5XGTX4YRH
aksjdf_lit
| #
Witamy w SlottyWay Kasyno – wiodącej, w pełni licencjonowanej platformie gier online, łączącej najnowocześniejsze rozwiązania technologiczne z doskonałą i przyjazną obsługą klienta. Nasza biblioteka gier jest niezwykle bogata i zróżnicowana – od klasycznych automatów z klimatem tradycyjnych kasyn, przez najnowsze sloty wideo, aż po nowoczesne gry stołowe na żywo z krupierami, które zapewnią Ci wrażenia rodem z prestiżowych salonów gier: Slottyway Casino. Stawiamy na pełne bezpieczeństwo i transparentność. Nasze zaawansowane systemy ochrony, w połączeniu z szyfrowaniem SSL, gwarantują ochronę Twoich danych osobowych i finansowych. Współpracujemy wyłącznie z uznanymi dostawcami oprogramowania i stosujemy rygorystyczne standardy bezpieczeństwa.
w69 slot daftar
| #
w69 slot daftar ปรับปรุงฟังก์ชันแอปใหม่ ใช้งานง่ายกว่าเดิม สนุกกับคาสิโนสดและเกมสล็อตได้ทุกที่ทุกเวลา
Kxyufaita
| #
darknet drug store darknet markets onion address
FNDaviditabe
| #
darkmarket link dark web market list
Dnrtweg
| #
Где приобрести диплом специалиста?
Мы предлагаем дипломы любой профессии по выгодным ценам. Мы можем предложить документы техникумов, расположенных в любом регионе России. Можно приобрести диплом от любого заведения, за любой год, в том числе документы старого образца. Документы печатаются на “правильной” бумаге самого высокого качества. Это позволяет делать настоящие дипломы, не отличимые от оригинала. Они будут заверены всеми требуемыми печатями и подписями. Всегда стараемся поддерживать для заказчиков адекватную политику цен. Для нас важно, чтобы документы были доступными для большого количества наших граждан. diplom-profi.ru/kupit-diplom-v-samare-2-4
Diplomi_nzKt
| #
Купить диплом любого ВУЗа!
Мы готовы предложить документы институтов, которые находятся в любом регионе Российской Федерации.
diplomj-irkutsk.ru/kupit-attestat-za-9-klass-v-moskve-4
w69 app login
| #
ติดตั้งแอป w69 app login รับประสบการณ์เดิมพันที่เหนือกว่า พร้อมฟีเจอร์ใหม่ช่วยให้เล่นเกมได้ง่ายขึ้น
Josephkaw
| #
новости футбола сегодня
w69 login slot
| #
ติดตั้งแอป w69 login slot – https://zfqc3.com รับประสบการณ์เดิมพันที่เหนือกว่า พร้อมฟีเจอร์ใหม่ช่วยให้เล่นเกมได้ง่ายขึ้น
Sazrsrk
| #
Мы готовы предложить дипломы любой профессии по доступным ценам. Цена может зависеть от той или иной специальности, года выпуска и образовательного учреждения. Стараемся поддерживать для заказчиков адекватную политику цен. Важно, чтобы документы были доступны для большого количества наших граждан. купить диплом в тюмени
w69 mo
| #
w69 mo – https://kmib5.com ปรับปรุงฟังก์ชันแอปใหม่ ใช้งานง่ายกว่าเดิม สนุกกับคาสิโนสดและเกมสล็อตได้ทุกที่ทุกเวลา
Trefoks
| #
Купить документ института вы имеете возможность у нас в Москве. diplomc-v-ufe.ru/kupit-diplom-omsk
Rabyitabe
| #
dark web market darknet site
เว็บw69
| #
อัปเกรดแอป เว็บw69 วันนี้ เพื่อการเล่นเกมที่ราบรื่นกว่าเดิม พร้อมฟังก์ชันใหม่ที่ช่วยให้คุณสนุกยิ่งขึ้น
uşak taksi
| #
Your site is very nice, the articles are great, I wish you continued success, I love your site very much, I will visit it constantly
Pingrutty
| #
dark websites dark market onion
w69 top
| #
ติดตั้งแอป w69 top แล้วพบกับการแจ้งเตือนโปรโมชั่นและกิจกรรมใหม่ๆ ก่อนใคร
w69slotทางเข้า
| #
ดาวน์โหลดแอป w69slotทางเข้า ง่ายๆ เพียงไม่กี่คลิก สนุกกับการเดิมพันกีฬา
คาสิโน และสล็อตในที่เดียว
Ronaldbut
| #
МикоФарм Фунги
Jariorroa
| #
Где приобрести диплом по актуальной специальности?
Наши специалисты предлагают выгодно и быстро заказать диплом, который выполнен на оригинальном бланке и заверен печатями, водяными знаками, подписями официальных лиц. Диплом способен пройти лубую проверку, даже с использованием специальных приборов. Достигайте свои цели быстро с нашим сервисом.
Заказать диплом университета diplomt-v-chelyabinske.ru/kupit-diplom-v-bryanske-15/
Sazrzpi
| #
купить диплом политеха
w69 ถอนเงิน ไม่ได้
| #
สมัครสมาชิกผ่านแอป w69 ถอนเงิน ไม่ได้ รับโบนัสต้อนรับ 100$ และสิทธิพิเศษอีกมากมาย
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Мишень гафния
MarkNOlaf
| #
onion dark website darknet markets links
Jariorgtu
| #
Где приобрести диплом по нужной специальности?
Наша компания предлагает максимально быстро заказать диплом, который выполняется на оригинальном бланке и заверен мокрыми печатями, водяными знаками, подписями должностных лиц. Документ способен пройти любые проверки, даже с использованием профессиональных приборов. Решайте свои задачи быстро с нашим сервисом. Приобрести диплом любого ВУЗа! vseamoskva.flybb.ru/viewtopic.php?f=2&t=1120
DonDonfaita
| #
dark market onion dark market onion
EdwardBiorn
| #
Ткань “дышит”, не жарко!
женские костюмы томск
Mazrdnh
| #
Мы предлагаем дипломы психологов, юристов, экономистов и любых других профессий по выгодным ценам. Стараемся поддерживать для клиентов адекватную ценовую политику. Важно, чтобы документы были доступными для большого количества наших граждан.
Покупка документа, который подтверждает обучение в ВУЗе, – это выгодное решение. Купить диплом ВУЗа: diplomius-docs.com/kupit-diplom-texnikuma-ob-okonchanii-3/
Lazrbpv
| #
Приобрести диплом ВУЗа!
Мы можем предложить дипломы любой профессии по приятным тарифам— diplomgorkiy.com/kupit-diplom-o-srednem-meditsinskom-obrazovanii-bistro-2/
SamuelGet
| #
Postanowilem sprawdzic aseita – bylem zawiedziony natychmiast. Zasoby edukacyjne sa przestarzale, nauczyciele nawet nie reaguja na zgloszenia. Pieniadze zostaly pobrane od razu, a rzetelnej edukacji zupelnie nie uzyskalem. Obsluga klienta to prawdziwy dramat, czuje, ze tam siedza bezduszne systemy. Bezwzgledne oszustwo, odradzam absolutnie nikomu! Lepiej zainwestowac srodki na kurs wartosciowego, a nie na podobny smiec.
ekokixHon
| #
Chat-gptru – лаборатория умной нейросети. Загрузите фотографию – ИИ мгновенно определит, что на ней изображено, и предоставит пояснения либо рекомендации. GPT быстро подбирает необходимую информацию из актуальных источников. https://chat-gptru.com – тут с мнениями наших пользователей можете ознакомиться. Также здесь указаны цены. Для применения нейросети нужно в чат ввести ваш запрос и отправить нажать. Chat GPT генерирует тексты для любых целей, придерживается высоких стандартов безопасности и конфиденциальности.
WilliamIRrutty
| #
darkmarket list dark web sites
Jamietah
| #
Переделанные счетчики
FNDaviditabe
| #
dark market list darknet market lists
Kxyufaita
| #
bitcoin dark web darknet drug store
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица товара подтверждена соответствующими документами, подтверждающими их качество. Превосходное обслуживание – наш стандарт – мы на связи, чтобы улаживать ваши вопросы и предоставлять решения под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
поставляемая продукция:
Проволока висмутовая L54930 – UNS Проволока висмутовая L54930 – UNS является высококачественным материалом, используемым РІ различных промышленных Рё научных приложениях. Рзготавливается РёР· висмута, что гарантирует отличные антикоррозионные свойства Рё устойчивость Рє высоким температурам. Рта проволока идеальна для сварки, создания сплавов Рё использования РІ электротехнике. Благодаря СЃРІРѕРёРј уникальным характеристикам, РѕРЅР° находит применение РІ медицинских устройствах Рё электронике. Если РІС‹ хотите купить Проволока висмутовая L54930 – UNS, выбирайте надежных поставщиков, чтобы получить РїСЂРѕРґСѓРєС‚ наилучшего качества.
RobertDaw
| #
Found the best non Gamstop casino for blackjack—loving it so far!
non gamstop ukgc casino sites
WilliamDix
| #
Мы готовы предложить дипломы любых профессий по приятным тарифам. Дипломы производятся на подлинных бланках государственного образца Приобрести диплом ВУЗа diplomers.com
Diplomi_zoka
| #
купить диплом в тобольске diplomys-vsem.ru .
Gefevpek
| #
Visit https://bestcasinoideal.com/ and discover expert reviews, top strategies and essential tips. Explore online slots, table games, live dealer experiences. We have compiled a complete collection of the best online casinos and gambling sites for 2025, tested and rated by experts.
Toliknaf
| #
darkmarket 2025 dark web market links
Pingrutty
| #
darknet market list darkmarkets
avtolombardnovosibsig
| #
автоломбард птс
https://career.abuissa.com/employer/zaim-pod-zalog-pts-12174/
кредит залог птс наличные
zaimcaree
| #
Быстрые онлайн займы на карту, подробнее на официальном сайте на сайте
https://wiki.cosmaticdrift.net/view/Cs2case_16k
| #
Что такое “Кейсы КС” и “Кейсы КС ГО”?
“Кейсы КС” (или “Кейсы CS”) и “Кейсы КС ГО” (CS:GO) — это виртуальные контейнеры, которые используются в играх Counter-Strike (КС), включая Counter-Strike: Global Offensive (CS:GO) и его обновленную версию Counter-Strike 2 (CS2). Внутри этих кейсов находятся различные игровые предметы — скины для оружия, ножи, перчатки и другие косметические элементы, которые игроки могут получить и использовать в игре для украшения своего инвентаря.
Как это работает?
1. **Регистрация**: Вы заходите на сайт и регистрируетесь, обычно через Steam, чтобы связать свой игровой аккаунт.
2. **Пополнение баланса**: Нужно внести деньги на сайт (например, через карту, криптовалюту или электронные кошельки), чтобы купить кейсы или внутриигровую валюту.
3. **Выбор кейса**: На сайте представлены разные кейсы с указанием возможных наград (от дешевых скинов до редких ножей) и их стоимости.
4. **Открытие кейса**: Вы выбираете кейс, оплачиваете его и открываете. Система случайным образом определяет, какой предмет вы получите.
5. **Вывод выигрыша**: Если вам повезло, вы можете вывести скин в Steam и использовать его в игре.
Чем отличается “Кейсы КС” от “Кейсы КС ГО”?
– **Кейсы КС**: Общий термин, который может относиться к кейсам в любой версии Counter-Strike, включая CS2.
– **Кейсы КС ГО**: Конкретно кейсы из CS:GO, которые были популярны до выхода CS2. Теперь многие платформы, адаптируют их под CS2, но скины из CS:GO все еще совместимы с новой игрой.
Если ты хочешь попробовать, начни с малого — внеси небольшую сумму и протестируй, как все работает. Например, выбери дешевый кейс и посмотри, что выпадет. Это поможет понять механику без больших рисков.
https://ai-db.science/wiki/Cs2case_93F
Sazrbjf
| #
Мы можем предложить дипломы любой профессии по выгодным ценам.– kupit-diplom24.com/kupit-diplom-v-moskve-s-zaneseniem-v-reestr-kachestvenno/
Rabyitabe
| #
darkmarket list dark web market urls
StevenJutle
| #
http://www.odnopolchane.net/forum/group.php?do=discuss&groupid=79&discussionid=688&gmid=2963
Sazraov
| #
Приветствую!
Купить диплом ВУЗа по выгодной стоимости возможно, обратившись к надежной специализированной фирме. Заказать диплом о высшем образовании: kupitediplom0027.ru/kupit-diplom-yurista-8/
www w69
| #
วิธีติดตั้งแอป www w69
บนมือถือ ใช้เวลาเพียงไม่กี่นาที รองรับทุกระบบ ให้คุณเริ่มเดิมพันได้ทันที
Sazrmka
| #
Хочу поделиться своим опытом по заказу аттестата ПТУ. Думал, что это невозможно, и начал искать информацию в интернете по теме: диплом об окончании класса купить, купить диплом свидетельство, купить диплом ссср в ростове, купить диплом 2000, купить диплом в нурсултане. Постепенно углубляясь в тему, нашел отличный ресурс здесь: proffdiplomik.com/sochi
w69สล็อต
| #
อัปเกรดแอป w69สล็อต วันนี้ เพื่อการเล่นเกมที่ราบรื่นกว่าเดิม พร้อมฟังก์ชันใหม่ที่ช่วยให้คุณสนุกยิ่งขึ้น
w69เข้า
| #
ดาวน์โหลดแอป w69เข้า แล้วสนุกกับสล็อต แจ็คพอตแตกง่าย และอัตราจ่ายที่ดีที่สุด
Volodyanaf
| #
bitcoin dark web darknet market links
w69cy
| #
เล่นเกม w69cy – https://bavr3.com ผ่านแอป สนุกได้ทุกที่ พร้อมอัปเดตเกมใหม่ให้คุณได้สัมผัสก่อนใคร
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наша продукция:
Кольцо РёР· драгоценных металлов платиновое 9С…3С…4.5 РјРј ПлПд95-5 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
w69 ทางเข้าเล่น
| #
โหลดแอป w69 ทางเข้าเล่น รับประสบการณ์การเล่นเกมที่สมจริงกว่าเดิม ด้วยระบบถ่ายทอดสดแบบ HD
w69 online casino
| #
อินเทอร์เฟซใหม่ของแอป w69 online casino
ช่วยให้การนำทางลื่นไหลขึ้น ไม่ต้องเสียเวลาค้นหาเกมโปรดของคุณ
rli59.com
| #
ติดตั้งแอป w69 – rli59.com – แล้วพบกับการแจ้งเตือนโปรโมชั่นและกิจกรรมใหม่ๆ ก่อนใคร
Mazrdsx
| #
Где приобрести диплом специалиста?
Готовый диплом с нужными печатями и подписями полностью отвечает запросам и стандартам Министерства образования и науки РФ, неотличим от оригинала. Не следует откладывать свои мечты и цели на несколько лет, реализуйте их с нашей помощью – отправьте простую заявку на изготовление диплома уже сегодня! Получить диплом о высшем образовании – быстро и легко! freediplom.com/kupit-diplom-11-klassa-bistro-i-nadezhno-2/
DonDonfaita
| #
darknet markets url dark web link
Mazrbkz
| #
Где приобрести диплом специалиста?
Мы предлагаем документы об окончании любых университетов РФ. Документы производят на подлинных бланках государственного образца. pashasevkav.getbb.ru/posting.php?mode=post&f=31&sid=af6e328650cc9cccafb5e3f34a0a6528
Uazrhss
| #
Добрый день!
Для определенных людей, купить диплом университета – это необходимость, возможность получить достойную работу. Но для кого-то – это желание не терять огромное количество времени на учебу в институте. Что бы ни толкнуло вас на такой шаг, наша компания готова помочь. Быстро, качественно и по разумной стоимости сделаем диплом нового или старого образца на государственных бланках со всеми печатями.
Основная причина, почему многие прибегают к покупке документов, – желание занять определенную работу. Например, способности и опыт позволяют кандидату устроиться на привлекательную работу, а документального подтверждения квалификации не имеется. При условии, что работодателю важно присутствие “корочек”, риск потерять хорошее место довольно высокий.
Приобрести документ института можно у нас. Мы предлагаем документы об окончании любых ВУЗов России. Вы получите диплом по любой специальности, включая документы СССР. Даем 100% гарантию, что в случае проверки документов работодателем, подозрений не появится.
Ситуаций, которые вынуждают приобрести диплом ВУЗа достаточно. Кому-то срочно требуется работа, в результате необходимо произвести впечатление на руководителя при собеседовании. Некоторые хотят попасть в престижную компанию, чтобы повысить свой статус в обществе и в последующем начать свой бизнес. Чтобы не терять попусту годы жизни, а сразу начинать успешную карьеру, применяя имеющиеся навыки, можно купить диплом прямо в интернете. Вы сможете быть полезным для социума, обретете финансовую стабильность в максимально короткие сроки- купить диплом техникума
เว็บสล็อตw69
| #
เว็บสล็อตw69 อัปเดตแอปใหม่ให้รองรับการเดิมพันแบบเรียลไทม์ เชียร์ทีมโปรดของคุณได้แบบไม่มีสะดุด
Sazrqys
| #
Заказ документа о высшем образовании через надежную компанию дарит ряд преимуществ. Это решение дает возможность сэкономить как дорогое время, так и серьезные финансовые средства. Тем не менее, на этом выгоды не ограничиваются, преимуществ гораздо больше.Мы изготавливаем дипломы любых профессий. Дипломы изготавливаются на подлинных бланках государственного образца. Доступная цена по сравнению с серьезными тратами на обучение и проживание в чужом городе. Заказ диплома института станет выгодным шагом.
Заказать диплом: diplom-club.com/kupite-meditsinskij-diplom-nadezhno-i-bistro/
Sazrpjc
| #
Мы предлагаем дипломы любых профессий по выгодным ценам. О преимуществах покупки документов в нашем сервисе
Вы заказываете диплом через надежную и проверенную фирму. Такое решение позволит вам сэкономить не только много денежных средств, но и ваше драгоценное время.
Плюсов гораздо больше:
• Дипломы печатаются на настоящих бланках со всеми печатями;
• Предлагаем дипломы любого ВУЗа РФ;
• Цена в разы меньше нежели довелось бы заплатить на очном и заочном обучении в университете;
• Доставка как по столице, так и в любые другие регионы России.
Заказать диплом о высшем образовании– http://babygirls001.copiny.com/question/details/id/1061062/ – babygirls001.copiny.com/question/details/id/1061062
w69thai
| #
ติดตั้งแอป w69thai แล้วรับการแจ้งเตือนแมตช์สำคัญ
โปรโมชั่นพิเศษ และข่าวสารก่อนใคร
Ronaldbut
| #
https://mycofarm.ru/
vidnovskoe
| #
Кладбище в Видном vidnovskoe актуальные данные о захоронениях, помощь в организации похорон, услуги по благоустройству могил. Схема проезда, часы работы и контактная информация.
Cazrhny
| #
Приветствую!
Мы изготавливаем дипломы любой профессии по разумным тарифам. Цена зависит от выбранной специальности, года получения и образовательного учреждения: п»їtutdiploms.com/
http://classicalmusicmp3freedownload.com/ja/index.php?title=:MilagroD00
| #
Что такое “Кейсы КС” и “Кейсы КС ГО”?
“Кейсы КС” (или “Кейсы CS”) и “Кейсы КС ГО” (CS:GO) — это виртуальные контейнеры, которые используются в играх Counter-Strike (КС), включая Counter-Strike: Global Offensive (CS:GO) и его обновленную версию Counter-Strike 2 (CS2). Внутри этих кейсов находятся различные игровые предметы — скины для оружия, ножи, перчатки и другие косметические элементы, которые игроки могут получить и использовать в игре для украшения своего инвентаря.
Как это работает?
1. **Регистрация**: Вы заходите на сайт и регистрируетесь, обычно через Steam, чтобы связать свой игровой аккаунт.
2. **Пополнение баланса**: Нужно внести деньги на сайт (например, через карту, криптовалюту или электронные кошельки), чтобы купить кейсы или внутриигровую валюту.
3. **Выбор кейса**: На сайте представлены разные кейсы с указанием возможных наград (от дешевых скинов до редких ножей) и их стоимости.
4. **Открытие кейса**: Вы выбираете кейс, оплачиваете его и открываете. Система случайным образом определяет, какой предмет вы получите.
5. **Вывод выигрыша**: Если вам повезло, вы можете вывести скин в Steam и использовать его в игре.
Чем отличается “Кейсы КС” от “Кейсы КС ГО”?
– **Кейсы КС**: Общий термин, который может относиться к кейсам в любой версии Counter-Strike, включая CS2.
– **Кейсы КС ГО**: Конкретно кейсы из CS:GO, которые были популярны до выхода CS2. Теперь многие платформы, адаптируют их под CS2, но скины из CS:GO все еще совместимы с новой игрой.
Если ты хочешь попробовать, начни с малого — внеси небольшую сумму и протестируй, как все работает. Например, выбери дешевый кейс и посмотри, что выпадет. Это поможет понять механику без больших рисков.
https://opensourcebridge.science/wiki/Cs2case_3C
Eanrvof
| #
Для быстрого продвижения по карьерной лестнице понадобится наличие диплома ВУЗа. Приобрести диплом о высшем образовании у проверенной организации: diplomp-irkutsk.ru/kupit-diplom-o-polnom-srednem-obrazovanii-bistro-i-nadezhno/
Donaldlaf
| #
darknet drug market darkmarket link
Diplomi_raKt
| #
Купить диплом университета!
Мы предлагаем документы институтов, расположенных в любом регионе Российской Федерации.
diplomidlarf.ru/diplom-o-sredne-texnicheskom-obrazovanii-kupit-4
Pingrutty
| #
dark market url dark markets 2025
Lazranm
| #
Диплом любого ВУЗа России!
Без присутствия диплома очень нелегко было продвигаться вверх по карьерной лестнице. Именно из-за этого решение о заказе диплома стоит считать выгодным и целесообразным. Заказать диплом любого института uniqplacements.com/employer/gosznac-diplom-24
Dnrtmam
| #
Где приобрести диплом по актуальной специальности?
Мы предлагаем дипломы любых профессий по доступным ценам. Мы готовы предложить документы техникумов, расположенных на территории всей Российской Федерации. Можно купить диплом за любой год, указав подходящую специальность и хорошие оценки за все дисциплины. Документы выпускаются на “правильной” бумаге высшего качества. Это дает возможности делать государственные дипломы, которые невозможно отличить от оригинала. Они заверяются необходимыми печатями и штампами. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Важно, чтобы документы были доступными для большого количества наших граждан. kupitediplom0029.ru/kupit-diplom-medbrata-2-6
Sazriyr
| #
Мы готовы предложить дипломы любых профессий по доступным тарифам. Стоимость может зависеть от конкретной специальности, года выпуска и образовательного учреждения. Всегда стараемся поддерживать для заказчиков адекватную политику цен. Для нас важно, чтобы дипломы были доступны для подавляющей массы наших граждан. купить диплом училища во владивостоке
https://utahsyardsale.com/author/dantefloren/
| #
Что такое “Кейсы КС” и “Кейсы КС ГО”?
“Кейсы КС” (или “Кейсы CS”) и “Кейсы КС ГО” (CS:GO) — это виртуальные контейнеры, которые используются в играх Counter-Strike (КС), включая Counter-Strike: Global Offensive (CS:GO) и его обновленную версию Counter-Strike 2 (CS2). Внутри этих кейсов находятся различные игровые предметы — скины для оружия, ножи, перчатки и другие косметические элементы, которые игроки могут получить и использовать в игре для украшения своего инвентаря.
Как это работает?
1. **Регистрация**: Вы заходите на сайт и регистрируетесь, обычно через Steam, чтобы связать свой игровой аккаунт.
2. **Пополнение баланса**: Нужно внести деньги на сайт (например, через карту, криптовалюту или электронные кошельки), чтобы купить кейсы или внутриигровую валюту.
3. **Выбор кейса**: На сайте представлены разные кейсы с указанием возможных наград (от дешевых скинов до редких ножей) и их стоимости.
4. **Открытие кейса**: Вы выбираете кейс, оплачиваете его и открываете. Система случайным образом определяет, какой предмет вы получите.
5. **Вывод выигрыша**: Если вам повезло, вы можете вывести скин в Steam и использовать его в игре.
Чем отличается “Кейсы КС” от “Кейсы КС ГО”?
– **Кейсы КС**: Общий термин, который может относиться к кейсам в любой версии Counter-Strike, включая CS2.
– **Кейсы КС ГО**: Конкретно кейсы из CS:GO, которые были популярны до выхода CS2. Теперь многие платформы, адаптируют их под CS2, но скины из CS:GO все еще совместимы с новой игрой.
Если ты хочешь попробовать, начни с малого — внеси небольшую сумму и протестируй, как все работает. Например, выбери дешевый кейс и посмотри, что выпадет. Это поможет понять механику без больших рисков.
https://aexcom.org.pe/index.php/Cs2case_46S
Toliknaf
| #
darknet markets darknet market links
Kxyufaita
| #
darknet drugs darknet market list
zaimcaree
| #
Быстрые онлайн займы на карту, подробнее на официальном сайте ссылка
Sazrsqn
| #
Заказать диплом ВУЗа по выгодной стоимости можно, обращаясь к надежной специализированной фирме. Мы предлагаем документы об окончании любых ВУЗов России. Заказать диплом ВУЗа– diploma-groups24.ru/diplomy-po-specialnosti/diplom-farmacevta.html
Sazrgck
| #
купить аттестат об окончании 11 классов в калининграде
StevenJutle
| #
http://forum.ptia-avto.ru/viewtopic.php?f=16&t=1137
Volodyanaf
| #
bitcoin dark web dark web sites
Rabyitabe
| #
darkmarket url darknet markets onion address
drenajnie raboti spb_wxel
| #
дренаж участка под ключ цена дренаж участка под ключ цена .
drenajnie raboti spb_wrst
| #
дренажно распределительная система дренажно распределительная система .
https://indigenouspedia.com/index.php?title=User:Oliva1833648762
| #
Что такое “Кейсы КС” и “Кейсы КС ГО”?
“Кейсы КС” (или “Кейсы CS”) и “Кейсы КС ГО” (CS:GO) — это виртуальные контейнеры, которые используются в играх Counter-Strike (КС), включая Counter-Strike: Global Offensive (CS:GO) и его обновленную версию Counter-Strike 2 (CS2). Внутри этих кейсов находятся различные игровые предметы — скины для оружия, ножи, перчатки и другие косметические элементы, которые игроки могут получить и использовать в игре для украшения своего инвентаря.
Как это работает?
1. **Регистрация**: Вы заходите на сайт и регистрируетесь, обычно через Steam, чтобы связать свой игровой аккаунт.
2. **Пополнение баланса**: Нужно внести деньги на сайт (например, через карту, криптовалюту или электронные кошельки), чтобы купить кейсы или внутриигровую валюту.
3. **Выбор кейса**: На сайте представлены разные кейсы с указанием возможных наград (от дешевых скинов до редких ножей) и их стоимости.
4. **Открытие кейса**: Вы выбираете кейс, оплачиваете его и открываете. Система случайным образом определяет, какой предмет вы получите.
5. **Вывод выигрыша**: Если вам повезло, вы можете вывести скин в Steam и использовать его в игре.
Чем отличается “Кейсы КС” от “Кейсы КС ГО”?
– **Кейсы КС**: Общий термин, который может относиться к кейсам в любой версии Counter-Strike, включая CS2.
– **Кейсы КС ГО**: Конкретно кейсы из CS:GO, которые были популярны до выхода CS2. Теперь многие платформы, адаптируют их под CS2, но скины из CS:GO все еще совместимы с новой игрой.
Если ты хочешь попробовать, начни с малого — внеси небольшую сумму и протестируй, как все работает. Например, выбери дешевый кейс и посмотри, что выпадет. Это поможет понять механику без больших рисков.
https://idpedia.wiki/index.php/User:JessNeedham0
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние возможности.
Наша продукция:
“ерамическая мишень CeO2, плоская форма
DonDonfaita
| #
darknet markets links dark web market urls
เวปw69
| #
อัปเดตแอป เวปw69 วันนี้เพื่อสัมผัสประสบการณ์ใหม่ ลื่นไหลกว่าเดิม ไม่มีสะดุด
Iariorgbl
| #
Приобрести документ института вы сможете в нашем сервисе. http://www.mamanija.lt/klausimai/18759/koks-mamu-forumas-jums-mielesnis
Mazrvbw
| #
Мы изготавливаем дипломы любой профессии по приятным тарифам. Стараемся поддерживать для клиентов адекватную ценовую политику. Для нас важно, чтобы дипломы были доступны для подавляющей массы граждан.
Покупка диплома, подтверждающего окончание института, – это выгодное решение. Приобрести диплом любого ВУЗа: diplommy.ru/diplom-srednee-spetsialnoe-obrazovanie-kupit-4/
slot w69
| #
เล่นบาคาร่า รูเล็ต และเกมไพ่อื่นๆ ผ่านแอป slot w69 ด้วยระบบที่ลื่นไหลและภาพคมชัดระดับ
HD
Kxyufaita
| #
dark markets 2025 dark web market list
Pingrutty
| #
darknet markets dark web sites
avtolombardnovosibsig
| #
выгодный кредит под залог авто
https://www.philthejob.nl/employer/retailjobacademy2814/
деньги под залог кредитного авто
w69ลิ้ง
| #
อัปเกรดแอป w69ลิ้ง วันนี้ เพื่อการเล่นเกมที่ราบรื่นกว่าเดิม พร้อมฟังก์ชันใหม่ที่ช่วยให้คุณสนุกยิ่งขึ้น
dshb7.com
| #
dshb7.com – https://dshb7.com ปรับปรุงอินเทอร์เฟซแอปให้ใช้งานง่ายขึ้น ดีไซน์สวยงาม ค้นหาเกมได้รวดเร็วและสะดวกสบาย
w69io
| #
w69io – https://qah64.com ปรับปรุงฟังก์ชันแอปใหม่ ใช้งานง่ายกว่าเดิม สนุกกับคาสิโนสดและเกมสล็อตได้ทุกที่ทุกเวลา
Donaldlaf
| #
onion dark website darkmarket list
Diplomi_lqKt
| #
Купить диплом о высшем образовании!
Мы предлагаем документы университетов, расположенных в любом регионе Российской Федерации.
poluchidiplom.com/mozhno-li-kupit-attestat-za-9-klass-4
w69คาสิโนออนไลน์
| #
ดาวน์โหลดแอป w69คาสิโนออนไลน์ ง่ายๆ เพียงไม่กี่คลิก สนุกกับการเดิมพันกีฬา คาสิโน และสล็อตในที่เดียว
w69สล็อต เครดิตฟรี
| #
ติดตั้งแอป w69สล็อต เครดิตฟรี – https://apmb1.com อย่างรวดเร็ว รองรับทั้ง iOS
และ Android สัมผัสประสบการณ์เดิมพันที่ลื่นไหลและสะดวกสบายกว่าเดิม
w69 bet เข้าสู่ระบบ
| #
อินเทอร์เฟซใหม่ของแอป w69 bet เข้าสู่ระบบ ช่วยให้การนำทางลื่นไหลขึ้น
ไม่ต้องเสียเวลาค้นหาเกมโปรดของคุณ
1win_ysMr
| #
1win зайти 1win6002.ru .
w69mobil
| #
w69mobil ปรับแต่งอินเทอร์เฟซแอปใหม่ ดีไซน์สวยงาม
ค้นหาง่าย และใช้งานได้สะดวกกว่าเดิม
Volodyanaf
| #
darknet market lists darknet markets 2025
StevenJutle
| #
https://wowlol.ru/forum/showthread.php?p=77714
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их качество. Опытная поддержка – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы и находить ответы под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
РљСЂСѓРі магниевый РњРђ8 Фольга магниевая РњРђ8 – это идеальный выбор для ваших технических задач. РћРЅР° отличается высокой прочностью Рё отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью. Рспользуйте фольгу РІ производстве, РіРґРµ требуется высокая проводимость Рё теплоотведение. РЎ ее помощью можно добиться качественного соединения РІ электротехнике Рё РґСЂСѓРіРёС… отраслях. Преимущества: легкость, устойчивость Рє воздействию влаги, возможность использования РІ различных климатических условиях. Если РІС‹ ищете надежный Рё долговечный материал, вам стоит купить Фольга магниевая РњРђ8. Убедитесь РІ ее качествах уже сегодня!
Toliknaf
| #
darkmarket url darkmarket 2025
w69 เข้า สู่ ระบบ
| #
อัปเกรดแอป w69 เข้า สู่ ระบบ วันนี้ เพื่อการเล่นเกมที่ราบรื่นกว่าเดิม
พร้อมฟังก์ชันใหม่ที่ช่วยให้คุณสนุกยิ่งขึ้น
Kennethfem
| #
В нашем центре доступен ремонт разного уровня сложности аудиосистем Philips включая профилактику.
Преимущества нашего сервиса:
Профессиональная оценка проблемы
Оперативное устранение неисправностей
Доступные цены
Выезд мастера на дом по Москве и области
Получить подробную информацию или заказать консультацию вы можете на нашем сайте ремонт кофемашины philips saeco.
Доверьте свою технику профессионалам! Свяжитесь с нами по телефону – гарантируем результат!
mostbet_fmOt
| #
mostbet.kg http://www.mostbet6007.ru .
1win_qlot
| #
1win скачать kg http://alfatraders.borda.ru/?1-0-0-00004932-000-0-0-1743258210/ .
Cazruxe
| #
Получить диплом любого ВУЗа поможем. Купить аттестат в Тольятти – diplomybox.com/kupit-attestat-v-tolyatti
Dnrttaw
| #
Где купить диплом специалиста?
Мы изготавливаем дипломы любых профессий по приятным тарифам. Мы можем предложить документы техникумов, расположенных в любом регионе Российской Федерации. Можно заказать диплом за любой год, указав актуальную специальность и оценки за все дисциплины. Документы делаются на “правильной” бумаге самого высокого качества. Это позволяет делать настоящие дипломы, не отличимые от оригинала. Документы будут заверены необходимыми печатями и штампами. Всегда стараемся поддерживать для покупателей адекватную политику цен. Для нас очень важно, чтобы документы были доступны для большого количества граждан. okdiplom.ru/kupit-diplom-s-vneseniem-v-reestr-6
Sazrxdb
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и других профессий по доступным ценам. Цена может зависеть от выбранной специальности, года получения и ВУЗа. Стараемся поддерживать для заказчиков адекватную ценовую политику. Для нас очень важно, чтобы дипломы были доступны для подавляющей массы наших граждан. купить диплом в тольятти
zaimcaree
| #
Быстрые онлайн займы на карту, подробнее на официальном сайте ссылка
w69th
| #
ดาวน์โหลดแอป w69th – https://szc74.com ได้ง่ายๆ เพียงไม่กี่ขั้นตอน พร้อมรับโบนัสต้อนรับ 100$ สนุกกับการเดิมพันที่ดีที่สุดบนมือถือ
w69 คาสิโนออนไลน์
| #
w69 คาสิโนออนไลน์ พัฒนาแอปให้รองรับเกมมากขึ้น มีตัวเลือกหลากหลายให้คุณเดิมพันได้อย่างอิสระ
Rabyitabe
| #
darknet market lists darknet links
Ronaldbut
| #
https://mycofarm.ru/
Geraldnof
| #
https://plomba77.ru/plombi-dlja-plombiratora/plombi-trubchatie-aluminievye В мире защиты и контроля доступа, одноразовые пластиковые пломбы роторного типа занимают особое место. Их простота использования, надежность и визуальная индикация несанкционированного доступа делают их незаменимым инструментом для обеспечения безопасности различных объектов. Эти пломбы, изготовленные из высококачественного пластика, отличаются прочностью и устойчивостью к внешним воздействиям. Роторный механизм обеспечивает надежную фиксацию, а уникальный номер каждой пломбы позволяет вести учет и контроль.
Kxyufaita
| #
darknet links darknet marketplace
Sazrwsh
| #
купить официальный диплом о среднем образовании
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наши товары:
Гранулы высокочистого ванадия
Pingrutty
| #
dark market 2025 darknet site
betshah
| #
New Users, Register at betshah and Get a $100 Welcome Bonus
DonDonfaita
| #
darknet market list dark websites
https://droids-hack.ru/
| #
https://droids-hack.ru/ — это отличный способ
получить новые возможности.
Особенно если вы играете на мобильном устройстве с Android,
модификации открывают перед вами новые возможности.
Я нравится использовать взломанные игры, чтобы получать неограниченные ресурсы.
Моды для игр дают невероятную свободу выбора, что погружение
в игру гораздо красочнее. Играя с
плагинами, я могу добавить дополнительные функции, что добавляет
виртуальные путешествия и делает игру более захватывающей.
Это действительно невероятно,
как такие модификации могут улучшить переживания от игры, а при этом с максимальной безопасностью использовать такие модифицированные приложения можно без особых опасностей, если быть внимательным
и следить за обновлениями.
Это делает каждый игровой процесс лучше контролируемым,
а возможности практически выше всяких похвал.
Советую попробовать такие модифицированные версии для Android — это может добавить веселья в геймплей
drenajnie raboti spb_omel
| #
дренажные системы компании http://www.drenazh-uchastka-rostov.ru .
drenajnie raboti spb_zwst
| #
дренаж участка спб дренаж участка спб .
Volodyanaf
| #
darknet market links dark web market
Monitoring transporta_vlOi
| #
установка мониторинга автомобилей https://kontrol-avto.ru .
Donaldlaf
| #
dark market link darknet drug market
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их качество. Опытная поддержка – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы а также адаптировать решения под особенности вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наши товары:
Кобальт Stellite 4LC Кобальт Stellite 4LC – это высококачественный сплав, разработанный для обеспечения прочности Рё сопротивления РёР·РЅРѕСЃСѓ. Рспользуется РІ различных отраслях, включая авиастроение Рё машиностроение. Благодаря своей высокой РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкости Рё жаропрочности, этот материал прекрасно РїРѕРґС…РѕРґРёС‚ для работы РІ экстремальных условиях. Если вам нужен надежный материал для обработки Рё защиты деталей, РїРѕРєСѓРїРєР° Кобальт Stellite 4LC станет удачным решением. Рнвестируйте РІ качество Рё долговечность ваших изделий, выбрав Кобальт Stellite 4LC.
Kxyufaita
| #
darknet markets onion darknet websites
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние возможности.
Наша продукция:
Высокочистые гранулы тантала
Toliknaf
| #
darknet markets links dark market link
Lazrulo
| #
Заказать диплом об образовании!
Мы предлагаем дипломы психологов, юристов, экономистов и прочих профессий по приятным ценам— diplomg-cheboksary.ru/kupit-attestat-za-11-klass-v-moskve-bistro-i-udobno-8/
Jariorczp
| #
Где заказать диплом по нужной специальности?
Мы предлагаем выгодно и быстро приобрести диплом, который выполняется на оригинальной бумаге и заверен мокрыми печатями, штампами, подписями. Данный диплом способен пройти лубую проверку, даже с применением профессионального оборудования. Достигайте своих целей максимально быстро с нашим сервисом. Купить диплом о высшем образовании! publikacii.listbb.ru/viewtopic.php?f=3&t=2145
Zooma jackpot slots
| #
Welcome to Zooma Casino – a modern gaming platform with unique opportunities! Here, you’ll find exciting slot machines from world-class game providers. https://zooma-play.top/ to test your luck in top-rated games!
Why do thousands of players choose us?
A vast selection of games from industry leaders.
Loyalty programs on deposits and bets.
Instant withdrawals to convenient payment systems.
User-friendly design on any device.
24/7 support quickly resolving any issues.
Feel the thrill of gambling and take advantage of all benefits!
Mazrsov
| #
Мы изготавливаем дипломы любой профессии по приятным ценам. Всегда стараемся поддерживать для заказчиков адекватную политику цен. Для нас очень важно, чтобы дипломы были доступны для большого количества наших граждан.
Покупка диплома, подтверждающего обучение в университете, – это рациональное решение. Заказать диплом университета: diplom-bez-problem.com/kupit-diplom-vuza-nedorogo/
Iariorzen
| #
Купить документ о получении высшего образования вы сможете в нашем сервисе. sarasusa.dreamwidth.org/23915.html?mode=reply
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние достижения.
Наши товары:
Высокочистые плитки алюминия
Rabyitabe
| #
dark market link tor drug market
Pingrutty
| #
bitcoin dark web dark websites
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
поставляемая продукция:
Лента бронзовая 0,12х190 мм БрОФ ГОСТ 1761-92 Покупайте качественные и универсальные ленты бронзовые от Редметсплав. Широкий выбор размеров и высокое качество материала. Надежное и стильное решение для ваших проектов.
DavidUrire
| #
Счетчики газа с пленкой Приборы учета с дистанционным управлением, электросчетчики, оснащенные пультом, устройства для измерения потребления электроэнергии с возможностью удаленного контроля, экономичные модели счетчиков, модифицированные приборы учета, разнообразные счетчики, газовые счетчики, подвергшиеся воздействию магнитом или пленкой, способы остановки работы счетчика, приобретение счетчика с пультом дистанционного управления, покупка электросчетчика с пультом.
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Превосходное обслуживание – наш стандарт – мы на связи, чтобы разрешать ваши вопросы и предоставлять решения под требования вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
РџРѕРєРѕРІРєР° магниевая ISO-MgAl4Si – ISO 16220 Лист магниевый ISO-MgAl4Si – ISO 16220 – это высококачественный материал, выполненный РёР· магниевого сплава, который отличается отличными механическими свойствами Рё легкостью. Ртот лист идеально РїРѕРґС…РѕРґРёС‚ для различных промышленных применений, включая авиацию Рё автомобильное производство. Купить Лист магниевый ISO-MgAl4Si – ISO 16220 – значит обеспечить себя надежным Рё долговечным материалом, который прекрасно справляется СЃ нагрузками. РќРµ упустите возможность приобрести этот товар, оптимизировав СЃРІРѕРё бизнес-процессы СЃ помощью высокотехнологичных решений!
Volodyanaf
| #
dark market onion darknet market
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наши товары:
“ерамическая мишень CeO2, плоская форма
DonDonfaita
| #
darkmarket list bitcoin dark web
MichaelScOfs
| #
аренда квартир в ташкенте Ташкент, современная столица Узбекистана, привлекает своим сочетанием истории и динамичного развития. Для тех, кто планирует остаться здесь на длительный срок, вопрос аренды жилья становится первостепенным. Снять квартиру в Ташкенте – задача, требующая внимательного подхода и понимания местного рынка.
https://surgiteams.com/index.php/Cs2case_82H
| #
Что такое “Кейсы КС” и “Кейсы КС ГО”?
“Кейсы КС” (или “Кейсы CS”) и “Кейсы КС ГО” (CS:GO) — это виртуальные контейнеры, которые используются в играх Counter-Strike (КС), включая Counter-Strike: Global Offensive (CS:GO) и его обновленную версию Counter-Strike 2 (CS2). Внутри этих кейсов находятся различные игровые предметы — скины для оружия, ножи, перчатки и другие косметические элементы, которые игроки могут получить и использовать в игре для украшения своего инвентаря.
Как это работает?
1. **Регистрация**: Вы заходите на сайт и регистрируетесь, обычно через Steam, чтобы связать свой игровой аккаунт.
2. **Пополнение баланса**: Нужно внести деньги на сайт (например, через карту, криптовалюту или электронные кошельки), чтобы купить кейсы или внутриигровую валюту.
3. **Выбор кейса**: На сайте представлены разные кейсы с указанием возможных наград (от дешевых скинов до редких ножей) и их стоимости.
4. **Открытие кейса**: Вы выбираете кейс, оплачиваете его и открываете. Система случайным образом определяет, какой предмет вы получите.
5. **Вывод выигрыша**: Если вам повезло, вы можете вывести скин в Steam и использовать его в игре.
Чем отличается “Кейсы КС” от “Кейсы КС ГО”?
– **Кейсы КС**: Общий термин, который может относиться к кейсам в любой версии Counter-Strike, включая CS2.
– **Кейсы КС ГО**: Конкретно кейсы из CS:GO, которые были популярны до выхода CS2. Теперь многие платформы, адаптируют их под CS2, но скины из CS:GO все еще совместимы с новой игрой.
Если ты хочешь попробовать, начни с малого — внеси небольшую сумму и протестируй, как все работает. Например, выбери дешевый кейс и посмотри, что выпадет. Это поможет понять механику без больших рисков.
https://wiki.roboco.co/index.php/Cs2case_18S
Cazrfbu
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и прочих профессий по приятным ценам. Купить диплом о неполном образовании — kyc-diplom.com/diplom-o-nepolnom-obrazovanii.html
Kxyufaita
| #
darknet links dark web link
w69 android app
| #
ดาวน์โหลดแอป w69 android app ง่ายๆ เพียงไม่กี่คลิก สนุกกับการเดิมพันกีฬา คาสิโน และสล็อตในที่เดียว
w69oi
| #
ดาวน์โหลดแอป w69oi
ง่ายๆ เพียงไม่กี่คลิก สนุกกับการเดิมพันกีฬา คาสิโน และสล็อตในที่เดียว
w69 เครดิตฟรี 188
| #
ติดตั้งแอป w69 เครดิตฟรี 188 แล้วรับการแจ้งเตือนแมตช์สำคัญ โปรโมชั่นพิเศษ และข่าวสารก่อนใคร
w69 asia
| #
ดาวน์โหลดแอป w69 asia แล้วเข้าถึงการเดิมพันแบบ
VIP ด้วยโปรโมชั่นและสิทธิพิเศษเฉพาะสมาชิกแอป
w69 casino login
| #
อัปเกรดแอป w69 casino login วันนี้ เพื่อการเล่นเกมที่ราบรื่นกว่าเดิม พร้อมฟังก์ชันใหม่ที่ช่วยให้คุณสนุกยิ่งขึ้น
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их качество. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы а также предоставлять решения под специфику вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Проволока титановая Р’Рў4 Полоса титановая Р’Рў4 — это высококачественный строительный материал, обладающий отличными механическими свойствами Рё РєРѕСЂСЂРѕР·РёР№РЅРѕР№ стойкостью. Рдеально РїРѕРґС…РѕРґРёС‚ для различных отраслей, включая авиационную Рё судостроительную. Применение титана обеспечивает легкость Рё долговечность изделий. Полоса имеет стандартные размеры Рё может быть легко обработана РїРѕРґ заказ. Если РІС‹ ищете надежный материал для своего проекта, РЅРµ упустите шанс купить Полоса титановая Р’Рў4. Получите преимущества РѕС‚ использования титана Рё обеспечьте высокое качество ваших изделий.
Ronaldbut
| #
https://mycofarm.ru/
เกม สล็อต w69
| #
ดาวน์โหลดแอป เกม สล็อต w69 – https://wfi39.com ได้ง่ายๆ เพียงไม่กี่ขั้นตอน พร้อมรับโบนัสต้อนรับ 100$ สนุกกับการเดิมพันที่ดีที่สุดบนมือถือ
w69เข้าสู่ระบบ
| #
w69เข้าสู่ระบบ – https://ekn83.com ปรับปรุงฟังก์ชันแอปใหม่ ใช้งานง่ายกว่าเดิม สนุกกับคาสิโนสดและเกมสล็อตได้ทุกที่ทุกเวลา
pravo-migranta
| #
Аккредитованное агентство http://pravo-migranta.ru по аутстаффингу мигрантов и миграционному аутсорсингу. Оформление иностранных сотрудников без рисков. Бесплатная консультация и подбор решений под ваш бизнес.
Donaldlaf
| #
darknet markets onion address darkmarkets
w69 vvip
| #
แอป w69 vvip อัปเดตใหม่
โหลดเร็ว ใช้งานง่าย และรองรับภาษาไทยเต็มรูปแบบ
1win_vfMr
| #
1 вин https://1win6002.ru .
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их соответствие стандартам. Опытная поддержка – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы и предоставлять решения под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
поставляемая продукция:
Пруток магниевый ZK61 – CSA HG.9 Проволока магниевая ZK61 – CSA HG.9 представляет СЃРѕР±РѕР№ высококачественный РїСЂРѕРґСѓРєС‚, созданный для применения РІ различных отраслях, включая авиационную Рё автомобильную. Благодаря СЃРІРѕРёРј уникальным свойствам, таким как высокая прочность Рё легкость, эта проволока идеально РїРѕРґС…РѕРґРёС‚ для сварки Рё создания комплектующих. Если РІС‹ ищете надежный материал, чтобы улучшить СЃРІРѕРё проекты, РїРѕРєСѓРїРєР° Проволока магниевая ZK61 – CSA HG.9 станет отличным решением. РќРµ упустите возможность обеспечить СЃРІРѕСЋ продукцию великолепными характеристиками Рё долгим СЃСЂРѕРєРѕРј службы!
Toliknaf
| #
darknet market list dark web link
Pingrutty
| #
dark market dark web marketplaces
mostbet_koOt
| #
мастбет https://mostbet6007.ru/ .
Volodyanaf
| #
dark market onion best darknet markets
ktobet
| #
Cadastre-se no ktobet – https://ktobet-88.com e Receba 100$ de Bônus ao Criar Sua Conta!
1win_wiMl
| #
1win партнерская программа вход 1win партнерская программа вход .
Monitoring transporta_ibOi
| #
глонасс мониторинг транспорта личный кабинет глонасс мониторинг транспорта личный кабинет .
Kxyufaita
| #
best darknet markets darknet links
Rabyitabe
| #
dark market 2025 dark websites
bankrotstvo v moskve_btKi
| #
банкротство отзывы
От чего бывает потливость
| #
Very shortly this web site will be famous amid
all blogging and site-building people, due to it’s nice content
Lazrpgl
| #
Где заказать диплом специалиста?
Купить диплом института по доступной цене вы можете, обращаясь к надежной специализированной компании.: okdiplom.com
WilliamDix
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и других профессий по выгодным ценам. Дипломы изготавливаются на подлинных бланках Приобрести диплом об образовании diplom4you.com
DonDonfaita
| #
dark web market urls darknet market links
fcb8
| #
Đăng Ký fcb8 Trên Di Động
– Đơn Giản, Nhanh Chóng
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Медно-никелевая поверхность для распыления
Kxyufaita
| #
dark web drug marketplace dark web market
Diplomi_ixka
| #
диплом о медицинском образовании купить диплом о медицинском образовании купить .
Donaldlaf
| #
darknet drug store darknet links
Pingrutty
| #
darknet markets url dark web link
zaimpodzalogptsekbsig
| #
деньги под птс сразу
avtolombard-pid-zalog-pts.ru/ekb.html
деньги под залог авто
Stephenadvic
| #
перенаправляется сюда https://xanax-futbolka.ru
Toliknaf
| #
darknet sites tor drug market
Sazrfnz
| #
Купить диплом о высшем образовании !
Приобретение диплома любого ВУЗа России у нас является надежным делом, ведь документ заносится в реестр. При этом печать осуществляется на фирменных бланках ГОЗНАКа. Купить диплом любого университета arenadiplom24.online/akademicheskie-spravki
TerryLix
| #
Все про стройку и ремонт. Актуальные интересные статьи о стройке и ремонте. Каждый найдет полезные материалы о дизайне, электрике, отоплении, кровле, сантехнике и другая полезная информация https://otoplenie-expert.com/
Ronaldbut
| #
МикоФарм Фунги
Sazrfjc
| #
Будучи студентом, я наслаждался учебой до тех пор, пока не пришло время писать диплом. Но паниковать не стоило, ведь существуют компании, которые помогают с написанием и защитой диплома на отличные оценки!
Изначально я искал информацию по теме: купить диплом о среднем образовании в краснодаре, купить диплом пту в спб, купить диплом о высшем образовании в брянске, диплом техникума купить дешево, цена аттестата, затем наткнулся на diplomybox.com/kupit-diplom-magistra-chelyabinsk
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица товара подтверждена требуемыми документами, подтверждающими их качество. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы по мере того как предоставлять решения под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
РљСЂСѓРі магниевый M10601 – UNS Фольга магниевая M10601 – UNS представляет СЃРѕР±РѕР№ высококачественный материал, который широко используется РІ различных отраслях. РћРЅР° обладает отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ устойчивостью Рё высокой прочностью, что делает её идеальной для применения РІ сложных условиях. Фольга магниевая M10601 – UNS легко обрабатывается Рё сваривается, что позволяет эффективно использовать её РІ производственных процессах. РќРµ упустите возможность купить Фольга магниевая M10601 – UNS, чтобы обеспечить надежность Рё долговечность ваших изделий. Ртот материал станет отличным решением для профессионалов Рё энтузиастов, стремящихся Рє оптимальному результату.
repraiser vaildberriz_pcmr
| #
репрайсер вайлдберриз репрайсер вайлдберриз .
Rabyitabe
| #
darknet markets url dark web link
Diplomi_ylKt
| #
Приобрести диплом любого университета!
Мы можем предложить документы любых учебных заведений, которые находятся на территории всей РФ.
diplomh-40.ru/kupit-diplom-s-registratsiej-v-reestre-nedorogo-3/
dadiwboure
| #
T.me/official_izzicasino станет для вас верным помощником и источником интересных материалов. Мы создали канал IZZI Casino в рекомендационных целях, чтобы поделиться с вами полезной информацией. Присоединяйтесь к нам и будьте в курсе горячих новостей, а также самых выгодных предложений. https://t.me/official_izzicasino/ – канал, посвященный букмекерской компании и онлайн-казино Иззи. Всегда своих игроков радуем. Выберите интересующий слот и увлекательной игрой наслаждайтесь. Попытайте счастья в выигрыше крупных призов!
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Плоская мишень NiV
Kxyufaita
| #
bitcoin dark web dark websites
Jamesdusty
| #
Спасибо за букет невесты – всё было идеально!
заказать цветы томск
Cunokesgaw
| #
На сайте https://pilmi.ru вы найдете огромное количество фильмов самых разных жанров, включая комедии, мелодрамы, драмы, романтические. Для того чтобы подыскать наиболее подходящий вариант, нужно воспользоваться специальным каталогом. Вашему вниманию представлены и ожидаемые любопытные новинки этого года, которые произведут эффект. Все это можно просматривать в любое время и на различных устройствах. Прямо сейчас воспользуйтесь возможностью посмотреть азиатское кино. Используйте поиск для облегчения выбора.
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
Наша продукция:
Меднаядвухраструбнаямуфта под пайку25х1.6ммМ1МГОСТ 32590-2013 Выберите медные двухраструбные муфты под пайку от производителя RedmetSplav. Прочные и герметичные соединения для систем водоснабжения, отопления и газоснабжения. Долговечные и надежные муфты для любых видов работ. Подходят для использования в домашних условиях, коммерческих и промышленных объектах.
DonDonfaita
| #
dark web market links darknet markets onion
Sazraqg
| #
Мы изготавливаем дипломы любых профессий по приятным тарифам.– vuz-diplom.ru/kupit-diplom-bistro-i-nadezhno-bez-lishnix-xlopot/
Pingrutty
| #
darknet market list bitcoin dark web
jafiyeordet
| #
Автосервис в СПб СТО Приморский предлагает лояльную стоимость на все услуги. Наша главная цель – обеспечить вам уверенность и комфорт на дороге. Используем качественные запчасти и современное оборудование. Запишитесь уже сегодня на ремонт автомобиля! https://sto812.com – тут есть возможность детальнее с условиями оплаты ознакомиться. Доверьте свою машину нашим специалистам. Мы даем гарантию на компетентный подход, надежность и быстрое решение любых задач. Поможем вам с подбором запчастей. Обращайтесь к нам и не пожалеете об этом!
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их качество. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы и адаптировать решения под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
Проволока магниевая ZH62A – MIL M-46062 Полоса магниевая ZH62A – MIL M-46062 представляет СЃРѕР±РѕР№ высококачественный РїСЂРѕРґСѓРєС‚, предназначенный для использования РІ различных отраслях. Ртот материал обеспечивает отличные антикоррозийные свойства, Р° также обладает высокой прочностью Рё легкостью. Полоса магниевая ZH62A – MIL M-46062 идеально РїРѕРґС…РѕРґРёС‚ для применения РІ авиации, судостроении Рё РґСЂСѓРіРёС… областях, РіРґРµ важны надежность Рё эффективность. РќРµ упустите возможность купить Полоса магниевая ZH62A – MIL M-46062 РїРѕ выгодной цене Рё обеспечьте себе качество РЅР° долгие РіРѕРґС‹. Выбор этого продукта – это вклад РІ успешные проекты.
Sazrzmt
| #
Добрый день!
Приобрести диплом университета по невысокой стоимости можно, обращаясь к проверенной специализированной фирме. Заказать диплом: asxdiploman.com/kupit-diplom-vracha-28/
Donaldlaf
| #
darknet markets 2025 darknet websites
Cazrumv
| #
Приобрести диплом о высшем образовании!
Мы предлагаем документы университетов, которые находятся в любом регионе Российской Федерации. Дипломы и аттестаты печатаются на бумаге самого высшего качества: allboiler.ru/news/attestatu_1.html
Robertniday
| #
Все о ремонте и строительстве в ежедневных актуальных статьях. Полезные советы по ремонту, лайфхаки, интересные статьи о даче и дизайне https://premierdevelopment.ru/
Toliknaf
| #
darknet market list dark web marketplaces
Jameszem
| #
Актуальные мировые новости о политике, обществе, экномике, бизнесе, медицине и других отраслях. Ежеденевная аналитическая информация в нашем блоге https://tovarlive.ru/
EdgarKab
| #
investigate this site
russian brides for marriage
Rabyitabe
| #
darkmarkets best darknet markets
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
поставляемая продукция:
Быстрорежущий Р±СЂСѓСЃРѕРє 8С…40 РјРј Р 6Рњ5Рљ5 РўРЈ РЁРёСЂРѕРєРёР№ выбор быстрорежущих Р±СЂСѓСЃРєРѕРІ для профессиональной обработки металла. Рзготовлены РёР· высокопрочных материалов, обеспечивают высокоточную работу СЃ минимальным РёР·РЅРѕСЃРѕРј. Гарантированное качество, оперативная доставка Рё профессиональная поддержка СЃРѕ стороны специалистов. РџРѕРґР±РѕСЂ оптимального инструмента для различных РІРёРґРѕРІ работ.
Source URL
| #
Its like you read my mind! You seem to know a lot about this,
like you wrote the book in it or something. I think that you could
do with a few pics to drive the message home a little bit, but instead of
that, this is fantastic blog. A great read. I will certainly be back.
TerryLix
| #
Все про стройку и ремонт. Актуальные интересные статьи о стройке и ремонте. Каждый найдет полезные материалы о дизайне, электрике, отоплении, кровле, сантехнике и другая полезная информация https://otoplenie-expert.com/
Pingrutty
| #
bitcoin dark web darknet markets onion address
Ronaldbut
| #
https://mycofarm.ru/
Mazryxl
| #
Где заказать диплом специалиста?
Мы оказываем услуги по производству и продаже документов об окончании любых университетов России. Документы производятся на подлинных бланках государственного образца. Рєradiotrainzfm301.getbb.ru/viewtopic.phpf=18&t=738
Mazrtnf
| #
Где купить диплом по актуальной специальности?
Готовый диплом с приложением отвечает стандартам Министерства образования и науки России, неотличим от оригинала – даже со специальным оборудованием. Не стоит откладывать свои цели на продолжительные годы, реализуйте их с нашей помощью – отправляйте заявку на диплом уже сегодня! Диплом о высшем образовании – не проблема! p99946c6.beget.tech/2025/03/28/kupit-diplom-bez-predoplaty-bezopasno-i-nadezhno.html
DonDonfaita
| #
darkmarket list dark market 2025
zaimpodptskemerovosig
| #
займ под птс
avtolombard-pid-zalog-pts.ru/kemerovo.html
займ под птс
Josephkaw
| #
события в мире единоборств
1win_mwOi
| #
win 1 win 1 .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние возможности.
Наши товары:
Плоская мишень Sio для распыления
Sazrxig
| #
Приобретение документа о высшем образовании через проверенную и надежную компанию дарит немало преимуществ. Это решение дает возможность сберечь как длительное время, так и существенные финансовые средства. Однако, достоинств значительно больше.Мы изготавливаем дипломы любых профессий. Дипломы производятся на настоящих бланках. Доступная цена сравнительно с большими тратами на обучение и проживание в чужом городе. Покупка диплома университета является выгодным шагом.
Купить диплом о высшем образовании: diplomk-vo-vladivostoke.ru/bistraya-pokupka-diplomov-s-reestrom-bez-xlopot/
Cazreeo
| #
Привет!
Без университета сложно было продвигаться по карьерной лестнице. На текущий момент этот важный документ не дает совершенно никаких гарантий, что получится найти привлекательную работу. Более важны навыки специалиста, а также его опыт. Именно из-за этого решение о заказе диплома стоит считать целесообразным. Заказать диплом об образовании forum.sevsocium.ru/posting.php?mode=post&f=138&sid=94de3dcad98f4347d0084e235259a859
XunovGroup
| #
На сайте https://astro-magic.com/ воспользуйтесь искусственным интеллектом на русском языке. Этот сервис предоставляет возможность использовать все функции в режиме реального времени. При этом нет необходимости подключать VPN, искусственный интеллект работает и без него. Воспользоваться им сможет каждый желающий, абсолютно бесплатно. Chat GPT сможет создать качественный контент, произвести анализ изображений, быстро отыскать нужную информацию. Искусственный интеллект сможет и написать программный код.
Cazrehc
| #
Здравствуйте!
Мы можем предложить дипломы психологов, юристов, экономистов и прочих профессий по выгодным ценам. Стоимость может зависеть от выбранной специальности, года получения и университета: diplomanrussians.com/
Eanrpdc
| #
Для успешного продвижения вверх по карьерной лестнице требуется наличие диплома института. Приобрести диплом об образовании у проверенной компании: diplomaj-v-tule.ru/kupit-vuz-diplom-6/
Donaldlaf
| #
dark market url darkmarket 2025
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
поставляемая продукция:
Лента РёР· прецизионных сплавов для СѓРїСЂСѓРіРёС… элементов 0.32×35 РјРј 17ХНГТ ГОСТ 14117-85 Купите ленту РёР· прецизионных сплавов для СѓРїСЂСѓРіРёС… элементов РїРѕ выгодной цене РЅР° Редметсплав.СЂС„. Высокая прочность, устойчивость Рє РєРѕСЂСЂРѕР·РёРё Рё отличная эластичность делают этот материал идеальным для различных отраслей. Предлагаем широкий ассортимент продукции Рё РїРѕРґСЂРѕР±РЅСѓСЋ информацию для правильного выбора.
MartinSef
| #
Официальный сайт 1win http://1win.kykyryza.ru ставки на спорт, киберспорт, казино, live-игры и слоты от лучших провайдеров. Моментальные выплаты, круглосуточная поддержка, щедрые акции и удобное мобильное приложение. Делай ставки и играй в казино на 1win — быстро, безопасно и выгодно!
w69 com
| #
ดาวน์โหลดแอป w69 com – https://lvm60.com วันนี้ รับโปรโมชั่นพิเศษมากมาย ใช้งานสะดวก ปลอดภัย
และรองรับทุกอุปกรณ์
Diplomi_bfKt
| #
Купить диплом о высшем образовании!
Мы готовы предложить документы университетов, расположенных на территории всей РФ.
diplomk-vo-vladivostoke.ru/bistroe-i-nadezhnoe-vnesenie-diploma-v-reestr-2/
RogerDup
| #
Swiat emocji z 1win https://1win-pl.com Zaklady sportowe i e-sportowe, kasyno online, poker, gry wirtualne i wiele wiecej. Szybka rejestracja, bonus powitalny i natychmiastowa wyplata wygranych. 1win – wszystko, czego potrzebujesz do gry w jednym miejscu!
Lazrgsz
| #
Диплом любого ВУЗа России!
Без наличия диплома очень сложно было продвинуться вверх по карьерной лестнице. Поэтому решение о заказе диплома следует считать рациональным. Приобрести диплом о высшем образовании toolsrepair.ru/forum/viewtopic.php?f=5&t=2977031
สล็อต w69 เว็บตรง
| #
อินเทอร์เฟซใหม่ของแอป
สล็อต w69 เว็บตรง ช่วยให้การนำทางลื่นไหลขึ้น ไม่ต้องเสียเวลาค้นหาเกมโปรดของคุณ
Toliknaf
| #
darknet markets onion address dark web markets
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
Наши товары:
Медный стандартный двухраструбный отвод 90 градусов под пайку 14х10х0.6 мм 10.6х12.6 мм мягкая пайка М3РМ ГОСТ 32590-2013 Покупайте медные двухраструбные отводы 90 градусов под пайку на Редметсплав.рф. Надежные и долговечные отводы из высококачественной меди для систем водоснабжения, отопления и кондиционирования воздуха. Гарантированно надежные соединения, простой монтаж и оптимальное распределение потока.
Xazrhfa
| #
Заказать диплом университета!
Мы изготавливаем дипломы психологов, юристов, экономистов и любых других профессий по доступным ценам. Вы заказываете документ в надежной и проверенной компании. : myworldavto.ru/byistroe-poluchenie-diploma-ot-zakaza-do-gotovnosti
Sazrpto
| #
Мы изготавливаем дипломы любых профессий по приятным тарифам. Преимущества приобретения документов у нас
Вы приобретаете диплом через надежную и проверенную временем компанию. Такое решение сэкономит не только деньги, но и ваше драгоценное время.
На этом преимущества не заканчиваются, их гораздо больше:
• Документы изготавливаем на подлинных бланках со всеми печатями;
• Дипломы любых учебных заведений России;
• Стоимость значительно меньше чем потребовалось бы платить за обучение в ВУЗе;
• Максимально быстрая доставка в любые регионы РФ.
Купить диплом университета– http://msfo-soft.ru/msfo/forum/user/54980/ – msfo-soft.ru/msfo/forum/user/54980
Josephkaw
| #
новости шахматных турниров
MetunwAwalk
| #
На сайте https://xn—-8sbaaajsa0adcpbfha1bdjf8bh5bh7f.xn--p1ai/ ознакомьтесь со всеми вариантами антиквариата, который вы сможете продать по честной стоимости. Станислав Бабкин, являясь частным коллекционером, скупает предметы старины дорого. Оценка происходит в режиме реального времени, а средства выплачиваются на карту либо выдаются наличными. Вы сможете продать следующие вещи: серебро, картины, иконы, золотые, серебряные монеты, ордена, часы, самовары и многое другое. Вся сумма выплачивается полностью во время оформления сделки.
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена требуемыми документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы по мере того как предоставлять решения под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
Пруток молибденовый РњРџР§ Пруток молибденовый РњРџР§ – это высококачественный металл, обладающий уникальными свойствами, такими как высокая температура плавления Рё стойкость Рє РєРѕСЂСЂРѕР·РёРё. Рти характеристики делают его незаменимым РІ различных отраслях, включая производство электроники Рё аэрокосмическую промышленность. Благодаря СЃРІРѕРёРј механическим свойствам, пруток идеально РїРѕРґС…РѕРґРёС‚ для изготовления деталей, подвергающихся значительным нагрузкам. Если РІС‹ хотите обеспечить надежность Рё долговечность СЃРІРѕРёС… изделий, рекомендую купить Пруток молибденовый РњРџР§, чтобы воспользоваться всеми преимуществами этого материала. Работая СЃ молибденом, РІС‹ получаете гарантированно качественный РїСЂРѕРґСѓРєС‚, который оправдает РІСЃРµ ваши ожидания.
casino Vodka официальный
| #
Добро пожаловать в Водка Казино — место, где
восторг сочетается с щедрыми бонусами и
шансами для выигрыша. У нас каждый игрок найдет свои любимые игры, будь то
начинающий или опытный игрок.
Здесь вы не только получите удовольствие, но и невероятные предложения.
Весь ассортимент игр в Водка Казино имеет высокие коэффициенты возврата, что увеличивает
шансы на выигрыш. Мы подготовили интерактивные игровые автоматы
и классические настольные игры, которые делают игровой процесс еще
более занимательным и прибыльным.
Не теряйте время! В Водка Казино вы можете быстро зарегистрироваться и легко пополнить свой счет, не тратя время на лишние шаги,
чтобы начать играть и выигрывать.
Мы всегда готовы предложить вам красивые бонусы для того, чтобы вы могли получить
больше шансов на выигрыш.
Присоединяйтесь к Водка Казино, чтобы погрузиться в мир азартных игр с высочайшими шансами на успех!
Мгновенная регистрация.
Привлекательные бонусы для новичков.
Регулярные турниры и акции для тех, кто
хочет повысить свои шансы на победу.
Круглосуточная поддержка, чтобы помочь вам в любой момент.
Играйте в любое время и в любом месте.
Погрузитесь в азартный мир Водка
Казино и проверьте свою удачу! https://vodka-casinosphere.space/
Rabyitabe
| #
darknet markets 2025 dark market
Robertniday
| #
Все о ремонте и строительстве в ежедневных актуальных статьях. Полезные советы по ремонту, лайфхаки, интересные статьи о даче и дизайне https://premierdevelopment.ru/
Pingrutty
| #
dark web market links darknet websites
Uazrckk
| #
Доброго времени суток!
Для многих людей, заказать диплом о высшем образовании – это необходимость, шанс получить отличную работу. Но для кого-то – это желание не терять время на учебу в универе. Что бы ни толкнуло вас на такой шаг, мы готовы помочь вам. Быстро, профессионально и выгодно изготовим документ нового или старого образца на подлинных бланках с реальными подписями и печатями.
Ключевая причина, почему люди покупают документы, – желание занять хорошую должность. Допустим, способности и опыт позволяют специалисту устроиться на работу, но подтверждения квалификации нет. Когда для работодателя важно наличие “корочек”, риск потерять место работы достаточно высокий.
Приобрести документ о получении высшего образования вы имеете возможность у нас в Москве. Мы предлагаем документы об окончании любых университетов России. Вы сможете получить необходимый диплом по любым специальностям, любого года выпуска, включая документы образца СССР. Гарантируем, что при проверке документа работодателями, подозрений не появится.
Обстоятельств, которые вынуждают купить диплом ВУЗа немало. Кому-то срочно требуется работа, а значит, нужно произвести впечатление на руководителя на протяжении собеседования. Другие планируют устроиться в большую компанию, для того, чтобы повысить свой статус и в будущем начать свой бизнес. Чтобы не тратить драгоценное время, а сразу начать успешную карьеру, используя врожденные таланты и приобретенные знания, можно заказать диплом через интернет. Вы станете полезным в обществе, обретете финансовую стабильность в кратчайшие сроки- диплом купить о среднем образовании
Lazropk
| #
Где приобрести диплом специалиста?
Купить диплом института по выгодной стоимости возможно, обратившись к проверенной специализированной компании.: kupite-diplom0024.ru
WilliamDix
| #
Мы изготавливаем дипломы любой профессии по приятным ценам. Дипломы производят на фирменных бланках государственного образца Приобрести диплом университета diplomd-magazinp.ru
Sazrkht
| #
Заказать диплом института по доступной цене возможно, обратившись к надежной специализированной фирме. Мы оказываем услуги по продаже документов об окончании любых университетов Российской Федерации. Заказать диплом ВУЗа– rusd-diplomj.ru/kupite-diplom-visshego-obrazovaniya-s-zaneseniem/
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
поставляемая продукция:
Титановая труба 23х1.6х3000 мм ПТ7М ГОСТ 22897-86 Откройте мир прочных и надежных титановых труб с компанией Редметсплав. Наши высококачественные титановые трубы сочетают в себе устойчивость к коррозии, износостойкость и легкий вес, делая их незаменимым материалом для применения в различных сферах промышленности. Широкий выбор различных диаметров и толщин, а также индивидуальные решения для вашего производства. Гарантированное качество и надежность поставок.
Sazrugl
| #
Купить документ университета вы имеете возможность в нашем сервисе. Мы оказываем услуги по продаже документов об окончании любых университетов России. Вы сможете получить диплом по любой специальности, включая документы образца СССР. Даем гарантию, что в случае проверки документа работодателем, подозрений не появится. diplomgorkiy.com/kupit-diplom-s-reestrom-bistro-i-udobno-3/
DonDonfaita
| #
darknet markets 2025 darknet market lists
SejehTot
| #
Welcome to the official Joy Casino blog https://officialjoycasino.net/ — a place where you can explore the fascinating world of online gambling from different perspectives: game mechanics, psychology, responsible gaming and industry analytics. If you are looking for a deep dive into gaming strategies, casino technology or responsible gaming tips, you have come to the right place.
Tuhonzoobers
| #
https://topsale.md/ – ваш надежный онлайн-гипермаркет в Молдове! Здесь вы найдете бытовую технику, электронику, товары для дома, детей и животных по выгодным ценам. Быстрая доставка по всей стране, удобные способы оплаты и акции для покупателей! Покупайте легко, экономьте время и деньги с TOPSALE.MD – ассортимент, который вам нужен, в одном месте!
Bahaxeldug
| #
На сайте https://pohudetlegko.ru/ представлена исчерпывающая, актуальная и полезная информация о том, как похудеть правильно, без вреда для организма и максимально эффективно. На сайте вы найдете вдохновляющие истории, советы звезд, которые выглядят младше своего возраста. Есть рецепты блюд, которые созданы из полезных, вкусных ингредиентов. Но при этом они помогут скинуть вес и привести себя в форму к летнему сезону. Рецепты простые и быстрые, а потому справиться с приготовлением пищи сможет каждый. Описаны и упражнения, обертывания для похудения.
1win_dvot
| #
один вин официальный сайт https://alfatraders.borda.ru/?1-0-0-00004932-000-0-0-1743258210 .
Donaldlaf
| #
darknet drug store darknet market list
Ronaldbut
| #
https://mycofarm.ru/
1win_ixOi
| #
1вин партнерка balashiha.myqip.ru/?1-12-0-00000437-000-0-0-1743258848 .
Toliknaf
| #
dark web drug marketplace dark market list
Pingrutty
| #
darknet market list dark web market urls
zaimpodptsmsksig
| #
деньги под залог автомобиля авто остается
avtolombard-pid-zalog-pts.ru
деньги под птс авто
Diplomi_poKt
| #
Заказать диплом о высшем образовании!
Мы предлагаем документы институтов, которые расположены в любом регионе Российской Федерации.
good-diplom.ru/kupit-diplom-s-zaneseniem-v-reestr-rossii-vigodno-3/
Rabyitabe
| #
dark market darkmarket
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наши товары:
Латуное наружное стопорное пружинное кольцо 88С…3 РјРј ЛС59 ГОСТ 13942-86 Купить латунные наружные стопорные кольца РїРѕ ГОСТ РІ Р РѕСЃСЃРёРё. Надежная фиксация элементов конструкции. Высокое качество РїРѕ доступной цене. Рспользуются РІ строительстве, машиностроении Рё производстве. Заказывайте сейчас РЅР° сайте Редметсплав.
DonDonfaita
| #
dark web sites onion dark website
Jameszem
| #
Актуальные мировые новости о политике, обществе, экномике, бизнесе, медицине и других отраслях. Ежеденевная аналитическая информация в нашем блоге https://tovarlive.ru/
1win_suMl
| #
1 win pro 1 win pro .
Donaldlaf
| #
dark market url dark web market
Pingrutty
| #
darknet drug links darknet site
1win_voot
| #
1 вин вход 1 вин вход .
WilliamFum
| #
Ты игрок в CS2 или КС ГО? Если тебе надоели твои обыные скины и ты хочешь продать их и вывести деньги себе на карту, то для этого существуют специальные сайты, которые помогают с этим. ТУТ: вывод скинов кс ты сможешь реализовать свои скины вывести деньги на карту в течение 10 минут.
Sazrvfl
| #
Заказать диплом университета !
Приобретение диплома любого университета России в нашей компании является надежным процессом, ведь документ будет заноситься в реестр. Печать выполняется на официальных бланках ГОЗНАКа. Купить диплом об образовании arenadiplom24.online/vuzy/lipetskogo-filiala-immf
Toliknaf
| #
dark market bitcoin dark web
Sazrsjl
| #
Задался вопросом: можно ли на самом деле купить диплом государственного образца в Москве? Был приятно удивлен — это реально и легально!
Сначала искал информацию в интернете на тему: купить диплом о высшем в тюмени, купить диплом о среднем образовании в новокузнецке, купить диплом строительного техникума, купить дипломы о высшем образовании в тюмени, петербург купить диплом о высшем образовании и получил базовые знания. В итоге остановился на материале: diplomybox.com/kupit-diplom-bakalavra-v-novokuznetske
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наши товары:
Латуное внутреннее стопорное пружинное кольцо 48х1.7 мм ЛЦ40С ГОСТ 13943-86 Купить латунные внутренние стопорные кольца по ГОСТ от производителя. Надежное крепление и удержание элементов механизмов. Широкий выбор размеров и конфигураций. Высокое качество и надежность. Применение в машиностроении, автомобильной и других отраслях. Обращайтесь к нашим специалистам для получения более подробной информации.
Rabyitabe
| #
dark websites best darknet markets
Ronaldbut
| #
https://mycofarm.ru/
Diplomi_ulka
| #
купить аттестат за 10 купить аттестат за 10 .
Sazrdgv
| #
Мы можем предложить дипломы психологов, юристов, экономистов и других профессий по невысоким ценам.– diplomc-v-ufe.ru/kupit-diplom-o-visshem-obrazovanii-reestr-7/
1win_fyOi
| #
1win прямой эфир 1win прямой эфир .
DonDonfaita
| #
dark web market links darknet market list
Cazrpkd
| #
Приобрести диплом любого ВУЗа!
Мы предлагаем документы институтов, которые расположены в любом регионе России. Дипломы и аттестаты делаются на бумаге самого высокого качества: s-klinika.ru/js/pgs/kupit_diplom_o_srednem_obrazovanii_30.html
Sazrxpi
| #
Добрый день!
Приобрести диплом университета по невысокой цене возможно, обратившись к проверенной специализированной компании. Купить диплом о высшем образовании: diplom-zentr.com/kupit-diplom-kemerovo-3/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наши товары:
Высокочистая металлическая мишень для распыления кремния
Pingrutty
| #
dark web drug marketplace darknet marketplace
Oscarciz
| #
Lause — fatalne miejsce do stawiania zakladow! Zgarnac nagrode jest szansa, ale uzyskac wygrana — granica nie do przekroczenia. Po uzyskaniu duzej glownej nagrody po prostu zamrozili moje konto bez zadnej informacji. Obsluga klienta milczy, a na zapytania przesylaja szablonowe odpowiedzi. Przeczytalem opinie — takich zawiedzionych sa setki! Jesli nie planujesz zmarnowac oszczednosci, omijaj szerokim lukiem na te strone!
zaimpodptsnovosibsig
| #
деньги в кредит под залог машины
avtolombard-pid-zalog-pts.ru/nsk.html
кредит под залог машины птс
Donaldlaf
| #
dark web market list darknet markets links
Toliknaf
| #
darknet sites darkmarket 2025
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их качество. Опытная поддержка – наш стандарт – мы на связи, чтобы разрешать ваши вопросы и адаптировать решения под требования вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
РљСЂСѓРі магниевый AZ91HP Фольга магниевая AZ91HP представляет СЃРѕР±РѕР№ легкий Рё прочный материал, идеально подходящий для различных промышленных Рё бытовых нужд. РћРЅР° обладает высокой РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью Рё отличной пластичностью, что делает ее подходящей для изготовления сложных деталей Рё конструкций. Благодаря СЃРІРѕРёРј характеристикам, фольга широко применяется РІ авиастроении, автомобильной промышленности Рё домашних проектах. Если РІС‹ хотите обеспечить надежность Рё прочность СЃРІРѕРёС… изделий, стоит купить Фольга магниевая AZ91HP. Ртот товар станет отличным выбором для профессионалов Рё любителей.
Rabyitabe
| #
darknet market darknet markets
Pingrutty
| #
darknet drug links darknet sites
DonDonfaita
| #
darkmarket link dark web sites
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наша продукция:
Однораструбный медный РѕР±РІРѕРґ РїРѕРґ пайку 22С…18С…0.9 РјРј 8С…10 РјРј твердая пайка Рњ1Рњ ГОСТ Р 52922-2008 Выбирайте высококачественные медные однораструбные РѕР±РІРѕРґС‹ РїРѕРґ пайку для надежных соединений. Рзготовлены РёР· меди высокой чистоты СЃ отличной теплопроводностью Рё устойчивостью Рє РєРѕСЂСЂРѕР·РёРё. Рдеальны для систем водоснабжения Рё отопления.
JuwadTib
| #
Спецкор Дніпро: Новини міста Дніпра і Дніпропетровської області – https://spetskor.dp.ua/ . Актуальні новини Дніпра і Дніпропетровської області. Довідкова, та корисна інформація про місто.
Ronaldbut
| #
https://mycofarm.ru/
MatthewMAind
| #
Иногда не хочется шума — просто открыть игру и разгрузить голову.
Платформа интуитивный, ничего не мешает. Выбор игр приличный, можно сменить жанр под настроение.
Депозиты заходят сразу, выплаты приходят нормально. Саппорт на связи — без лишней воды.
В целом, Игровые автоматы казино — это именно то, что надо, когда хочется просто поиграть без напряга.
Cazrwsn
| #
Здравствуйте!
Мы предлагаем дипломы любой профессии по приятным тарифам. Стоимость зависит от определенной специальности, года выпуска и ВУЗа: rdiplomm24.com/
arenda lofta v spb_bzMi
| #
лофты спб аренда для вечеринки http://www.snyat-loft-v-spb.ru .
detskaya odejda s nadpisyami_xuma
| #
одежда с надписями https://dbkids.ru/ .
Eanruve
| #
Для успешного продвижения вверх по карьерной лестнице понадобится наличие диплома о высшем образовании. Заказать диплом о высшем образовании у надежной организации: diplomg-kurerom.ru/gde-kupit-attestati-za-11-klass-nedorogo-i-bistro/
Donaldlaf
| #
tor drug market darknet markets onion address
augmented reality platform_wkSi
| #
ar gallery augmented-reality-platform.ru .
Sazraev
| #
Мы изготавливаем дипломы любой профессии по приятным тарифам. Основные преимущества заказа документов в нашем сервисе
Вы заказываете документ в надежной и проверенной компании. Такое решение позволит вам сэкономить не только много средств, но и ваше драгоценное время.
Плюсов гораздо больше:
• Документы изготавливаем на оригинальных бланках с печатями и подписями;
• Дипломы любого ВУЗа России;
• Стоимость значительно меньше той, которую понадобилось бы платить за обучение в университете;
• Доставка в любые регионы РФ.
Купить диплом о высшем образовании– http://sakhd.3nx.ru/viewtopic.php?p=3259#3259/ – sakhd.3nx.ru/viewtopic.php?p=3259#3259
Toliknaf
| #
darknet links onion dark website
Xazrgkj
| #
Заказать диплом о высшем образовании!
Мы изготавливаем дипломы любой профессии по приятным ценам. Вы заказываете диплом через надежную компанию. : matchfishing.ru/forumtest/1/member.php?u=23110
KevinKaf
| #
https://provereno-rf.ru/
Uazrkca
| #
Приветствую!
Для определенных людей, купить диплом о высшем образовании – это необходимость, уникальный шанс получить достойную работу. Но для кого-то – это желание не терять время на учебу в ВУЗе. С какой бы целью вам это не понадобилось, наша компания готова помочь вам. Быстро, профессионально и по доступной цене изготовим документ нового или старого образца на государственных бланках с реальными печатями.
Главная причина, почему люди покупают документы, – желание занять хорошую должность. Например, знания позволяют кандидату устроиться на работу, а документального подтверждения квалификации нет. В том случае если работодателю важно наличие “корочек”, риск потерять место работы очень высокий.
Заказать документ института можно в нашей компании. Мы предлагаем документы об окончании любых университетов России. Вы сможете получить необходимый диплом по любой специальности, включая документы Советского Союза. Даем 100% гарантию, что в случае проверки документа работодателем, никаких подозрений не возникнет.
Ситуаций, которые вынуждают приобрести диплом о среднем образовании очень много. Кому-то очень срочно нужна работа, и нужно произвести особое впечатление на руководителя при собеседовании. Другие желают устроиться в престижную компанию, для того, чтобы повысить собственный статус и в будущем начать свой бизнес. Чтобы не тратить драгоценное время, а сразу начать успешную карьеру, используя врожденные таланты и приобретенные навыки, можно купить диплом через интернет. Вы станете полезным в обществе, обретете денежную стабильность очень быстро и просто- купить аттестат
Pingrutty
| #
dark markets dark market link
Rabyitabe
| #
dark web market darknet drug store
ScottWeish
| #
Игровое заведение Вулкан – это захватывающий мир азарта, который поражает с первого взгляда. Благодаря прекрасному дизайну и интуитивно понятному интерфейсу, пользователи быстро оказываются в этом игровом пространстве.
Среди плюсов казино игровые автоматы следует отметить широкий ассортимент игр, включая слоты и настольные азартные игры, которые придутся по вкусу каждому игроку. Качество графики и анимации делает игровой процесс дополнительно увлекательным и завораживающим.
Однако больше всего поразил подход казино к безопасности игроков. Они используют новейшие технологии шифрования, чтобы обеспечить приватность личных и финансовых данных пользователей. Это делает казино Вулкан отличным выбором для онлайн-игры.
CumowBaify
| #
On the website https://za-forex.com/ you will find information about blockchain technology. The blog of an experienced mathematician, the one who accepted bitcoin and blockchain technology, realizing the potential of digital assets long before mainstream investors paid attention to it. He shares his knowledge to develop automated trading algorithms and risk models that can bring you profit.
KevinPiorn
| #
https://corfu-tours.ru/
zaimpodzalogptsekbsig
| #
взять деньги под птс
zaim-pod-zalog-pts1.ru/ekb.html
взять займ под залог птс
DonDonfaita
| #
darknet markets onion address dark web drug marketplace
arenda lofta v spb_ftMi
| #
аренда лофт аренда лофт .
detskaya odejda s nadpisyami_cpma
| #
одежда с надписями брендов http://dbkids.ru/ .
1win_ovpl
| #
1win казино http://1win6050.ru .
augmented reality platform_luSi
| #
augmented reality story augmented reality story .
hatori-bet
| #
hatori-bet
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Керамическая мишень для распыления РѕРєСЃРёРґР° олова – 4N, Плоская форма
2-bet
| #
2-bet
blogcaree
| #
Подробнее, читайте в блоге – ссылка
Donaldlaf
| #
bitcoin dark web https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark web marketplaces
rezeki-bet
| #
rezeki-bet
1win_joEa
| #
1win казино http://www.1win6003.ru .
nissin-bet
| #
nissin-bet
mostbet_oiSr
| #
мостбет кыргызстан https://mostbet6008.ru/ .
arti-bet
| #
arti-bet
saldo-bet
| #
saldo-bet
mazda-bet
| #
http://hornoscasaemilio-valoriani.com/br/index.php?id=mazda-bet
Mazrcec
| #
Где заказать диплом специалиста?
Мы оказываем услуги по производству и продаже документов об окончании любых университетов России. Документы изготавливаются на подлинных бланках государственного образца. Рєmagnusrecruitment.com.au/employer/premialnie-diplom-24
Toliknaf
| #
onion dark website https://github.com/abacuslink4jjku/abacuslink – dark web market links
Pingrutty
| #
dark markets https://github.com/aresonioncq0a7/aresonion – dark web market list
Mazrmro
| #
Где купить диплом специалиста?
Готовый диплом с приложением 100% отвечает стандартам, никто не отличит его от оригинала. Не откладывайте свои мечты на несколько лет, реализуйте их с нашей компанией – отправьте заявку на изготовление диплома уже сегодня! Диплом о среднем образовании – не проблема! bisound.com/forum/showthread.phpp=1849429#post1849429
arenda lofta v spb_cxMi
| #
аренда лофтов спб аренда лофтов спб .
detskaya odejda s nadpisyami_iqma
| #
детская одежда с надписями http://www.dbkids.ru .
augmented reality platform_wpSi
| #
turn photo into augmented reality http://www.augmented-reality-platform.ru .
Rabyitabe
| #
darknet drugs https://github.com/darkwebsitesyhshv/darkwebsites – dark web sites
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог высококачественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы по мере того как предоставлять решения под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
поставляемая продукция:
Фольга магниевая Р›2 – РўРЈ 1714-002-00545484-99 РўСЂСѓР±Р° магниевая Р›2 – РўРЈ 1714-002-00545484-99 является РѕРґРЅРёРј РёР· самых надежных материалов для различных отраслей. РћРЅР° обладает отличной прочностью Рё лёгким весом, что делает её идеальной для применения РІ строительстве Рё промышленности. Данная труба изготовлена СЃ соблюдением строгих стандартов, что гарантирует её качество Рё долговечность. Если РІС‹ ищете эффективное решение для вашего проекта, купить РўСЂСѓР±Р° магниевая Р›2 – РўРЈ 1714-002-00545484-99 – отличный выбор. РћРЅР° идеально РїРѕРґС…РѕРґРёС‚ для создания конструкций, требующих высокой устойчивости.
rival-bet-303
| #
http://pribehyfotek.cz/br/index.php?id=rival-bet-303
DonDonfaita
| #
dark market list https://github.com/nexusdarkrtv1u/nexusdark – dark websites
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние достижения.
Наши товары:
Металлическая мишень РРЅРґРёСЏ 5N плоская для распыления
Sazrdbg
| #
Приобретение документа о высшем образовании через проверенную и надежную компанию дарит ряд достоинств. Это решение помогает сберечь как длительное время, так и значительные финансовые средства. Тем не менее, плюсов гораздо больше.Мы предлагаем дипломы любой профессии. Дипломы производят на оригинальных бланках государственного образца. Доступная цена по сравнению с крупными издержками на обучение и проживание. Покупка диплома института будет выгодным шагом.
Приобрести диплом о высшем образовании: diplomgorkiy.com/bistraya-pokupka-diplomov-s-reestrom-legko-i-udobno/
betflix
| #
I don’t even know the way I finished up right here, but I assumed this put up was good.
I don’t recognize who you’re but definitely you are going to a famous blogger for those who are not already.
Cheers!
blogcaree
| #
Подробнее, читайте в блоге – ссылка
1win_kgEa
| #
1win.online 1win6003.ru .
Pingrutty
| #
darkmarkets https://github.com/aresonioncq0a7/aresonion – onion dark website
Donaldlaf
| #
dark web market links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark market 2025
mostbet_naSr
| #
скачать mostbet http://www.mostbet6008.ru .
Toliknaf
| #
darknet markets https://github.com/abacuslink4jjku/abacuslink – dark market list
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние возможности.
Наши товары:
Высокочистый тигель из вольфрама
zaimpodptskazancoN
| #
кредит под птс
avtolombard-pid-zalog-pts.ru/kazan.html
займ под залог птс авто
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – наш стандарт – мы на связи, чтобы ответить на ваши вопросы по мере того как адаптировать решения под особенности вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
Молибден МЧВП Молибден МЧВП — это высококачественный металл, востребованный в различных отраслях благодаря своим уникальным свойствам. Он обладает высокой температурной устойчивостью и прочностью, что делает его идеальным выбором для производства комплектующих в атомной, аэрокосмической и медной промышленности. Если вы ищете надежный материал, который обеспечит долговечность и эффективность в работе, то стоит купить Молибден МЧВП. Не упустите возможность пополнить склад данным уникальным продуктом, который станет залогом вашего успеха. Обращайтесь к нам, чтобы узнать о доступных вариантах и ценах.
Jamescoaft
| #
Find the Perfect Clock http://www.clocks-top.com for Any Space! Looking for high-quality clocks? At Top Clocks, we offer a wide selection, from alarm clocks to wall clocks, mantel clocks, and more. Whether you prefer modern, vintage, or smart clocks, we have the best options to enhance your home. Explore our collection and find the perfect timepiece today!
Sazrynr
| #
Заказать документ о получении высшего образования можно в нашей компании. Мы оказываем услуги по изготовлению и продаже документов об окончании любых ВУЗов РФ. Вы сможете получить необходимый диплом по любым специальностям, любого года выпуска, в том числе документы Советского Союза. Даем 100% гарантию, что при проверке документов работодателями, никаких подозрений не возникнет. asxdiplommy.com/kupit-diplom-s-zaneseniem-v-reestr-po-nizkoj-tsene-4/
Rabyitabe
| #
dark web marketplaces https://github.com/darkwebsitesyhshv/darkwebsites – darknet websites
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние возможности.
Наши товары:
Распыляемая мишень из эрбия, плоская
1win_teEa
| #
1wi http://1win6003.ru .
1win_hbpl
| #
1win кейсы http://www.1win6050.ru .
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
поставляемая продукция:
Мишень РёР· драгоценного металла или его сплава кольцевая золото 200С…2 РјРј ЗлСрМ95-2.5 РўРЈ Выберите уникальные мишени РёР· драгоценных металлов для напыления РѕС‚ Редметсплав.СЂС„. Обеспечивают высокую степень защиты Рё долговечность. Рдеальный выбор для промышленности, строительства Рё производства.
NurayVoing
| #
Компания «КАРДЕКС» https://card-oil.ru/ — надежный партнер на рынке топливных решений, работающий более 12 лет. Специализируется на выпуске и обслуживании топливных карт для юридических лиц и индивидуальных предпринимателей. Преимущество КАРДЕКС — доступ к обширной сети из более чем 13000+ АЗС по всей России, включая федеральные и региональные заправочные станции. Это позволяет клиентам заправляться без привязки к конкретному бренду, оптимизируя расходы и логистику. Топливные карты КАРДЕКС обеспечивают удобный контроль затрат, предоставляют детальную аналитику и помогают экономить за счет персонализированных условий и скидок. Компания зарекомендовала себя как стабильный и технологичный игрок, предлагающий инновационные решения для бизнеса.
vavadaukr__jrPr
| #
Добро пожаловать на vavadaukr.kiev.ua, полезные ресурсы.
лучшие советы, для того чтобы.
Познакомьтесь с vavadaukr.kiev.ua, доступны.
текущих событиях.
инсайдерская информация.
обширную информацию о.
Развивайтесь вместе с vavadaukr.kiev.ua, идеями.
vavadaukr.kiev.ua предоставляет, которые.
На сайте vavadaukr.kiev.ua вы увидите, новыми ресурсами.
С vavadaukr.kiev.ua вы открываете.
vavadaukr.kiev.ua – ваш надежный партнер, открывать новое.
С vavadaukr.kiev.ua вы имеете, обогатит ваш опыт.
Сайт vavadaukr.kiev.ua уникален тем, все самое лучшее.
поиска знаний.
vavadaukr.kiev.ua – это площадка для, обсуждения.
Узнайте о возможностях vavadaukr.kiev.ua, которые.
Как vavadaukr.kiev.ua может помочь вам, вызывая интерес.
vavadaukr.kiev.ua vavadaukr.kiev.ua .
DonDonfaita
| #
darknet marketplace https://github.com/nexusdarkrtv1u/nexusdark – dark web link
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Керамическая мишень Zno 3N5
Pingrutty
| #
onion dark website https://github.com/aresonioncq0a7/aresonion – darknet market links
mostbet_fzSr
| #
aviator mostbet http://www.mostbet6008.ru .
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – наш стандарт – мы на связи, чтобы разрешать ваши вопросы и предоставлять решения под специфику вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Рзделия РёР· вольфрама W-Al Рзделия РёР· вольфрама W-Al представляют СЃРѕР±РѕР№ высококачественные компоненты, изготовленные РёР· уникального сплава вольфрама. Рти изделия идеально РїРѕРґС…РѕРґСЏС‚ для применения РІ различных отраслях, включая машиностроение Рё электронику. РС… высокая прочность Рё стойкость Рє температурам делают РёС… незаменимыми РІ условиях жесткой эксплуатации. РњС‹ предлагаем широкий ассортимент изделий, что позволяет выбрать именно то, что вам нужно. Если РІС‹ ищете надежные Рё долговечные решения, вам стоит купить Рзделия РёР· вольфрама W-Al. Убедитесь РІ РёС… превосходных характеристиках Рё высоком качестве. Приобретите РёС… уже сегодня!
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их качество. Дружелюбная помощь – наш стандарт – мы на связи, чтобы улаживать ваши вопросы а также находить ответы под требования вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Рлектрод вольфрамовый Р’Р§-1 Рлектрод вольфрамовый Р’Р§-1 — это важный элемент для высокочастотной сварки. РћРЅ обеспечивает надежный процесс Рё высокое качество соединения. РЎ помощью этого электрода можно добиться отличных результатов даже РїСЂРё сложных условиях работы. Если вам нужно купить Рлектрод вольфрамовый Р’Р§-1, РјС‹ предлагаем широкий выбор продуктов РїРѕ конкурентным ценам. Обратите внимание РЅР° его долговечность Рё надежность РІ эксплуатации. Рнвестируйте РІ качество — ваш проект этого заслуживает!
1win_efmr
| #
сайт 1win obovsem.myqip.ru/?1-9-0-00000059-000-0-0-1743051936 .
유로247주소
| #
유로247주소
A Brief History of Elemental Analysis » Artemis Analytical
Toliknaf
| #
darknet websites https://github.com/abacuslink4jjku/abacuslink – dark websites
mostbet_ixmt
| #
мостюет https://svstrazh.forum24.ru/?1-18-0-00000136-000-0-0-1743260517 .
Williamzig
| #
pop over to these guys
Get SMS online
슬롯사이트
| #
슬롯사이트
A Brief History of Elemental Analysis » Artemis Analytical
JasonRom
| #
selector casino
MichaelPiEft
| #
Калининград куршская коса экскурсии http://www.kurshskaya-kosa-ekskursii.ru
sikiş
| #
sikiş
A Brief History of Elemental Analysis » Artemis Analytical
Rutusssycle
| #
На сайте https://italsvet.ru/shop/CID_1.html изучите большой выбор изысканных и роскошных люстр популярной марки «ARTBRONZE». Это оптимальное решение для тех, кто хочет ежедневно наслаждаться красивым стилем, дизайном, а также привлекательным, теплым и приятным светом. Каждое изделие выполнено в особом и уникальном стиле, а потому обязательно впишется в любую концепцию. Все изделия произведены из практичных и надежных материалов, поэтому не потеряют своей привлекательности. Приобрести люстру получится в пару кликов.
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица товара подтверждена требуемыми документами, подтверждающими их происхождение. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы по мере того как предоставлять решения под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
поставляемая продукция:
Проволока магниевая EN-MB10030 – EN 12421:1998 Полоса магниевая EN-MB10030 – EN 12421:1998 представляет СЃРѕР±РѕР№ высококачественный материал, используемый РІ различных отраслях, включая авиацию Рё автомобилестроение. Ртот РїСЂРѕРґСѓРєС‚ отличается отличной прочностью Рё легкостью, что делает его идеальным выбором для конструктивных элементов. Убедитесь РІ надежности Рё стойкости полосы магниевой РїСЂРё использовании РІ различных условиях. Если РІС‹ ищете эффективное Рё надежное решение, то купить Полоса магниевая EN-MB10030 – EN 12421:1998 будет отличным вкладом РІ ваш проект. РќРµ упустите возможность приобрести этот уникальный материал для СЃРІРѕРёС… нужд!
wbc247사이트
| #
wbc247사이트
A Brief History of Elemental Analysis » Artemis Analytical
Rabyitabe
| #
darkmarket url https://github.com/darkwebsitesyhshv/darkwebsites – darkmarket link
turk porno
| #
turk porno
A Brief History of Elemental Analysis » Artemis Analytical
에그벳주소
| #
에그벳주소
A Brief History of Elemental Analysis » Artemis Analytical
Jamesdusty
| #
Ваши флористы понимают с полуслова – описали идею, и её воплотили!
цветы
Pingrutty
| #
darknet markets links https://github.com/aresonioncq0a7/aresonion – dark web market urls
Bosoxltut
| #
На сайте https://autopiter.kg/ получится приобрести как оригинальные, так и неоригинальные запчасти, комплектующие для ТО, автохимию и автомасла, аккумуляторы, все для спецтехники. Представлен и каталог ГАЗ, ВАЗ, КАМАЗ. Есть возможность подобрать фильтр для автомобиля. Также представлены и автолампочки. Изучите раздел с рекомендуемыми товарами. Для того чтобы подобрать необходимую деталь, нужно воспользоваться поиском – в строке указать название детали. Все запчасти хранятся на складе и в нужном количестве.
DonDonfaita
| #
dark web drug marketplace https://github.com/nexusdarkrtv1u/nexusdark – darknet markets onion
forced bi onlyfans
| #
forced bi onlyfans
A Brief History of Elemental Analysis » Artemis Analytical
Sazrbrw
| #
Купить диплом ВУЗа по выгодной цене возможно, обратившись к надежной специализированной компании. Мы оказываем услуги по продаже документов об окончании любых университетов России. Приобрести диплом о высшем образовании– diplomh-40.ru/diplom-s-zaneseniem-v-reestr-po-nizkoj-tsene-2/
Josephkaw
| #
последние спортивные новости
zaimpodptskemerovosig
| #
деньги под залог авто
zaim-pod-zalog-pts1.ru/kemerovo.html
займ под залог птс авто
blogcaree
| #
Подробнее, читайте в блоге – на сайте
Donaldlaf
| #
dark web market links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet links
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние достижения.
Наша продукция:
Керамическая мишень Licoo2 3N, плоская
double website power
| #
double website power
A Brief History of Elemental Analysis » Artemis Analytical
Toliknaf
| #
dark web market links https://github.com/abacuslink4jjku/abacuslink – dark market list
GeorgeTom
| #
Source:
https://smo-nso.ru/
Pingrutty
| #
dark web market links https://github.com/aresonioncq0a7/aresonion – dark market list
Rabyitabe
| #
dark web link https://github.com/darkwebsitesyhshv/darkwebsites – dark web market list
GeraldFup
| #
The new TR Energy https://robotech.com/forums/viewthread/2237212 service aims to optimize both energy and bandwidth usage, solving the problem of high transaction fees that have previously hampered TRON’s scalability.
Start A franchise business
| #
Start A franchise business
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их происхождение. Дружелюбная помощь – наш стандарт – мы на связи, чтобы разрешать ваши вопросы по мере того как адаптировать решения под требования вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наша продукция:
РџРѕРєРѕРІРєР° кобальтовая ЮНДКТ5БА – ГОСТ 17809-72 РџРѕРєРѕРІРєР° кобальтовая ЮНДКТ5БА – ГОСТ 17809-72 представляет СЃРѕР±РѕР№ высококачественный материал, идеально подходящий для создания инструментов Рё деталей, требующих высокой прочности Рё РєРѕСЂСЂРѕР·РёР№РЅРѕР№ стойкости. Рта РїРѕРєРѕРІРєР° обладает отличными механическими свойствами Рё легкостью РІ обработке, что делает ее незаменимой РІ различных отраслях, таких как авиационно-космическая Рё машиностроительная. Если РІС‹ ищете надежное решение для СЃРІРѕРёС… производственных нужд, РЅРµ упустите возможность купить РџРѕРєРѕРІРєР° кобальтовая ЮНДКТ5БА – ГОСТ 17809-72. Ртот товар станет отличным выбором для повышения эффективности вашего производства.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наша продукция:
Распылительная мишень IGZO 4N – Плоская
Richarddig
| #
Ежедневные актуальные новости уходящего дня. Мы говорим о медицине, автопроме, обществе, науке и многом ином https://sp-piter.ru/
유로247가입코드
| #
유로247가입코드
A Brief History of Elemental Analysis » Artemis Analytical
DonDonfaita
| #
darknet marketplace https://github.com/nexusdarkrtv1u/nexusdark – dark web market links
Josephkaw
| #
хоккейные события
Willieavamy
| #
aviator
Willieavamy
| #
sugar rush
porno izle
| #
porno izle
A Brief History of Elemental Analysis » Artemis Analytical
Mazrkhv
| #
Где заказать диплом по нужной специальности?
Мы предлагаем документы об окончании любых ВУЗов РФ. Документы изготавливаются на оригинальных бланках. Рєaboutalltour.ru/diplom-na-zakaz-ofitsialnyie-dokumentyi-pod-lyubyie-nuzhdyi
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
поставляемая продукция:
Никелевая капиллярная трубка 2.4х0.1х5000 мм НМг0.05в ГОСТ 13548-2016 Приобретите никелевые капиллярные трубки от Редметсплав.рф для прочных, высокотехнологичных и надежных решений в различных отраслях.
Mazrznq
| #
Где приобрести диплом специалиста?
Готовый диплом со всеми печатями и подписями полностью отвечает стандартам Министерства образования и науки России, никто не сможет отличить его от оригинала – даже со специальным оборудованием. Не откладывайте свои мечты и цели на продолжительные годы, реализуйте их с нашей помощью – отправьте быструю заявку на изготовление документа сегодня! Диплом о среднем специальном образовании – быстро и легко! apkjobs.site/companies/premialnie-diplom-24
MarkNOlaf
| #
dark market url best darknet markets
betflix
| #
betflix
A Brief History of Elemental Analysis » Artemis Analytical
1win_tbmr
| #
1win ваучер https://www.obovsem.myqip.ru/?1-9-0-00000059-000-0-0-1743051936 .
Albertrog
| #
oxys crema Uruguay
Kxyufaita
| #
darknet links darknet drug store
i thought about this
| #
i thought about this
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их качество. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы и находить ответы под специфику вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
РљСЂСѓРі висмутовый CACIn911 – JIS H 2202-2016 РљСЂСѓРі висмутовый CACIn911 – JIS H 2202-2016 – это уникальный материал, который находит широкое применение РІ различных отраслях. РћРЅ обладает высокой прочностью Рё стабильностью РІ условиях повышенных температур. Ртот висмутовый РєСЂСѓРі идеально РїРѕРґС…РѕРґРёС‚ для сварки Рё пайки, обеспечивая качественный Рё надежный результат.Если РІС‹ ищете надежное решение для вашего проекта, то вам стоит рассмотреть данный РїСЂРѕРґСѓРєС‚. Купить РљСЂСѓРі висмутовый CACIn911 – JIS H 2202-2016 можно РїСЂСЏРјРѕ сейчас, Рё это отличный выбор для профессионалов Рё любителей. Ртот РєСЂСѓРі станет незаменимым инструментом РІ вашей работе.
Franksor
| #
взять займ без процентов
WilliamIRrutty
| #
onion dark website dark market
FurontDus
| #
Looking for flower delivery in Ukraine? Visit https://ukraineflora.com/ where you will find the largest selection of fresh flowers and exclusive gifts. Look through the catalog and you will definitely find not only luxurious flowers and bouquets, but also gift baskets, perfumes, balloons and even cakes.
FNDaviditabe
| #
darkmarket 2025 dark web markets
Willieavamy
| #
sugar rush
Willieavamy
| #
plinko
Nikigboith
| #
На сайте https://antipoligraf.ru/ почитайте про Институт прикладной психофизиологии. Здесь ведется профессиональная, комплексная подготовка к тому, чтобы пройти полиграф. Учат и тому, чтобы быстро, безопасно и комплексно снять стресс при помощи уникальных и инновационных приборов обратной связи. И самое важное, что все это абсолютно безопасно, при этом гарантированно поможет. Кроме того, ведется обучение саморегуляции при помощи обратной связи. У института огромное количество партнеров, которые стараются работать в комплексе.
Willieavamy
| #
mines
webexalinG
| #
Looking for connect crypto exchange on the site? Youtube.com/@Quickex_io, here you will find a video about how the online cryptocurrency exchanger works, how to connect the exchanger to your site and start earning, as well as various videos related to cryptocurrencies and their profitable exchange. You will be able to learn everything about the cryptocurrency market in one place.
mostbet_jhmt
| #
mostbet скачать на телефон бесплатно андроид https://www.svstrazh.forum24.ru/?1-18-0-00000136-000-0-0-1743260517 .
Sazraxh
| #
Покупка документа о высшем образовании через надежную фирму дарит много достоинств. Это решение дает возможность сберечь время и серьезные средства. Однако, преимуществ гораздо больше.Мы можем предложить дипломы любой профессии. Дипломы изготавливаются на настоящих бланках государственного образца. Доступная цена в сравнении с крупными издержками на обучение и проживание в чужом городе. Приобретение диплома университета станет рациональным шагом.
Заказать диплом о высшем образовании: poluchidiplom.com/kupit-diplom-s-zaneseniem-v-reestr-bistro-i-nadezhno-32/
1win_bbmr
| #
1win личный кабинет obovsem.myqip.ru/?1-9-0-00000059-000-0-0-1743051936 .
MarkNOlaf
| #
dark markets dark web marketplaces
Willieavamy
| #
gates of olympus
Willieavamy
| #
mines
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
поставляемая продукция:
Никелевая капиллярная трубка 3.15х0.1х5000 мм НВМг3-0.08в ГОСТ 13548-2016 Приобретите никелевые капиллярные трубки от Редметсплав.рф для прочных, высокотехнологичных и надежных решений в различных отраслях.
blogcaree
| #
Подробнее, читайте в блоге – на сайте
sikis izle
| #
sikis izle
A Brief History of Elemental Analysis » Artemis Analytical
Kxyufaita
| #
darknet websites darknet site
Click here
| #
Click here
A Brief History of Elemental Analysis » Artemis Analytical
Trefolq
| #
Купить документ института вы можете в нашей компании. diplomass.com/kupit-diplom-o-srednem-obrazovanii-22
CarozrdAsymn
| #
На сайте https://bitovayatechnika.blogspot.com/2025/03/blog-post.html изучите полезную, актуальную информацию, которая касается сплит-систем и того, как правильно их выбрать домой либо в офис. Эти функциональные и умные устройства нацелены на то, чтобы поддержать приятную температуру в помещении. Но для того, чтобы воспользоваться всеми функциями, необходимо правильно выбрать устройство. К важным преимуществам такой техники относят то, что она является энергоэффективной, работает практически бесшумно.
Willieavamy
| #
plinko
Ronaldbut
| #
https://mycofarm.ru/
Jariorgok
| #
Где заказать диплом по необходимой специальности?
Наша компания предлагает максимально быстро приобрести диплом, который выполняется на оригинальном бланке и заверен печатями, штампами, подписями. Документ пройдет лубую проверку, даже при помощи профессиональных приборов. Достигайте цели быстро и просто с нашей компанией.
Приобрести диплом университета diplomidlarf.ru/kuplyu-diplom-s-zaneseniem-8/
porna
| #
porna
A Brief History of Elemental Analysis » Artemis Analytical
Sazrwlj
| #
купила диплом как защитить
Albertrog
| #
oxys precio ecuador
WilliamIRrutty
| #
darknet site darknet drug store
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние достижения.
Наша продукция:
Высокочистые гранулы Y2O3
FNDaviditabe
| #
darknet links bitcoin dark web
Cazrvhi
| #
Получить диплом о высшем образовании можем помочь. Купить диплом о высшем образовании в Ставрополе – diplomybox.com/kupit-diplom-o-vysshem-obrazovanii-v-stavropole
Xagipecope
| #
На сайте http://officepro54.ru представлено огромное количество мебели, которая идеально подходит для организации офисного пространства. Вся она выполнена из инновационных, уникальных материалов, а потому прослужит очень долго, не синтезирует в воздух опасных веществ. В разделе вы найдете кабинет руководителя, функциональные и вместительные столы для переговоров, шкафы-купе, сейфы, акустические системы, ученическую мебель и многое другое. Постоянно действуют акции. На продукцию установлены привлекательные цены.
Sazrrnu
| #
Купить документ о получении высшего образования вы имеете возможность у нас. Мы оказываем услуги по продаже документов об окончании любых университетов РФ. Вы получите диплом по любым специальностям, любого года выпуска, в том числе документы старого образца. Даем гарантию, что в случае проверки документов работодателями, каких-либо подозрений не появится. diplomgorkiy.com/kupit-diplom-o-visshem-obrazovanii-otzivi/
Willieavamy
| #
plinko
PrestonWag
| #
Женские хитрости и секреты красоты и моды, психологии отношений и кулинарии. Также наши статьи направлены на родителей и педагогов, интересующихся вопросами воспитания и обучения детей https://mentalar.ru/
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наша продукция:
Латуное наружное стопорное пружинное кольцо M40 36.5С…1.75 РјРј ЛСч DIN 471 Латунные наружные стопорные кольца РїРѕ DIN – широкий выбор высококачественных крепежных изделий РёР· латуни для промышленных Рё строительных применений. Большой ассортимент размеров. Высокая надежность Рё долговечность. Заказывайте РїСЂСЏРјРѕ сейчас РЅР° сайте Редметсплав.
RobertDaw
| #
Non Gamstop casino UK made my day with a quick withdrawal—nice!
non gamstop casinos uk 10 deposit
Willieavamy
| #
plinko
MarkNOlaf
| #
darknet markets onion darknet sites
PrestonWag
| #
Женские хитрости и секреты красоты и моды, психологии отношений и кулинарии. Также наши статьи направлены на родителей и педагогов, интересующихся вопросами воспитания и обучения детей https://mentalar.ru/
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наша продукция:
Заготовка из конструкционной стали листовая 30 мм 45ХН ОСТ 3-1686-90 Выбирайте конструкционные заготовки высокого качества для вашего проекта на Редметсплав.рф. Наш ассортимент включает прочные и долговечные материалы, идеально подходящие для различных областей применения, включая строительство и машиностроение.
Kxyufaita
| #
darknet site darknet markets onion address
internetCah
| #
интернет провайдер екатеринбург
domashij-internet-ekaterinburg001.ru
дешевый интернет екатеринбург
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог высококачественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их качество. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы а также предоставлять решения под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в гибкости нашего предложения
поставляемая продукция:
Проволока магниевая MAG 10 – BS 2970 Полоса магниевая MAG 10 – BS 2970 представляет СЃРѕР±РѕР№ высококачественный металлический РїСЂРѕРґСѓРєС‚, обладающий отличными механическими свойствами Рё РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью. РћРЅР° идеально РїРѕРґС…РѕРґРёС‚ для использования РІ строительстве, Р° также РІ производстве различных конструкций. Рспользование магниевых полос обеспечивает легкость Рё прочность, что делает изделия более надежными Рё долговечными. РќРµ упустите возможность улучшить СЃРІРѕРё проекты! Купить Полоса магниевая MAG 10 – BS 2970 можно РІ нашем интернет-магазине, РіРґРµ РІС‹ найдете только качественные товары РїРѕ лучшим ценам.
WilliamIRrutty
| #
dark market 2025 best darknet markets
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наша продукция:
Лента РёР· прецизионных сплавов для СѓРїСЂСѓРіРёС… элементов 1.4×35 РјРј РРџ414 ГОСТ 14117-85 Купите ленту РёР· прецизионных сплавов для СѓРїСЂСѓРіРёС… элементов РїРѕ выгодной цене РЅР° Редметсплав.СЂС„. Высокая прочность, устойчивость Рє РєРѕСЂСЂРѕР·РёРё Рё отличная эластичность делают этот материал идеальным для различных отраслей. Предлагаем широкий ассортимент продукции Рё РїРѕРґСЂРѕР±РЅСѓСЋ информацию для правильного выбора.
Акне на лице
| #
Акне на лице
A Brief History of Elemental Analysis » Artemis Analytical
FNDaviditabe
| #
darknet drug store bitcoin dark web
Акне фото
| #
Акне фото
A Brief History of Elemental Analysis » Artemis Analytical
Botox от потливости ног
| #
Botox от потливости ног
A Brief History of Elemental Analysis » Artemis Analytical
Willieavamy
| #
pin up
Jariorpsy
| #
Где заказать диплом по необходимой специальности?
Мы предлагаем максимально быстро купить диплом, который выполняется на оригинальной бумаге и заверен мокрыми печатями, водяными знаками, подписями. Данный диплом пройдет лубую проверку, даже при использовании профессиональных приборов. Достигайте своих целей быстро и просто с нашей компанией. Приобрести диплом любого ВУЗа! gess.flybb.ru/viewtopic.php?f=2&t=1175
Iarioreyo
| #
Заказать документ университета вы имеете возможность в нашем сервисе. kidzgetdown.com
Косметика при акне
| #
Косметика при акне
A Brief History of Elemental Analysis » Artemis Analytical
Проблемная кожа акне
| #
Проблемная кожа акне
A Brief History of Elemental Analysis » Artemis Analytical
Lazremo
| #
Заказать диплом о высшем образовании!
Мы предлагаем дипломы любой профессии по разумным тарифам— diplomt-nsk.ru/diplom-kupit-krasnoyarsk-2/
Mazrsys
| #
Мы изготавливаем дипломы любых профессий по выгодным тарифам. Стараемся поддерживать для заказчиков адекватную политику тарифов. Важно, чтобы дипломы были доступными для большого количества наших граждан.
Покупка документа, подтверждающего окончание ВУЗа, – это грамотное решение. Купить диплом о высшем образовании: nsk-diplom.com/kupit-texnicheskij-diplom-6/
augmented reality platform_odSi
| #
augmented reality wallpapers augmented reality wallpapers .
detskaya odejda s nadpisyami_tema
| #
одежда с надписями http://www.dbkids.ru .
arenda lofta v spb_oiMi
| #
аренда лофтов спб аренда лофтов спб .
Ronaldbut
| #
МикоФарм Фунги
MarkNOlaf
| #
dark web market list dark markets
Willieavamy
| #
super hot fruits
Cazrzms
| #
Мы предлагаем дипломы психологов, юристов, экономистов и прочих профессий по приятным тарифам. Купить диплом в городе Люберцы — kyc-diplom.com/geography/lyubercy.html
Причины акне
| #
Причины акне
A Brief History of Elemental Analysis » Artemis Analytical
Jamesgam
| #
Добрый день!
Клининг квартиры в СПб стоимость – это доступные тарифы на уборку жилья. Мы предлагаем генеральную, регулярную и срочную уборку. Наши специалисты работают с современными чистящими средствами. Мы гарантируем чистоту и порядок. Узнайте клининг квартиры в СПб стоимость и закажите услугу.
Влажная уборка офиса – это обязательная услуга для поддержания чистоты на рабочем месте. Мы обеспечим уборку вашего офиса, включая мытье полов и чистку мебели. Влажная уборка офиса с гарантией высокого качества. Наши специалисты используют только безопасные моющие средства и оборудование. Закажите уборку и создайте комфортные условия для работы в своем офисе.
Вся информация на сайте – https://service-cleanspb.ru/
мойка окон и витрин, услуги клининга в спб для квартиры, клининг на дом срочно
клининг спб срочно, клининг спб дешево, срочная уборка
Удачи!
Kxyufaita
| #
darknet drug links dark market
Крем анти акне
| #
Крем анти акне
A Brief History of Elemental Analysis » Artemis Analytical
index backlink free
| #
index backlink free
A Brief History of Elemental Analysis » Artemis Analytical
Dnrtnoi
| #
Где купить диплом специалиста?
Мы предлагаем дипломы психологов, юристов, экономистов и прочих профессий по невысоким тарифам. Мы предлагаем документы ВУЗов, расположенных в любом регионе РФ. Можно приобрести диплом от любого учебного заведения, за любой год, указав подходящую специальность и хорошие оценки за все дисциплины. Документы печатаются на “правильной” бумаге самого высокого качества. Это позволяет делать государственные дипломы, которые не отличить от оригиналов. Документы заверяются всеми необходимыми печатями и подписями. Стараемся поддерживать для заказчиков адекватную политику цен. Для нас важно, чтобы документы были доступны для подавляющей массы граждан. nsk-diplom.com/kupit-diplom-v-permi-8
WilliamIRrutty
| #
onion dark website tor drug market
Willieavamy
| #
plinko
FNDaviditabe
| #
dark market url darknet markets onion address
Willieavamy
| #
how to play aviator game
Против акне
| #
Против акне
A Brief History of Elemental Analysis » Artemis Analytical
Bevaylem
| #
Ищете оборудование из Китая по ценам производителя? Посетите сайт https://totem28.ru/ где вы найдете широкий ассортимент не только оборудования, но и спец техники. Наш каталог постоянно пополняется, мы тщательно отбираем новинки китайского машиностроения. Посмотрите наш каталог и узнайте подробнее на сайте.
xonuCunny
| #
Looking for prediccion del precio de ripple? Тhetradable.com/es/ es un poderoso portal de informacin que le trae a su atencin las ltimas noticias del mundo de los negocios, las criptomonedas, todo sobre Forex y la economa global. Aqu encontrar informacin actualizada sobre criptomonedas, bolsas de valores, metales preciosos como el oro y otros activos clave.
Willieavamy
| #
craps
RobertDaw
| #
Non Gamstop casino UK is super fast—really impressed!
non gamstop boku casino sites
aleyna tilki por
| #
aleyna tilki por
A Brief History of Elemental Analysis » Artemis Analytical
am ylma
| #
am ylma
A Brief History of Elemental Analysis » Artemis Analytical
yaşlı kadın po
| #
yaşlı kadın po
A Brief History of Elemental Analysis » Artemis Analytical
sekse po
| #
sekse po
A Brief History of Elemental Analysis » Artemis Analytical
Бухгалтер удаленно
| #
Бухгалтер удаленно
A Brief History of Elemental Analysis » Artemis Analytical
erotık konulu
| #
erotık konulu
A Brief History of Elemental Analysis » Artemis Analytical
Средство от гипергидроза ног
| #
Средство от гипергидроза ног
A Brief History of Elemental Analysis » Artemis Analytical
Willieavamy
| #
legacy of dead
lizewaded
| #
Сделать мероприятие незабываемым – это главная цель компании АРЕНДА КОМФОРТА. Предлагаем доступные цены и лояльные условия аренды шатров. Готовы помочь с выбором подходящей конструкции. Позвоните нам уже сейчас! xn--https://xn--80aaaaaaxhx2a2ai2af3a8grd.xn--p1ai/ – здесь размещены отзывы наших клиентов, ознакомиться с ними можно в любое время. Если ищете компанию, которая предоставляет высококачественные шатры, то АРЕНДА КОМФОРТА является одним из наилучших выборов. Обращайтесь к нам, мы гарантируем вам отличный уровень обслуживания.
sks vidolari
| #
sks vidolari
A Brief History of Elemental Analysis » Artemis Analytical
türkçe konuşmalı sek
| #
türkçe konuşmalı sek
A Brief History of Elemental Analysis » Artemis Analytical
ücretsiz sek
| #
ücretsiz sek
A Brief History of Elemental Analysis » Artemis Analytical
roket por
| #
roket por
A Brief History of Elemental Analysis » Artemis Analytical
ertk izle
| #
ertk izle
A Brief History of Elemental Analysis » Artemis Analytical
Причины возникновения акне
| #
Причины возникновения акне
A Brief History of Elemental Analysis » Artemis Analytical
por filimler
| #
por filimler
A Brief History of Elemental Analysis » Artemis Analytical
Лечение псориаза таблетками
| #
Лечение псориаза таблетками
A Brief History of Elemental Analysis » Artemis Analytical
Treatment of acne comedones
| #
Treatment of acne comedones
A Brief History of Elemental Analysis » Artemis Analytical
tiürk sikiiş
| #
tiürk sikiiş
A Brief History of Elemental Analysis » Artemis Analytical
MarkNOlaf
| #
darknet drug market darknet markets onion address
internetCah
| #
какие провайдеры на адресе в екатеринбурге
domashij-internet-ekaterinburg002.ru
проверить провайдеров по адресу екатеринбург
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
Наша продукция:
Медный однораструбный отвод 45 градусов под пайку 28х23х0.9 мм 18.4х20.4 мм мягкая пайка Cu-DHP ГОСТ 32590-2013 Приобретите высококачественные Медные отводы 45 градусов под пайку от компании Редметсплав.рф и улучшите вашу трубопроводную систему. Прочные, долговечные и устойчивые к коррозии отводы обеспечат надежное соединение под нужным углом, обеспечивая оптимальное распределение потока и высокую производительность вашей системы.
hikayeler ero
| #
hikayeler ero
A Brief History of Elemental Analysis » Artemis Analytical
Kxyufaita
| #
dark market link darkmarket 2025
Rylonnie shtori s elektroprivodom_lcEa
| #
рулонные электрошторы рулонные электрошторы .
Чистка акне
| #
Чистка акне
A Brief History of Elemental Analysis » Artemis Analytical
porno
| #
porno
A Brief History of Elemental Analysis » Artemis Analytical
porna izleme sitesi
| #
porna izleme sitesi
A Brief History of Elemental Analysis » Artemis Analytical
binance
| #
Can you be more specific about the content of your article? After reading it, I still have some doubts. Hope you can help me.
Willieavamy
| #
Jackpot Raider
augmented reality platform_dcSi
| #
turn photo into augmented reality https://www.augmented-reality-platform.ru .
detskaya odejda s nadpisyami_rsma
| #
детская одежда с надписями https://www.dbkids.ru .
Willieavamy
| #
fortune mouse
WilliamIRrutty
| #
darknet sites darknet market list
Xagipecope
| #
На сайте http://officepro54.ru представлено огромное количество мебели, которая идеально подходит для организации офисного пространства. Вся она выполнена из инновационных, уникальных материалов, а потому прослужит очень долго, не синтезирует в воздух опасных веществ. В разделе вы найдете кабинет руководителя, функциональные и вместительные столы для переговоров, шкафы-купе, сейфы, акустические системы, ученическую мебель и многое другое. Постоянно действуют акции. На продукцию установлены привлекательные цены.
Ботокс в платизму
| #
Ботокс в платизму
A Brief History of Elemental Analysis » Artemis Analytical
Sazrifq
| #
Заказать диплом института по выгодной стоимости возможно, обращаясь к надежной специализированной компании. Мы предлагаем документы об окончании любых ВУЗов РФ. Купить диплом о высшем образовании– poluchidiplom.com/diplom-s-vneseniem-v-reestr-dlya-vashej-budushej-kareri/
Jackpot bet
| #
Jackpot bet
A Brief History of Elemental Analysis » Artemis Analytical
FNDaviditabe
| #
dark web market dark web market urls
arenda lofta v spb_fpMi
| #
аренда лофтов аренда лофтов .
Willieavamy
| #
plinko
turkce altyazılı po
| #
turkce altyazılı po
A Brief History of Elemental Analysis » Artemis Analytical
Richarddig
| #
Ежедневные актуальные новости уходящего дня. Мы говорим о медицине, автопроме, обществе, науке и многом ином https://sp-piter.ru/
https://thietkeinanbanghieu.com
| #
https://thietkeinanbanghieu.com
A Brief History of Elemental Analysis » Artemis Analytical
1win_uoEa
| #
1win kg скачать http://1win6003.ru/ .
pono sks
| #
pono sks
A Brief History of Elemental Analysis » Artemis Analytical
DavidGag
| #
https://farexpo.ru/club/user/40148/forum/message/6217/6254/#message6254 Переезжаете и уже ненавидите этот процесс? Стоп! В этой статье — гайд для тех, кто хочет сделать всё легко: как упаковать вещи за час, перевезти их без повреждений и даже сэкономить. Хотите переехать без лишних заморочек? Тогда вам сюда!
Акне на плечах
| #
Акне на плечах
A Brief History of Elemental Analysis » Artemis Analytical
MarkNOlaf
| #
darknet drug store onion dark website
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наши товары:
Быстрорежущая пластина форма 50 номер 5004 левая 12С…16С…6 РјРј сечение резца 25×16 РјРј Р 2РђРњ9Рљ5 ГОСТ 2379-77 Выберите лучшие быстрорежущие пластины Рє резцам онлайн. Отличная износостойкость, высокая точность обработки Рё широкий выбор размеров. Узнайте, как наши пластины РјРѕРіСѓС‚ улучшить вашу металлообработку.
FrankMam
| #
Твой предпочтение для азартных игр – казино Слотозал!
Слотозал предлагает широкий набор игровых автоматов, выгодные бонусные программы и высокий уровень безопасности. Интерфейс веб-сайта интуитивно понятен, навигация удобна, что дает возможность скоро обнаружить любимые игры. Команда поддержки постоянно готова помочь с всеми вопросами. Зарегистрируйтесь на основном веб-сайте https://slotozal56.com/ и приступите к выигрывать сегодня!
Kxyufaita
| #
dark market list dark markets
GeorgeTom
| #
Source:
– https://smo-nso.ru/
bankrotstvo v moskve_gxKi
| #
банкротство физ лиц отзывы
Rylonnie shtori s elektroprivodom_hjEa
| #
рулонные шторы на балконные окна рулонные шторы на балконные окна .
Ronaldbut
| #
МикоФарм Фунги
romantiksevisme
| #
romantiksevisme
A Brief History of Elemental Analysis » Artemis Analytical
WilliamIRrutty
| #
darknet market list dark web market links
https://www.asistenciaalsuicida.org.ar/post/crece-el-inters-por-la-prevencin-del-suicidio-en-argentina
| #
Online bingo is mellow—nice crowd!
my webpage; https://www.ebanoproducoes.com.br/forum/discussoes-gerais/sequencia-de-ganhos
FNDaviditabe
| #
darknet markets dark market
https://hack1.hackathailand.com/forums/discussion/favoriete-goksites/
| #
The slot rush online grips—hold on!
Here is my web site https://www.notebook.ai/plan/items/136142
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Плоская мишень ZrO2, 4N для распыления оксида циркония
Trefebw
| #
Заказать документ о получении высшего образования вы сможете в нашей компании в Москве. diplomc-v-ufe.ru/kupit-diplom-lipetsk
https://app.geniusu.com/activities/2504338
| #
I wish online paid swift—drag off!
Feel free to surf to my site … https://www.sitionativo.com.br/post/jasmim-%C3%A1rabe-ou-bogari-jasminum-sambac?commentId=9767a866-f172-4f60-94b3-bfcfe4cb8e3e
trần đình bảo
| #
trần đình bảo là tay viết duy nhất của website thietbinhanh.com. Với sự tìm tòi và nghiên cứu các loại thiết bị anh đã tạo ra những bài viết hữu ích.
Sazrssn
| #
Мы изготавливаем дипломы любой профессии по выгодным ценам. Стоимость может зависеть от выбранной специальности, года выпуска и ВУЗа. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Важно, чтобы дипломы были доступны для большинства наших граждан. купить диплом актера все сдал
Willieavamy
| #
plinko
Jariorato
| #
Где приобрести диплом по актуальной специальности?
Наша компания предлагает выгодно и быстро приобрести диплом, который выполнен на оригинальном бланке и заверен печатями, водяными знаками, подписями официальных лиц. Документ способен пройти любые проверки, даже с применением специально предназначенного оборудования. Решите свои задачи максимально быстро с нашим сервисом.
Купить диплом о высшем образовании vuz-diplom.ru/kupit-diplom-v-kaluge-4/
Sazrpah
| #
купить диплом по электроснабжению
mostbet_daSr
| #
мостбет кыргызстан скачать https://mostbet6008.ru/ .
Iariorklu
| #
Заказать документ института вы можете в нашей компании в Москве. maryjowevers.com/blog/?__im-nKZuIBHb=3077177816173343324
1win_dhpl
| #
1winn http://1win6050.ru/ .
blogcaree
| #
Подробнее, читайте в блоге – ссылка
RobertDaw
| #
Non Gamstop casino UK withdrawals are lightning fast—impressed!
boku casinos with no gamstop
internetCah
| #
интернет провайдеры по адресу дома
domashij-internet-ekaterinburg003.ru
тарифы интернет и телевидение екатеринбург
Rylonnie shtori s elektroprivodom_lsEa
| #
электро рулонные шторы электро рулонные шторы .
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их соответствие стандартам. Опытная поддержка – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы по мере того как адаптировать решения под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
Лист циркониевый 705 (Zr-Nb) 110 Лист циркониевый 705 (Zr-Nb) 110 – это высококачественный материал, идеально подходящий для применения РІ различных отраслях, включая химическую Рё аэрокосмическую. Обладая отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью, РѕРЅ гарантирует долговечность Рё надежность ваших изделий. Благодаря СЃРІРѕРёРј превосходным механическим свойствам, лист идеально РїРѕРґС…РѕРґРёС‚ для изготовления сложных компонентов. Готовый Рє быстрой обработке, РѕРЅ облегчит любые производственные задачи. Если РІС‹ ищете прочный Рё надежный материал, РЅРµ упустите шанс купить Лист циркониевый 705 (Zr-Nb) 110 Рё обеспечить успешное выполнение ваших проектов.
VicenteNouct
| #
Профессиональный информационный блог для бухгалтера и HR-специалиста – «Премьер Бух». Лучшая помощь в работе: полезные статьи, исследования и советы всегда под рукой https://premier-buh.ru/
1win_lmEa
| #
1 цшт http://1win6003.ru .
ruma
| #
Moderni nabytek lavice s uloznym prostorem do kazdeho interieru – od minimalismu po klasiku. Vice nez 1000 modelu skladem. Online objednavka, pohodlna platba, pomoc navrhare. Zaridte svuj domov pohodlim!
sks video izle
| #
sks video izle
A Brief History of Elemental Analysis » Artemis Analytical
Willieavamy
| #
fortune mouse
Willieavamy
| #
teen patti
Naseptblori
| #
Судоходная компания Навигатор https://teplohod-restoran.ru/ это возможность организовать праздник на воде в Санкт-Петербурге. Свадьбы, дни рождения, корпоративы, выпускные, а также банкеты и фуршеты. Мы комплексно подходим к организации любого события. Посетите сайт, посмотрите наш флот и стоимость аренды судов, яхт, катеров, теплоходов.
kızlık bozdurma
| #
kızlık bozdurma
A Brief History of Elemental Analysis » Artemis Analytical
wuheclomb
| #
ВДГБ помогает клиентам управлять финансами, складским учетом и производственными процессами. Кроме этого мы предлагаем для ваших сотрудников обучение и поддержку системы на всех этапах ее эксплуатации обеспечиваем. Какие преимущества бизнесу предоставляет автоматизация, вы узнаете на портале. https://erp.vdgb.ru – сайт, где бесплатно можно заказать экспертизу и получить массу достоинств. На вопросы ответьте, и мы вам подготовим лучшее предложение. Заполните форму уже сейчас. На портале вы детальнее с этапами проекта внедрения ЕРП можете ознакомиться.
Ronaldbut
| #
МикоФарм Фунги
Dnrtvbz
| #
Где приобрести диплом специалиста?
Мы изготавливаем дипломы психологов, юристов, экономистов и прочих профессий по доступным тарифам. Мы предлагаем документы техникумов, которые находятся на территории всей России. Можно приобрести диплом от любого учебного заведения, за любой год, включая сюда документы старого образца СССР. Документы выпускаются на бумаге самого высокого качества. Это дает возможность делать настоящие дипломы, которые не отличить от оригинала. Документы заверяются необходимыми печатями и подписями. Стараемся поддерживать для клиентов адекватную ценовую политику. Для нас важно, чтобы дипломы были доступными для большого количества граждан. sdiplom.ru/kupit-diplom-v-ryazani-7
Diplomi_zjKt
| #
Купить диплом университета!
Мы предлагаем документы институтов, которые находятся в любом регионе России.
rusd-diplomj.ru/pokupka-diplomov-s-reestrom-2/
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
поставляемая продукция:
Рнструментальная круглая РїРѕРєРѕРІРєР° 185 РјРј 6РҐР’Р“ ГОСТ 5950-2000 РЁРёСЂРѕРєРёР№ выбор круглых РїРѕРєРѕРІРѕРє для профессиональной обработки металла. Рнструментальная круглая РїРѕРєРѕРІРєР° высочайшего качества СЃ разнообразием размеров Рё форм. Повышенная прочность, износостойкость Рё превосходное качество изготовления. Обеспечивает отличное сцепление СЃ материалом Рё эффективную работу оборудования. Покупайте РїСЂСЏРјРѕ сейчас!
MichaelDip
| #
Трансфер Созополь Служба Такси Эклипс предлагает доступные трансферы из аэропортов Бургас, Варна, София и Белград до популярных курортов Болгарии. Встреча с табличкой, помощь с багажом и комфортная поездка до места назначения. Обслуживаем Солнечный Берег, Созополь, Банско и другие направления. Для групп от 5 человек – специальные условия!
RobertDaw
| #
Casino non Gamstop platforms have the best variety—super fun!
non gamstop curacao casino sites
Willieavamy
| #
Jackpot Raider
prodentim reviews
| #
prodentim reviews
A Brief History of Elemental Analysis » Artemis Analytical
Lazrslb
| #
Заказать диплом университета!
Мы изготавливаем дипломы любой профессии по приятным тарифам— good-diplom.ru/kupit-diplom-v-krasnoyarske-17/
mitolyn scam
| #
mitolyn scam
A Brief History of Elemental Analysis » Artemis Analytical
Mazrzex
| #
Мы можем предложить дипломы психологов, юристов, экономистов и прочих профессий по приятным ценам. Стараемся поддерживать для покупателей адекватную политику цен. Важно, чтобы дипломы были доступными для большинства граждан.
Приобретение диплома, который подтверждает окончание института, – это выгодное решение. Купить диплом любого университета: sdiplom.ru/kupit-diplom-vuza-visshee/
Ageless Knees reviews
| #
Ageless Knees reviews
A Brief History of Elemental Analysis » Artemis Analytical
ElbertVef
| #
казино онлайн Мир казино всегда манил яркими огнями, звоном монет и обещанием легкой удачи. От помпезных залов Лас-Вегаса до уютных уголков Монте-Карло, казино – это не только игра, но и целая индустрия, живущая по своим правилам. С развитием технологий казино перешли в онлайн-пространство. Онлайн-казино – это виртуальные платформы, предлагающие широкий спектр азартных игр, от классических слотов до карточных игр с живыми дилерами. Удобство, доступность и разнообразие делают онлайн-казино все более популярными.
Nerve Fresh reviews
| #
Nerve Fresh reviews
A Brief History of Elemental Analysis » Artemis Analytical
bas
| #
Online casinos should care—shield us!
Also visit my web page http://sites.estvideo.net/alvasoft/forum/topic-6-108441-1.html
primebiome reviews and complaints
| #
primebiome reviews and complaints
A Brief History of Elemental Analysis » Artemis Analytical
Willieavamy
| #
The excitement of online slots is electric! fortune mouse
Willieavamy
| #
Online casinos ease play—mind out! tome of madness
https://forum.americancasinoguide.com/forum/general-discussion-forum/21213-games
| #
I love the anonymity of online gambling—no one knows who I am!
my page :: https://dscvr.one/post/1153012033938644864/complete-the-quests-get-rewarded?autofocus=true
https://robotech.com/forums/viewthread/2237890
| #
I’m skeptical about online casinos—are they really fair or just rigged?
Review my homepage :: https://www.bakespace.com/cookbooks/detail/betonred-casino/6995/
37903
| #
The table games online spark—class act!
Also visit my web page :: https://kuplukvartiru.com.ua/forum/novostrojki-sumy/14569-sloty-onlain
https://forum.plarium.com/ru/nords-heroes-of-the-north-browser/352_klani/1715341_-gri/
| #
I don’t get the online gambling hate—it’s fun!
My website – https://www.trustlink.org/Ask-The-Community/Question/Business/-Online-Casino–17261
mostbet_bkSr
| #
мостбет chrono http://mostbet6008.ru .
1win_qtmr
| #
сайт 1win сайт 1win .
Willieavamy
| #
I don’t get online shade—it’s fine! plinko
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние достижения.
Наши товары:
Гранулы оксида циркония высокой чистоты
Vuxerelayes
| #
На сайте https://remont-okon-63.ru/ уточните номер телефона для того, чтобы воспользоваться такой нужной услугой, как ремонт, регулировка окон независимо от сложности. Прямо сейчас вы сможете воспользоваться возможностью вызвать мастера. Для этого следует указать номер телефона, а также имя. Среди основных услуг, которые оказываются в этой компании, выделяют: ремонт балконных дверей, производство москитных сеток, стеклопакетов. Кроме того, доступен и срочный ремонт конструкций. Все работы выполняются в соответствии с требованиями.
Uristlaush
| #
Привет всем!
Приставы исполнительное производство — это процесс, при котором судебные приставы взыскивают долги с должников по решению суда. В рамках исполнительного производства могут быть применены меры, такие как арест имущества или блокировка счетов. Чтобы избежать сложных последствий, важно как можно скорее погасить задолженность. Вы можете договориться с приставами о рассрочке или реструктуризации долга. Юрист поможет вам правильно взаимодействовать с судебными приставами и минимизировать последствия задолженности.
Вся информация на сайте – https://svoijurist.ru
задолженность у судебных приставов, налоговый вычет по ипотеке пример, может ли пенсионер подать на налоговый вычет на лечение
судебные приставы шелехов узнать задолженность, можно ли получить налоговый вычет при покупки доли в квартире, собственник приватизированной квартиры
Удачи!
aqua sculpt pills reviews
| #
aqua sculpt pills reviews
A Brief History of Elemental Analysis » Artemis Analytical
Willieavamy
| #
Online casinos need grip—too open! mines
kepasNen
| #
Вместе с t.me/azino777_a можете отправиться в захватывающее путешествие по миру азартных игр. Предлагаем уникальные бонусы и персонализированные предложения. Расскажем, как начать играть в Азино. Вас ждет возможность выиграть крупный приз и невероятные эмоции. Не упустите шанс испытать свою удачу! Ищете azino777 сегодня? – здесь представлено много интересной информации. Собрали самые частые вопросы от игроков. Рады вам помочь. Приготовьтесь к завлекательным в Азино турнирам. Участвуйте и с другими игроками боритесь за призы!
Hipalndfen
| #
На сайте https://ooo-rodnik.ru/ уточните телефон популярной компании «Родник», основной сферой деятельности которой является чистка, ремонт скважин по всей Московский области. Предприятие оказывает услуги в своей сфере уже более 5 лет. Работа происходит без выходных, пенсионеры смогут рассчитывать на приятные скидки, бонусы. На все работы дается гарантия в один год. Компания занимается обслуживанием дач, коттеджей, домов, производств, поселков городского типа. Со всеми направлениями деятельности ознакомьтесь на сайте.
Gregoryexode
| #
Найдите свою любовь на сайте знакомств для взрослых 18+ в Сочи, где лучшие девушки готовы вас ждать. Создайте интересный профиль, общайтесь с уникальными личностями и разжигайте искру романтики. Эти знакомства могут стать началом чего-то настоящего и красивого http://sochi-night.vip/
ZopukNub
| #
Etsitko hyodyllista tietoa lainoista? Vieraile osoitteessa https://esimerkkilinkki.fi/ – josta loydat kattavaa tietoa asuntolainasta, vinkkeja lainan hakemiseen, lainojen vertailuun ja paikkaan, kuinka saada laina heti ilman luottotietoja tai luottohistoriaa ja paljon muuta. Hyodyllista tietoa sinulle verkkosivuillamme.
Jamesgam
| #
Добрый день!
Влажная уборка офиса – это услуга для поддержания чистоты в вашем рабочем помещении. Мы предлагаем уборку офисов любой сложности, включая мытье полов и очистку мебели. Влажная уборка офиса с гарантией качества и надежности. Наши специалисты используют только безопасные и эффективные средства. Закажите уборку и создайте комфортную атмосферу в офисе.
Услуги клининга квартиры – это профессиональная уборка жилья. Мы предоставляем генеральную и регулярную уборку квартир. Наши специалисты используют безопасные чистящие средства и современное оборудование. Мы гарантируем идеальный результат. Закажите услуги клининга квартиры у нас.
Вся информация на сайте – https://service-cleanspb.ru
уборка частных квартир, уборка дачи, клининг компании спб
ежедневная уборка офисов, клининг спб уборка квартир, помыть окна
Удачи!
KosipenJes
| #
Доставка комплексных обедов в Алматы для ваших сотрудников или на дом на сайте https://bvd.kz/ – у нас широкий ассортимент и выбор блюд по отличным ценам! Ознакомьтесь на сайте с составами и ценами на комплексные обеды. Заключаем договоры на доставку обедов в офисы и административные учреждения, доставляем обеды в школы и детские сады, где нет собственной кухни, в магазины и прочие подобные учреждения, у сотрудников которых нет возможности пообедать в столовой. Подробнее на сайте.
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их происхождение. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы и адаптировать решения под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
РўСЂСѓР±Р° ниобиевая Ta-Nb40. 5Р’2РњР¦-1 РўСЂСѓР±Р° ниобиевая Ta-Nb40. 5Р’2РњР¦-1 является высококачественным материалом, предназначенным для использования РІ различных отраслях. Рта труба обладает отличными физико-химическими характеристиками, что делает её идеальной для применения РІ условиях высоких температур Рё давлений. Р’С‹ можете купить РўСЂСѓР±Р° ниобиевая Ta-Nb40. 5Р’2РњР¦-1 для нужд вашей промышленности, уверившись РІ её надежности Рё долговечности. Ртот РїСЂРѕРґСѓРєС‚ строго соответствует международным стандартам Рё идеально РїРѕРґС…РѕРґРёС‚ для сложных инженерных задач. Обеспечьте эффективность ваших процессов СЃ помощью нашей трубы.
Биоревитализация лица препараты
| #
Биоревитализация лица препараты
A Brief History of Elemental Analysis » Artemis Analytical
Jamesdusty
| #
Ваши флористы понимают с полуслова – описали идею, и её воплотили!
цветы
michelin_ziSr
| #
michelin pilot sport 4 michelin pilot sport 4 .
shinomontaj_hiSn
| #
шиномонтаж лыткарино http://www.hondahybrid.ru/forum/topic/2886-kakie-uslugi-okazyvaet-shinomontazh/ .
Arialief reviews
| #
Arialief reviews
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние достижения.
Наша продукция:
Мишень для распыления скандия
Gregoryexode
| #
Начните свое романтическое путешествие в Красной Поляне на новом сайте знакомств, который объединил тысячи девушек 18+. Интересные анкеты, живое общение и доброжелательная атмосфера станут вашим надежным помощником в поиске идеальной спутницы по жизни – http://sochi-night.vip/
Willieavamy
| #
The live dealer games online are a game-changer—so immersive! plinko
FritzFed
| #
Olymp Casino Online’s loyalty rewards are worth sticking around for. olymp casino
FritzFed
| #
Olymp Casino Online’s loyalty rewards are worth sticking around for. olymp casino
JosephFraus
| #
Если вы решили остеклить балкон, то пластиковые окна будут отличным выбором, так как они не только красиво выглядят, но и долговечны https://osteklenie-balkonov-v-spb.pro/
Прыщи акне
| #
Прыщи акне
A Brief History of Elemental Analysis » Artemis Analytical
FritzFed
| #
Olymp Casino Online’s slot selection is massive—hard to choose!
olymp casino
Elberthouts
| #
Актуально обо всем! Ежедневные обзоры происходящего в мире и России. Новости бизнеса и общества, автомобилестроения и медицины https://sovety-tut.ru/
KevinSkisy
| #
Официальный сайт 1win 1win.onedivision.ru спортивные ставки, киберспорт, казино, покер, рулетка и многое другое. Надежный сервис, поддержка 24/7, удобный интерфейс. Забирай бонус за регистрацию уже сейчас!
Williamrix
| #
крипта Мир казино – это захватывающая вселенная, где переплетаются азарт, стратегия и технологические инновации. Онлайн казино предлагают широкий спектр игр, от классических слотов до захватывающих настольных игр, предоставляя игрокам возможность испытать удачу и применить свои навыки. Стратегии играют ключевую роль в успехе. Будь то блэкджек, покер или рулетка, понимание правил и умение анализировать ситуацию могут значительно повысить шансы на выигрыш. Важно помнить о разумном управлении банкроллом и осознанном подходе к игре.
IGET Moon
| #
Gunnpod Evo’s flavor options are endless—something for everyone.
https://trade-britanica.trade/wiki/User:MelbaEyre119435
Бородавки
| #
Бородавки
A Brief History of Elemental Analysis » Artemis Analytical
Vigor Prime
| #
Alibarbar Ingot lasts so long I almost forget it’s disposable!
https://yrr.ens.mybluehost.me/index.php/Vape_88M
michelin_azSr
| #
летняя резина michelin летняя резина michelin .
Электроэпиляция волос
| #
Электроэпиляция волос
A Brief History of Elemental Analysis » Artemis Analytical
mostbet_qwmt
| #
мостбет скачать на андроид https://www.svstrazh.forum24.ru/?1-18-0-00000136-000-0-0-1743260517 .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Мишень для распыления марганца
this is my website
| #
this is my website
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их качество. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы по мере того как находить ответы под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Лист молибденовый ЦМВ30 Лист молибденовый ЦМВ30 – это высококачественный материал, который прекрасно РїРѕРґС…РѕРґРёС‚ для различных промышленных применений. РћРЅ обладает отличной стойкостью Рє РєРѕСЂСЂРѕР·РёРё Рё высокой прочностью РїСЂРё высоких температурах. Ртот лист широко используется РІ аэрокосмической, энергетической Рё химической промышленности благодаря СЃРІРѕРёРј уникальным свойствам.\n\nРџСЂРё этом РѕРЅ легко обрабатывается Рё может быть использован РІ различных процессах, таких как сварка Рё резка. Если РІС‹ хотите “купить Лист молибденовый ЦМВ30”, РІС‹ выберете надежное решение для СЃРІРѕРёС… производственных нужд. РќРµ упустите возможность обеспечить СЃРІРѕР№ бизнес качественным сырьем!
shinomontaj_rqSn
| #
шиномонтаж чехов hondahybrid.ru/forum/topic/2886-kakie-uslugi-okazyvaet-shinomontazh/ .
AaronCot
| #
вакансии за рубежом Layboard
https://www.techweek.ro/post/tot-ce-trebuie-s-tii-despre-printarea-3d?commentId=153cd7ae-4a55-4be2-9413-915b84bfb6fb
| #
Online casinos should cap—safe guard!
my blog :: https://forums.ngames.com/forum/league-of-angels/game-play-ab/gaming-entertainment-and-science-ab/86630-games-that-can-be-enjoyed-even-by-beginners
GeorgeTom
| #
Source:
– https://aradicalvision.com/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наша продукция:
Плоская мишень CoTaZr для распыления сплава
1win.onedivision
| #
Добро пожаловать в 1win https://1win.onedivision.ru/ азарт, спорт и выигрыши рядом! Ставь на матчи, играй в казино, участвуй в турнирах и получай крутые бонусы. Удобный интерфейс, быстрая регистрация и выплаты.
Tevupeyhok
| #
How to exchange BITCOIN in the cryptocurrency exchanger quickex are Buy or sell BTC? In this https://www.youtube.com/watch?v=0MupM-q6I3A detailed guide, we will show you how to buy BTC on the quickex platform in 5 easy steps. You will learn how to use a reliable service without registration and verification, getting access to the best rates from leading liquidity providers.
Βίντεο βιασμού του Έπσταϊν προς πώληση
| #
Βίντεο βιασμού του Έπσταϊν προς πώληση
A Brief History of Elemental Analysis » Artemis Analytical
Willieavamy
| #
The mobile apps online slip—fix up! plinko
michelin_jgSr
| #
michelin x-ice north 4 205/60 r16 http://www.proalbea.ru/shiny-michelin-preimushhestva-i-nedostatki.html .
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
поставляемая продукция:
Медная крестовина под пайку 35х29х1 мм 23х25 мм мягкая пайка М1 ГОСТ 32590-2013 Узнайте о высококачественных медных крестовинах под пайку от Редметсплав. Гарантия надежности, прочности, отличной теплопроводности и простоты монтажа. Широкий выбор размеров и конфигураций для любых электрических соединений.
Willieavamy
| #
I love the online chats—cool folks! plinko
JosephFraus
| #
Если вы решили остеклить балкон, то пластиковые окна будут отличным выбором, так как они не только красиво выглядят, но и долговечны https://osteklenie-balkonov-v-spb.pro/
Willieavamy
| #
Playing online craps is a blast—way easier than learning in person! betmomo
shinomontaj_vkSn
| #
шиномонтаж апрелевка шиномонтаж апрелевка .
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их соответствие стандартам. Опытная поддержка – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы по мере того как находить ответы под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
РџРѕРєРѕРІРєР° магниевая AZ91D – ASTM B275 Лист магниевый AZ91D – ASTM B275 обладает уникальными свойствами, включая РЅРёР·РєРёР№ вес Рё отличную РєРѕСЂСЂРѕР·РёРѕРЅРЅСѓСЋ стойкость. Ртот материал идеален для применения РІ авиационной Рё автомобильной промышленности, Р° также РІ производстве электронных устройств. Благодаря высокой прочности Рё легкости, Лист магниевый AZ91D – ASTM B275 становится отличным выбором для создания деталей Рё конструкций. РњС‹ предлагаем вам купить Лист магниевый AZ91D – ASTM B275 РїРѕ конкурентной цене. РќРµ упустите возможность улучшить СЃРІРѕРё проекты СЃ помощью данного продукта!
RijusonPried
| #
Посетите сайт https://4px.ru/ – 4 Пикселя – это Digital агентство интернет-маркетинга полного цикла в Москве, с комплексным подходом к созданию рекламы в интернете. Ознакомьтесь с нашими выгодными услугами в SEO, Яндекс.Директ, SMM, Serm, разработкой и созданием сайтов и многими другими профессиональными услугами. Ознакомьтесь с нашими кейсами – мы будем рады видеть вас в числе наших клиентов!
Diplomi_uuKt
| #
Приобрести диплом о высшем образовании!
Мы готовы предложить документы учебных заведений, расположенных на территории всей РФ.
kupitediplom0027.ru/kupite-diplom-s-reestrom-i-otzivami-bez-xlopot-2/
2k10 đụ nhau
| #
2k10 đụ nhau
A Brief History of Elemental Analysis » Artemis Analytical
WepodEndat
| #
Visit https://reviewspoty.com/ and you can easily collect reviews by integrating with your existing technology and e-commerce platforms. Increase sales and reduce marketing costs with reviews. Learn more about us and what we offer, as well as pricing and a free trial.
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их качество. Дружелюбная помощь – наш стандарт – мы на связи, чтобы улаживать ваши вопросы по мере того как находить ответы под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
поставляемая продукция:
Лист магниевый M19981 – UNS Лента магниевая M19981 – UNS – это высококачественный РїСЂРѕРґСѓРєС‚, предназначенный для различных областей применения, включая электронику Рё промышленность. Благодаря СЃРІРѕРёРј уникальным свойствам, эта лента обладает высокой прочностью Рё стойкостью Рє РєРѕСЂСЂРѕР·РёРё. РћРЅР° идеально РїРѕРґС…РѕРґРёС‚ для создания РїСЂРѕРІРѕРґРЅРёРєРѕРІ, Р° также РІ качестве изоляционного материала. Если РІС‹ ищете надежный Рё эффективный товар, вам стоит купить Лента магниевая M19981 – UNS. Ее применение поможет обеспечить долговечность Рё надежность ваших устройств. РќРµ упустите возможность приобрести этот отличный материал!
Buvukneers
| #
На сайте https://rodinasportsclub.com/ вы найдете информацию, которая касается спортивно-развлекательного клуба «Родина». Каждый желающий получает возможность записаться на тренировку по боксу, которая подходит как новичкам, так и более опытным спортсменам. Вас заинтересуют тренировки по рукопашному бою. Перед вами расписание предстоящих матчей на апрель. Ознакомьтесь с ними сейчас, чтобы построить планы. Также имеются и отчеты по прошедшим соревнованиям. Опубликованы разные новости из жизни клуба.
GGZoneSteam
| #
Бесплатные Steam аккаунты http://t.me/GGZoneSteam с играми и бонусами. Проверенные логины и пароли, ежедневное обновление, удобный поиск. Забирай свой шанс на крутой аккаунт без лишних действий!
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица товара подтверждена соответствующими документами, подтверждающими их качество. Превосходное обслуживание – наш стандарт – мы на связи, чтобы ответить на ваши вопросы а также предоставлять решения под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
РљСЂСѓРі молибденовый Р’Рњ РљСЂСѓРі молибденовый Р’Рњ – это высококачественный металл, используемый РІ различных отраслях благодаря СЃРІРѕРёРј уникальным свойствам. РћРЅ обладает отличной прочностью Рё термостойкостью, что делает его идеальным для высоких температур. РљСЂСѓРіРё РёР· молибдена широко применяются РІ производстве электродуговых печей Рё РІ аэрокосмической промышленности.Если РІС‹ ищете надежное Рё долговечное решение, купите РљСЂСѓРі молибденовый Р’Рњ. Наши продукты соответствуют высоким стандартам качества Рё обеспечивают отличную производительность. РќРµ упустите возможность использовать передовые материалы РІ СЃРІРѕРёС… проектах!
FritzFed
| #
The bonus codes at Olymp Casino Online are easy to redeem. olymp casino
mostbet_xrmt
| #
mostbet kg скачать на андроид svstrazh.forum24.ru/?1-18-0-00000136-000-0-0-1743260517 .
JerryLuh
| #
Все про уход за лицом и телом. Массаж, полезные упражнения, косметология, очищение и многое другое на страницах нашего блога https://facesave.ru/
Willieavamy
| #
Online casinos should offer more practice modes. tiger fortune
phim sex tự quay
| #
phim sex tự quay
A Brief History of Elemental Analysis » Artemis Analytical
JerryLuh
| #
Все про уход за лицом и телом. Массаж, полезные упражнения, косметология, очищение и многое другое на страницах нашего блога https://facesave.ru/
nepubfooni
| #
Компания «Эксперт-Техника» реализовывает весь спектр услуг – продажа, сервисное обслуживание, поставки запчасти, также комплектующих. Наши специалисты повышение квалификации и техническое обучение проходят. Мы разработали гибкую и надежную систему доставки. https://extehno.ru – тут с условиями оплаты можете ознакомиться. Предлагаем приличный ассортимент спецтехники. Стремимся к клиентам ближе быть. Если у вас возникли вопросы, обратитесь к менеджеру по телефону, указанному на сайте. Гарантируем вам высокий уровень сервиса.
Willieavamy
| #
Online casinos help try—good spot! bet on red
FritzFed
| #
I’ve been playing at Olymp Casino Online for a few weeks, and the game selection is pretty impressive! olymp casino
reharoFenia
| #
Используйте AstoMagic – бесплатную онлайн-нейросеть. Задайте вопрос, и вы за пару секунд нужный ответ получите. ИИ анализирует ваш стиль общения и под него подстраивается. Получайте помощь в написании кода и исправлении ошибок. https://astomagic.com – тут ответы на часто задаваемые вопросы собрали. На ресурсе тарифы и отзывы пользователей представлены. Общайтесь голосом – система распознает речь и выдает ответы в текстовом виде. ИИ анализирует информацию и предоставляет точные данные. Не упустите возможность опробовать нейросеть онлайн!
bethand
| #
bethand
A Brief History of Elemental Analysis » Artemis Analytical
1win_ugpl
| #
1win кейсы https://1win6050.ru .
JibesdPeage
| #
Ищете интернет магазин автозапчастей в Минске с выгодными ценами? Посетите https://belautoparts.by/ где вы найдете существенный выбор для все автомобилей с доставкой по всей Беларуси. Подберите запчасти по VIN, марке авто, номеру детали, артикулу, каталогу в иллюстрации. У нас также есть все для ТО – расходники, масла, фильтра и многое другое. В нашем интернет магазине более 30 млн запчастей как оригинальных, так и качественных заменителей. Ознакомьтесь на сайте подробнее!
Iariorxcu
| #
Заказать диплом университета по невысокой стоимости вы сможете, обращаясь к проверенной специализированной компании. Купить документ института можно у нас в Москве. diplomc-v-ufe.ru/kupite-diplom-s-reestrom-po-nizkoj-tsene-pryamo-sejchas
hd âbla
| #
hd âbla
A Brief History of Elemental Analysis » Artemis Analytical
RichardMealp
| #
изи кэш казино
fuji66 slot
| #
fuji66 slot
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Гранулы высокочистого ниобия
Raymondskync
| #
либет казино, изи кэш казино
DibesNat
| #
На сайте https://boostclicks.ru/ вы сможете ознакомиться с рекомендациями, ценными и важными советами про API интеграцию, скрипты, а также любопытные сервисы из мира трафика. Для того чтобы вам было удобней ориентироваться, все материалы поделены на разделы, что позволит отыскать то, что действительно актуально и интересно в данный момент. Регулярно на портале появляется новый, интересный контент на данную тему. Имеется и список тех статей, которые пользуются особой популярностью. Все они созданы лучшими и высококлассными экспертами с огромным опытом.
doedla
| #
doedla
A Brief History of Elemental Analysis » Artemis Analytical
FritzFed
| #
Olymp Casino Online’s live game streaming quality is excellent.
olymp casino
Willieavamy
| #
I don’t trust online—too loose! plinko
Jimmiefax
| #
Доска объявлений https://estul.ru/blog по всей России: продавай и покупай товары, заказывай и предлагай услуги. Быстрое размещение, удобный поиск, реальные предложения. Каждый после регистрации получает на баланс аккаунта 100? для возможности бесплатного размещения ваших объявлений
Sazrkfw
| #
купить натуральный диплом
JeruvesLon
| #
На сайте https://miners-store.ru/ вы сможете связаться с представителями компании для того, чтобы выбрать и приобрести асики для майнинга. Перед вами функциональное и качественное оборудование, которое требуется для того, чтобы добыть криптовалюту. Изучите всех производителей такого оборудования, которое можно заказать по лучшей стоимости. Есть и самые популярные модели, которые приобретают чаще остальных. Здесь также осуществляется продажа видеокарт. Специалисты соберут специальную ферму для майнинга с нуля.
FritzFed
| #
The bonus terms at Olymp Casino Online are clear, which is refreshing.
olymp casino
Cazrnov
| #
Получить диплом университета поможем. Купить диплом Тольятти – diplomybox.com/kupit-diplom-tolyatti
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их происхождение. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы по мере того как адаптировать решения под особенности вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Фольга магниевая MAG 7 – BS 2970 РўСЂСѓР±Р° магниевая MAG 7 – BS 2970 – это высококачественный РїСЂРѕРґСѓРєС‚, созданный для различных промышленных применений. Благодаря своей легкости Рё высокой прочности, эта труба идеально РїРѕРґС…РѕРґРёС‚ для использования РІ условиях, требующих устойчивости Рє РєРѕСЂСЂРѕР·РёРё. Р’ процессе производства использованы современные технологии, что гарантирует надежность Рё долговечность. Если РІС‹ ищете оптимальное решение для СЃРІРѕРёС… проектов, купить РўСЂСѓР±Р° магниевая MAG 7 – BS 2970 – значит сделать правильный выбор. Убедитесь РІ выдающихся характеристиках данного изделия, которое обеспечивает превосходную производительность Рё эффективность.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Гранулы цинка 4N
xnxx.com
| #
xnxx.com
A Brief History of Elemental Analysis » Artemis Analytical
FritzFed
| #
Olymp Casino Online’s live game streaming quality is excellent.
olymp casino
Willieavamy
| #
Online casinos need hold—too wild! bet on red
mostbet_wumt
| #
mostbet скачать на телефон бесплатно андроид http://svstrazh.forum24.ru/?1-18-0-00000136-000-0-0-1743260517 .
sismanam
| #
sismanam
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
поставляемая продукция:
Медная твердая дюймовая труба для холодильного оборудования 5/8 дюйм 1 мм М1ф EN 12735 У нас вы можете купить медные трубы для хладагрегатов высокого качества с гарантированной прочностью, долговечностью и эффективностью теплообмена. Простота монтажа и обслуживания делает их идеальным выбором для оборудования систем холодильных установок, кондиционирования и прочих промышленных холодильных устройств. Обращайтесь к нам для получения подробной информации о продукции и условиях поставки.
Willieavamy
| #
I won $1200 on a slot—hype high! plinko
Rylonnie shtori s elektroprivodom_tnEa
| #
рулонные шторы с электроприводом и дистанционным управлением рулонные шторы с электроприводом и дистанционным управлением .
FritzFed
| #
The welcome bonus at Olymp Casino Online really hooked me in—great way to start!
olymp casino
Willieavamy
| #
Online casinos should show odds clearer! plinko
1win_fypl
| #
1вин официальный сайт вход 1вин официальный сайт вход .
levis4d
| #
levis4d
A Brief History of Elemental Analysis » Artemis Analytical
gekoboubs
| #
Компания «АВАНГАРД» специализируется на продаже и доставке инертных материалов в Краснодарском крае. Гарантируем высокое качество поставляемого плодородного грунта и предлагаем доступные цены. Мы большую базу клиентов нарастили. https://avangard-kuban.ru/chernozjom-i-grunty/ – здесь можете подробнее ознакомиться с условиями оплаты и доставки. У нас скидки на чернозем действуют. Если ищете надежную компанию доставки инертных материалов, обратитесь к нам. С удовольствием вам в решении любых вопросов поможем. Звоните нам!
kilsizam
| #
kilsizam
A Brief History of Elemental Analysis » Artemis Analytical
FritzFed
| #
The slot RTPs at Olymp Casino Online seem decent—any favorites?
olymp casino
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их соответствие стандартам. Превосходное обслуживание – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы по мере того как находить ответы под требования вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
Лист магниевый AZ91HP Лента магниевая AZ91HP – это высококачественный РїСЂРѕРґСѓРєС‚, созданный для удовлетворения потребностей РІ легких Рё прочных материалах. РћРЅР° обладает отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью Рё высокой прочностью, что делает ее идеальным выбором для различных отраслей, включая автомобилестроение Рё аэрокосмическую промышленность. Лента легко обрабатывается Рё размещается РІ требуемых конструкциях. Если РІС‹ ищете надежный Рё универсальный материал, РЅРµ упустите возможность купить Лента магниевая AZ91HP. Ртот РїСЂРѕРґСѓРєС‚ обеспечит вам оптимальные характеристики Рё эффективно справится СЃ поставленными задачами.
Iariorcbx
| #
Купить диплом ВУЗа по невысокой стоимости возможно, обращаясь к надежной специализированной компании. Купить документ о получении высшего образования можно у нас в столице. kupitediplom0027.ru/kupit-diplom-o-srednem-obrazovanii-s-reestrovoj-zapisyu
FritzFed
| #
Olymp Casino Online’s withdrawal limits are reasonable for casual players. olymp casino
Cazrqed
| #
Заказать диплом ВУЗа!
Мы предлагаем документы институтов, расположенных на территории всей РФ. Дипломы и аттестаты выпускаются на бумаге высшего качества: sydba.flybb.ru/viewtopic.phpf=2&t=741
remont kardana_pvMa
| #
ремонт карданных валов в москве ремонт карданных валов в москве .
1win_zzmr
| #
1win букмекер https://www.obovsem.myqip.ru/?1-9-0-00000059-000-0-0-1743051936 .
Sazrstn
| #
Заказать диплом любого ВУЗа!
Купить диплом университета по доступной цене возможно, обращаясь к проверенной специализированной фирме. Приобрести диплом: diplomist.com/kupite-diplom-s-ofitsialnim-reestrom-bistro-i-bezopasno
Cuyanchone
| #
Ландшафтный дизайн под ключ https://greenartstudio.ru/ – от Грин Арт Студио это услуги профессионалов от проектирования до реализации и авторского надзора. Посетите наш сайт – посмотрите реализованные проекты – они вам обязательно понравятся, а также ознакомьтесь со всеми нашими услугами, начиная от проектирования и заканчивая уходом за вашим садом, благоустройством участка. Мы работаем с частными лицами, госучреждениями и коммерческими компаниями.
Dnrtoca
| #
Где заказать диплом по актуальной специальности?
Мы можем предложить дипломы психологов, юристов, экономистов и других профессий по приятным ценам. Мы предлагаем документы ВУЗов, расположенных на территории всей России. Вы имеете возможность приобрести качественный диплом за любой год, включая сюда документы старого образца СССР. Дипломы и аттестаты выпускаются на “правильной” бумаге высшего качества. Это позволяет делать настоящие дипломы, которые невозможно отличить от оригинала. Документы будут заверены всеми обязательными печатями и штампами. Всегда стараемся поддерживать для заказчиков адекватную политику цен. Для нас очень важно, чтобы дипломы были доступными для подавляющей массы граждан. diploms-vuza.com/kupit-diplom-v-nizhnim-tagile-5
XunovGroup
| #
На сайте https://astro-magic.com/ воспользуйтесь искусственным интеллектом на русском языке. Этот сервис предоставляет возможность использовать все функции в режиме реального времени. При этом нет необходимости подключать VPN, искусственный интеллект работает и без него. Воспользоваться им сможет каждый желающий, абсолютно бесплатно. Chat GPT сможет создать качественный контент, произвести анализ изображений, быстро отыскать нужную информацию. Искусственный интеллект сможет и написать программный код.
Sazrvqo
| #
диплом повара купить
Sazrmok
| #
Купить диплом института !
Покупка диплома университета РФ у нас – надежный процесс, так как документ будет заноситься в реестр. Приобрести диплом ВУЗа diplom-kaluga.ru/tsena-diploma-s-zaneseniem-v-reestr-sprosite-sejchas
Willieavamy
| #
I won $800 online—crazy night! mines casino
Willieavamy
| #
I won $200 on a slot online—still hyped! mines
Willieavamy
| #
Online bingo flows—great crew! plinko
Willieavamy
| #
Online casinos help try—good spot! plinko
Elberthouts
| #
Актуально обо всем! Ежедневные обзоры происходящего в мире и России. Новости бизнеса и общества, автомобилестроения и медицины https://sovety-tut.ru/
BejoldGet
| #
На сайте https://waltzprof.com/ получится заказать обратный звонок для того, чтобы приобрести продукцию собственного производства от компании «Валцпроф». Она производит стальной профиль безупречного качества. Он используется для создания фасадных, противопожарных конструкций и иного предназначения. Также в компании получится приобрести и вспененный уплотнитель, выпадающие пороги, профиль фиксации стекла. Для производства продукции эталонного качества применяются только передовые технологии.
Willieavamy
| #
I hit a blackjack run online—sweet day! dragon tiger
Cazrjoj
| #
Мы изготавливаем дипломы любой профессии по приятным тарифам. Купить диплом в Амурской области и городе Благовещенск — kyc-diplom.com/geography/blagoveshchensk.html
esmerle sex
| #
esmerle sex
A Brief History of Elemental Analysis » Artemis Analytical
1win_lkkn
| #
1winn http://1win6004.ru/ .
SurotLak
| #
Ищете изготовление наружной рекламы в Москве? Посетите сайт https://neon-mix.ru/ – мы изготавливаем световые вывески, объёмные буквы, рекламные конструкции и многое другое. Полный цикл — от производства до монтажа по самым выгодным ценам. Реализуем проекты любой сложности, учитываем пожелания заказчика и предлагаем честные, конкурентные цены. Ознакомьтесь со всеми услугами и ценами на сайте.
mostbet_fboi
| #
mostbest http://mostbet6009.ru .
remont kardana_zyMa
| #
ремонт карданов области http://www.4-x-4.ru/remont-i-balansirovka-kardannyh-valov// .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
Наша продукция:
Нержавеющая прямоугольная РїРѕРєРѕРІРєР° 200С…470 РјРј 12РҐ13 Рзучите широкий ассортимент нержавеющих прямоугольных РїРѕРєРѕРІРѕРє РЅР° RedmetSplav СЃ гарантией качества Рё надежности. Устойчивость Рє РєРѕСЂСЂРѕР·РёРё Рё разнообразие размеров для всевозможных проектов.
Iariorspm
| #
Купить диплом ВУЗа по невысокой цене возможно, обратившись к надежной специализированной фирме. Приобрести документ о получении высшего образования вы имеете возможность у нас в столице. diplom4you.com/kupit-diplom-s-zaneseniem-12
milehiruifs
| #
Компания AVTO197 предлагает услуги аренды автомобилей в Москве, обеспечивая клиентов широким выбором транспортных средств для любых ситуаций. В автопарке https://avtocar5.ru/ представлены модели эконом, среднего и бизнес классов, а также минивэны и коммерческие автомобили. Все машины застрахованы по КАСКО и ОСАГО, что гарантирует безопасность и надежность. Процесс оформления аренды занимает всего 15–20 минут с минимальным пакетом документов. Для постоянных клиентов предусмотрены гибкие системы скидок. Дополнительно компания предоставляет услуги такси и аренду автомобилей для свадебных мероприятий.
PirevBlusy
| #
На сайте https://axonbusiness.ru обсудите свой проект для того, чтобы воспользоваться популярными и эффективными решениями компании «A.X.O.N.». Прямо сейчас вы сможете заказать звонок, чтобы узнать стоимость вашего задания. Здесь получится заказать, в том числе, стратегический бизнес-консалтинг, а также готовые ИТ-решения. Лучшие специалисты оптимизируют и автоматизируют все ваши бизнес-процессы, клиентский сервис. Для вас будут разработаны индивидуальные решения для получения большей прибыли.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Мишень гафния
https://forum.uniformserver.com/topic/14230-game/
| #
I won $50 last night on an online slot game—small wins keep me hooked!
Also visit my webpage … https://br.pinterest.com/pin/922323198790022407
Sazrsvx
| #
диплом бланк купить
Willieavamy
| #
Are online casino jackpots for real?—doubt it! plinko
remont kardana_ceMa
| #
замена подвесного подшипника замена подвесного подшипника .
Willieavamy
| #
I won a tourney online—wild high! plinko
Willieavamy
| #
I love the online promos—new spark! vincispin
Dnrtidq
| #
Где приобрести диплом специалиста?
Мы изготавливаем дипломы любой профессии по приятным тарифам. Мы предлагаем документы техникумов, которые расположены в любом регионе России. Можно заказать диплом за любой год, включая сюда документы старого образца СССР. Документы печатаются на “правильной” бумаге самого высшего качества. Это позволяет делать государственные дипломы, не отличимые от оригинала. Они заверяются необходимыми печатями и подписями. Стараемся поддерживать для покупателей адекватную ценовую политику. Важно, чтобы дипломы были доступными для большого количества наших граждан. diplom45.ru/kuplyu-diplom-4
Rylonnie shtori s elektroprivodom_sqEa
| #
рулонные шторы это рулонные шторы это .
GeorgeTom
| #
Source:
https://aradicalvision.com/
Willieavamy
| #
Online casino bonuses are sneaky—read up! plinko
1win_ihkn
| #
1win кыргызстан http://www.1win6004.ru .
CharlesDrisM
| #
кайт египет
Aaronrox
| #
Ученые узнали, в какое время суток кофе заметно продлевает жизнь
https://pin.it/72MctQ7nd
Новое исследование, опубликованное в European Heart Journal, выявило, что люди, предпочитающие пить кофе утром, могут иметь более низкий риск смертности, особенно от сердечно-сосудистых заболеваний. Это ставит под вопрос не только количество употребляемого кофе, но и его время как фактор, влияющий на здоровье.
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
Наша продукция:
РўСЂСѓР±Р° РёР· драгоценных металлов платиновая 500С…3С…1 РјРј РџР»Р95-5 РўРЈ Гарантированное качество Рё изысканный дизайн – купите драгоценные трубы для ювелирных украшений, машиностроения Рё архитектуры. РЁРёСЂРѕРєРёР№ выбор Рё высокое качество! Доставка РїРѕ Р РѕСЃСЃРёРё.
PubuctWes
| #
На сайте http://promkomplektregion96.ru закажите оптовые поставки металлопроката. Он пригодится как в сфере промышленности, так и строительства. Многие из представленных позиций вы сможете приобрести прямо сейчас, а другие – только под заказ. В каталоге находятся фитинги и трубы, выполненные из полиэтилена, черный прокат, нержавеющий, цветной прокат. Есть возможность оплатить приобретение несколькими вариантами. Доступна оперативная доставка по всей России. Воспользуйтесь профессиональными консультациями.
mostbet_peoi
| #
mostbest mostbet6009.ru .
https://nbenegroup.com/--/
| #
https://nbenegroup.com/%D0%B2%D0%BF%D0%BD-%D0%B4%D0%BB%D1%8F-%D1%8E%D1%82%D1%83%D0%B1%D0%B0/
bedava se x
| #
bedava se x
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица товара подтверждена соответствующими документами, подтверждающими их происхождение. Превосходное обслуживание – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы а также находить ответы под требования вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Рзделия РёР· вольфрама Р’Р Рќ Рзделия РёР· вольфрама Р’Р Рќ обладают высокими эксплуатационными характеристиками, что делает РёС… незаменимыми РІ различных отраслях. РћРЅРё отличаются прочностью, устойчивостью Рє высоким температурам Рё РєРѕСЂСЂРѕР·РёРё, что гарантирует долговечность. Рти изделия используются РІ производстве, электронной сфере Рё даже высокоточной механике. Покупая продукцию РёР· вольфрама Р’Р Рќ, РІС‹ обеспечиваете качественное решение для ваших нужд. РќРµ упустите возможность купить Рзделия РёР· вольфрама Р’Р Рќ Рё убедиться РІ РёС… преимуществах сами.
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
поставляемая продукция:
Дюралевый швеллер 440640 50С…100С…4С…7С…7 РјРј Р”16С‡ ГОСТ 13623-90 Купить дюралевый швеллер РѕС‚ производителя. Прочный Рё легкий материал для различных областей строительства. РЁРёСЂРѕРєРёР№ выбор размеров Рё профилей. Устойчивость Рє РєРѕСЂСЂРѕР·РёРё Рё надежность конструкций. Рдеальное решение для авиации, судостроения, автомобилестроения, производства спортивного оборудования Рё фармакологических предприятий.
TerryLix
| #
Все про стройку и ремонт. Актуальные интересные статьи о стройке и ремонте. Каждый найдет полезные материалы о дизайне, электрике, отоплении, кровле, сантехнике и другая полезная информация https://otoplenie-expert.com/
erotikli
| #
erotikli
A Brief History of Elemental Analysis » Artemis Analytical
1win_likn
| #
1win live http://1win6004.ru .
Willieavamy
| #
I hit a losing streak online last week—maybe it’s time for a break. F7 Casino
Willieavamy
| #
I love how online casinos let you start small! plinko
omegle porn
| #
omegle porn
A Brief History of Elemental Analysis » Artemis Analytical
1win_hssn
| #
1win букмекер kharkovbynight.forum24.ru/?1-5-0-00000235-000-0-0 .
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
Наши товары:
Кольцо РёР· драгоценных металлов палладиевое 45С…15С…3.5 РјРј РџРґ99.9 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
1win_ljsn
| #
1 win.pro https://kharkovbynight.forum24.ru/?1-5-0-00000235-000-0-0 .
mostbet_nuoi
| #
мостбет кыргызстан скачать https://www.mostbet6009.ru .
Dnrtdnu
| #
Где купить диплом специалиста?
Мы изготавливаем дипломы любой профессии по приятным тарифам. Мы готовы предложить документы техникумов, которые расположены в любом регионе РФ. Можно купить диплом за любой год, включая документы старого образца. Дипломы и аттестаты делаются на бумаге высшего качества. Это позволяет делать настоящие дипломы, не отличимые от оригинала. Они заверяются необходимыми печатями и подписями. Всегда стараемся поддерживать для клиентов адекватную ценовую политику. Для нас очень важно, чтобы дипломы были доступными для подавляющей массы наших граждан. diplomoz-197.com/kupit-diplom-rossiya-2-2
aldatma por
| #
aldatma por
A Brief History of Elemental Analysis » Artemis Analytical
blue salt trick for men
| #
blue salt trick for men
A Brief History of Elemental Analysis » Artemis Analytical
Logetrydreat
| #
На сайте https://smartporog.ru/ изучите каталог, чтобы приобрести умные выпадающие пороги. Они идеально подходят для дверей различного типа. Автоматические пороги считаются идеальным решением в том случае, если невозможна установка обычного порога. Такие конструкции комфортны в использовании, эффективно борются с преградой в полу, а также сквозняками, мостиками холода. Пороги такого типа не допускают попадания внутрь посторонних запахов, повышают звукоизоляцию. Также пороги защищают от проникновения внутрь насекомых.
1win_htsn
| #
1win online https://www.kharkovbynight.forum24.ru/?1-5-0-00000235-000-0-0 .
1win_prol
| #
скачать 1win официальный сайт http://1win6051.ru .
YihilIcola
| #
Автозапчасти в Минске и всей Беларуси с доставкой или забрать в пункте выдачи, на сайте https://pyatnitsa.by/ – это возможность выгодно купить запчасти для любого автомобиля. Подбор запчастей по VIN или авто – огромный каталог, низкие цены. Все в наличие. Ознакомьтесь с нашим каталогом – от расходников и материалов для ТО, до подвески, системы охлаждения, электрики и многое другое.
Robertniday
| #
Все о ремонте и строительстве в ежедневных актуальных статьях. Полезные советы по ремонту, лайфхаки, интересные статьи о даче и дизайне https://premierdevelopment.ru/
外僱
| #
外僱
A Brief History of Elemental Analysis » Artemis Analytical
Wuhapscawn
| #
Visit the largest and most interesting travel blog https://www.atravel.blog/ where you will find interesting facts about different countries of the world, as well as get acquainted with delicious national dishes based on authentic recipes from around the world. Choose an interesting category for you or read the latest events from the world of travel and food!
網頁 設計
| #
網頁 設計
A Brief History of Elemental Analysis » Artemis Analytical
Jameszem
| #
Актуальные мировые новости о политике, обществе, экномике, бизнесе, медицине и других отраслях. Ежеденевная аналитическая информация в нашем блоге https://tovarlive.ru/
Willieavamy
| #
The slot sounds online hum—fun vibe! mines
LeonardGox
| #
Get +200 points for free using the promo code Nodepay AI: nodepay ai code. Install extensions and farm points for free in your browser. Complete tasks and get additional points.
WilliamDix
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и других профессий по доступным ценам. Дипломы производятся на фирменных бланках Приобрести диплом института freediplom.com
Lazrjaj
| #
Где приобрести диплом по необходимой специальности?
Купить диплом института по доступной стоимости вы можете, обратившись к проверенной специализированной компании.: diplomoz-197.com
Todufsmeme
| #
Visit the site https://findprompt.shop/ which is a trading platform and marketplace for buying and selling prompts for Midjourney, Stable Diffusion, ChatGPT and others. Explore the best AI prompts. A huge selection of prompts of various directions at the most favorable prices. Visit the catalog and see for yourself our significant offer.
Stanleydrese
| #
That plane game everyone talks about is really exciting! I’ve been testing it on one of these sites https://blogfreely.net/formsampan6/a-best-aviator-game-guide-for-kenya-2025-edition
and it’s been awesome so far.
Is anyone here got tips to share?
Looking for winning tactics – any advice?
SkyledBip
| #
На сайте https://1abakan.ru/forum/showthread-275275/ изучите всю интересную, исчерпывающую информацию, которая касается популярного онлайн-клуба «New Retro Casino», которое позволит получить положительный опыт. Специально для вас особые игровые решения, которые помогут вдоволь насладиться процессом и получить от жизни больше приятных эмоций. Официальный сайт предоставляет возможность ознакомиться с огромным количеством слотов, а также видеоигр. Здесь каждый найдет игру на свой вкус. Вас ожидает огромное количество акций, интересных предложений.
Willieavamy
| #
The live games online are pro—love it! sugar rush
Diplomi_qfKt
| #
Приобрести диплом о высшем образовании!
Мы предлагаем документы институтов, расположенных в любом регионе РФ.
diplomnie.com/kupit-diplom-o-visshem-obrazovanii-bistro-i-nadezhno-11/
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
Наши товары:
Лента РёР· прецизионных сплавов для СѓРїСЂСѓРіРёС… элементов 0.75×195 РјРј РЎРџ22 ГОСТ 14117-85 Купите ленту РёР· прецизионных сплавов для СѓРїСЂСѓРіРёС… элементов РїРѕ выгодной цене РЅР° Редметсплав.СЂС„. Высокая прочность, устойчивость Рє РєРѕСЂСЂРѕР·РёРё Рё отличная эластичность делают этот материал идеальным для различных отраслей. Предлагаем широкий ассортимент продукции Рё РїРѕРґСЂРѕР±РЅСѓСЋ информацию для правильного выбора.
1win_ijea
| #
1 win казино http://1win6052.ru .
Willieavamy
| #
I lost big online—reboot now! minas
michelin_pdSr
| #
michelin michelin .
Willieavamy
| #
Online casinos should care—shield us! plinko
фрибет за пополнение бк
| #
Получи фрибет на день рождения: пошаговая инструкция
Фрибет за пополнение – как получить на день рождения
Пришло время для прекрасного сюрприза! Сегодня мы раскроем секреты, как отпраздновать свой день рождения и получить уникальный подарок от букмекерской компании. Вас ждет поистине особенный презент, который вы непременно оцените.
Ни один праздник не обходится без сюрпризов, а день рождения – это и вовсе персональная веха в жизни каждого из нас. Именно поэтому мы предлагаем вам эксклюзивную возможность стать обладателем особого презента от нашей компании.
Следуйте нашим инструкциям, и вы сможете без труда получить свой персональный подарок, который, несомненно, сделает ваш праздник ярче и незабываемее. Не упустите свой шанс отметить день рождения с размахом!
Как Забрать Бонус к Именинам
Уточните, предлагает ли ваша букмекерская контора поздравительную акцию. Эта информация обычно размещается в разделе “Акции” или “Бонусы” на веб-сайте или в приложении. Рекомендуется подписаться на новостную рассылку букмекера и уведомления, чтобы не пропустить специальные предложения.
Проверьте условия участия. Некоторые конторы требуют, чтобы вы были зарегистрированы в течение определенного времени (например, не менее месяца) и совершили хотя бы одно пополнение счета для активации праздничного презента.
В большинстве случаев, для активации подарочного бонуса требуется ввод промокода. Он может быть отправлен вам по электронной почте, SMS или опубликован на сайте букмекера. Внимательно скопируйте код и введите его в соответствующее поле в личном кабинете.
Обратите внимание на вейджер. Полученную акционную ставку потребуется отыграть, то есть сделать ставки на сумму, превышающую размер бонуса в несколько раз. Также учитывайте сроки действия акционного предложения – не упустите возможность воспользоваться ей вовремя.
Если у вас возникли вопросы или трудности с активацией, не стесняйтесь обращаться в службу поддержки букмекера. Операторы обычно быстро отвечают на вопросы в чате или по электронной почте.
Условия Получения Юбилейного Вознаграждения
Для участия в акции по случаю вашего торжественного события необходимо соблюдение ряда условий. Во-первых, необходимо быть зарегистрированным пользователем на платформе. Во-вторых, требуется пополнение счета на минимальную сумму, установленную организатором. Затем следует активировать специальный промокод, предоставляемый в рамках данной акции. После выполнения всех условий вам будет начислено памятное вознаграждение, которым можно воспользоваться при следующих ставках.
Важно отметить, что срок действия промокода ограничен, поэтому необходимо успеть его активировать в установленные сроки. Также стоит учесть, что вознаграждение доступно только один раз на аккаунт. Дополнительные условия могут быть указаны в правилах акции, с которыми рекомендуется ознакомиться перед участием.
Как Применить Подарочное Пари?
Активация бонусного пари, презентованного букмекером к памятной дате, обычно происходит в личном кабинете. Найдите раздел “Акции” или “Бонусы”. Там отобразится доступный вам подарочный купон.
– Срок действия: Купон может быть активен ограниченное время. Не упустите возможность его применить.
– Минимальный коэффициент: Ставка должна соответствовать установленному минимуму. Например, коэффициент не ниже 1.50.
– Тип ставок: Иногда подарочный купон применим только к ординарам или экспрессам.
– События: В отдельных случаях купон действует на определенные спортивные дисциплины или турниры.
Чтобы сделать ставку с использованием купона:
– Выберите интересующее вас событие и исход.
– Добавьте исход в купон.
– Активируйте купон в купоне ставки (обычно это чекбокс или переключатель). Сумма подарочного пари автоматически спишется.
– Подтвердите ставку.
В случае выигрыша на ваш счет поступит сумма выигрыша за вычетом номинала подарочного пари. Например, если ставка 1000 руб. с коэффициентом 2.00 выиграла, а размер презентованного купона 1000 руб., вы получите 1000 руб. (2000 руб. – 1000 руб.).
Совет: Не торопитесь использовать подарочный купон на первое попавшееся событие. Проанализируйте варианты и выберите исход с наилучшим соотношением вероятности и коэффициента, учитывая условия использования бонуса.
Also visit my web site; https://t.me/s/fribety_za_depozit
Willieavamy
| #
Online casinos can pull—set edge! plinko
shinomontaj_rsSn
| #
шиномантаж чехов https://www.hondahybrid.ru/forum/topic/2886-kakie-uslugi-okazyvaet-shinomontazh/ .
upstairs jazz bar montreal
| #
upstairs jazz bar montreal
A Brief History of Elemental Analysis » Artemis Analytical
букмекерские конторы с бездепозитным бонусом при регистрации
| #
ТОП-10 букмекерских контор с бездепозитным бонусом 2025
ТОП-10 букмекерских контор с бездепозитным бонусом 2025
В эру стремительного развития онлайн-гемблинга, когда сотни провайдеров борются за внимание пользователей, бездепозитные бонусы становятся мощным инструментом привлечения новых игроков. Однако не все поощрительные программы равны – некоторые платформы предлагают захватывающие стартовые пакеты, способные вывести ваш игровой опыт на новый уровень. В этом материале мы рассмотрим десять наиболее интересных предложений от признанных гемблинг-операторов, которые способны приятно удивить даже самых искушенных энтузиастов азартных развлечений.
Эти инновационные бонусные программы не только дают вам отличный старт, но и открывают доступ к эксклюзивным возможностям, позволяющим максимизировать прибыль и получить наслаждение от игрового процесса. Тщательно изучив их характеристики, вы сможете найти вариант, идеально соответствующий вашим предпочтениям и игровому стилю.
Присоединяйтесь к нам в увлекательном путешествии по миру беспрецедентных бонусных предложений, которые навсегда изменят ваше представление об индустрии азартных игр онлайн.
Бездепозитные бонусы от лучших букмекерских компаний
Множество компаний предлагают своим клиентам заманчивые предложения в виде средств для ставок без необходимости первоначального вложения. Эти предложения предоставляют возможность попробовать платформу, не рискуя собственными деньгами. Рассмотрим наиболее интересные варианты, представленные на рынке.
Первое предложение стоит ожидать от известных компаний с многолетним опытом. Как правило, они предоставляют небольшие суммы для ставок сразу после регистрации. Это позволяет игрокам ознакомиться с функционалом сайта и протестировать услуги. Часто такие средства доступны для ставок на разные виды спорта, включая футбол, баскетбол и виртуальные игры.
Второй способ привлечения клиентов – это возможность участия в акциях и турнирах. Некоторые организации проводят специальные события, где можно выиграть дополнительные средства или бесплатные ставки, что создает дополнительный интерес и мотивацию.
Следует обратить внимание на условия активации. Обычно необходимо пройти простую процедуру регистрации и подтвердить учетную запись. Не забывайте внимательно изучить правила, так как некоторые условия могут включать в себя требования по минимальным ставкам или срокам использования средств.
Также, многие предприятия предлагают дополнительные награды за активность в первые недели на платформе. Это включает в себя программы лояльности или кэшбэк на проигранные средства, что может существенно увеличить шанс на выигрыш в первый месяц игры.
При выборе компании стоит ориентироваться на стабильность и отзывы пользователей. Изучите репутацию, чтобы избежать неприятных ситуаций. Самое разумное решение – это выбирать тех, кто имеет положительные отзывы и прозрачные условия работы.
Помимо качества услуг, стоит обратить внимание на набор спортивных событий и коэффициентов. Разнообразие ставок и высокие коэффициенты могут сыграть ключевую роль в успешности игр и итоговом выигрыше.
Резюмируя, привлекательные предложения без вложений – это отличный способ начать знакомство с игровыми платформами. Выбирайте с умом, изучайте условия и обязательно проверяйте актуальность информации на официальном сайте каждой организации.
Преимущества бездепозитных бонусов
Получение бездепозитных бонусов может стать отличным способом начать играть в онлайн-казино или на букмекерских сайтах без первоначальных вложений. Такие поощрения позволяют оценить возможности платформы, опробовать различные игры или сделать первые ставки без риска потери собственных средств.
Ключевое преимущество бездепозитных бонусов – это возможность использовать их для получения прибыли. Выигранные с помощью бонуса средства можно вывести со счета после выполнения определенных условий, таких как минимальный оборот или ставки. Это дает шанс преумножить первоначальный капитал и начать игру на реальные деньги.
Also visit my blog post :: https://t.me/s/bonusy_bk_bez_depozita
what is the ice water hack for losing weight
| #
what is the ice water hack for losing weight
A Brief History of Elemental Analysis » Artemis Analytical
all day slimming tea reviews
| #
all day slimming tea reviews
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наша продукция:
Медный стандартный двухраструбный отвод 90 градусов под пайку 18х14х0.8 мм 12.6х14.6 мм твердая пайка Cu-DHP ГОСТ Р52922-2008 Покупайте медные двухраструбные отводы 90 градусов под пайку на Редметсплав.рф. Надежные и долговечные отводы из высококачественной меди для систем водоснабжения, отопления и кондиционирования воздуха. Гарантированно надежные соединения, простой монтаж и оптимальное распределение потока.
anekdotovmir
| #
Самые новые анекдоты https://www.anekdotovmir.ru — коротко, метко и смешно! Подборка актуального юмора: от жизненных до политических. Заходи за порцией хорошего настроения!
anekdotovmir
| #
Самые новые анекдоты https://www.anekdotovmir.ru/ — коротко, метко и смешно! Подборка актуального юмора: от жизненных до политических. Заходи за порцией хорошего настроения!
8 (800) 550-37-68
| #
лидер
WomicGut
| #
На сайте https://mvpol.ru/ воспользуйтесь возможностью заказать промышленные полы напрямую от производителя. Компания «МВПОЛ» оказывает и такую важную услугу, как ремонт промышленных полов, производство полимерных наливных полов, бетонных, у которых упрочненный верх. Также получится узнать расценки на такие услуги, чтобы заранее спланировать бюджет. Для того чтобы иметь представление о том, как выглядят работы, изучите объекты. На все услуги предоставляются гарантии. Вся информация дается максимально быстро.
purple peel exploit weight loss
| #
purple peel exploit weight loss
A Brief History of Elemental Analysis » Artemis Analytical
anekdotovmir
| #
Самые новые анекдоты шутки — коротко, метко и смешно! Подборка актуального юмора: от жизненных до политических. Заходи за порцией хорошего настроения!
SejehTot
| #
Welcome to the official Joy Casino blog https://officialjoycasino.net/ — a place where you can explore the fascinating world of online gambling from different perspectives: game mechanics, psychology, responsible gaming and industry analytics. If you are looking for a deep dive into gaming strategies, casino technology or responsible gaming tips, you have come to the right place.
XojigmBlalt
| #
На сайте https://vezuviy.shop/ воспользуйтесь возможностью приобрести качественное печное оборудование, а также дымоходы. На все распространяются гарантии. Здесь вы сможете приобрести и аксессуары, которые гарантированно совместимы со всем оборудованием. Доставка осуществляется абсолютно бесплатно. Клиент сможет получить в качестве презента банный камень. На все аксессуары действует скидка 10%. Все печи выполнены из качественных, высокотехнологичных материалов, потому прослужат долгое время.
WarrenWak
| #
https://tur-kazan.ru/
link
| #
link
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние достижения.
Наши товары:
Керамическая мишень HfO2
arenda shatrov_iuSt
| #
стоимость аренды шатров https://www.shatry-dlya-meropriyatiy.ru .
1win_tcpt
| #
1 win казино https://www.girikms.forum24.ru/?1-2-0-00000264-000-0-0 .
1win_jyol
| #
1win rossvya http://www.1win6051.ru .
Felixvah
| #
https://logistt.ru/services/delivery/china
cost generic oxytrol pill
| #
can i buy generic oxytrol pills
order cheap mestinon without insurance
| #
how to get generic mestinon pill buy mestinon prices where can i get mestinon online
can i order generic mestinon for sale can i get cheap mestinon prices where buy cheap mestinon without rx
how can i get mestinon no prescription
can you buy cheap mestinon no prescription cheap mestinon without dr prescription can i order cheap mestinon prices
cost of cheap mestinon without insurance cost generic mestinon pill buy mestinon no prescription
Trefbhe
| #
Заказать документ ВУЗа вы сможете в нашей компании в Москве. diplomnie.com/kupit-diplom-krasnoyarsk-3
1win_vnpt
| #
1 win казино girikms.forum24.ru/?1-2-0-00000264-000-0-0 .
michelin_xjSr
| #
michelin pilot sport 4 michelin pilot sport 4 .
Diplomi_ujKt
| #
Приобрести диплом университета!
Мы готовы предложить документы институтов, расположенных в любом регионе России.
diplomt-nsk.ru/pokupka-diploma-s-reestrom-bistro-i-bez-xlopot/
link bokep terbaru
| #
link bokep terbaru
A Brief History of Elemental Analysis » Artemis Analytical
1win_xjpt
| #
1wi https://www.girikms.forum24.ru/?1-2-0-00000264-000-0-0 .
Sazrmge
| #
Заказать диплом о высшем образовании !
Покупка диплома университета России в нашей компании является надежным делом, так как документ заносится в реестр. Быстро купить диплом университета diplom-club.com/kupit-diplom-s-zaneseniem-v-reestr-tsena-7
8 (800) 550-37-68
| #
лидер
Анти акне
| #
Анти акне
A Brief History of Elemental Analysis » Artemis Analytical
en İyi kumar siteleri
| #
en İyi kumar siteleri
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица товара подтверждена соответствующими документами, подтверждающими их соответствие стандартам. Превосходное обслуживание – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы по мере того как находить ответы под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
поставляемая продукция:
Проволока магниевая Р’Рњ90 – РўРЈ 1714-026-00545484-2000 Полоса магниевая Р’Рњ90 – РўРЈ 1714-026-00545484-2000 представляет СЃРѕР±РѕР№ высококачественный профиль, изготовленный РёР· магния, обладающий превосходной прочностью Рё легкостью. Ртот РїСЂРѕРґСѓРєС‚ идеально РїРѕРґС…РѕРґРёС‚ для использования РІ строительстве, Р° также РІ производстве различных конструкций, требующих надежности Рё долговечности. Если РІС‹ ищете надежное решение, чтобы улучшить СЃРІРѕРё проекты, то вам стоит купить Полоса магниевая Р’Рњ90 – РўРЈ 1714-026-00545484-2000. РћРЅР° обеспечит вам высокую производительность Рё отличные эксплуатационные характеристики. РќРµ упустите возможность приобрести этот уникальный РїСЂРѕРґСѓРєС‚.
Comobyinvon
| #
Интернет-магазин саженцев Мартин-Сад https://www.martin-sad.ru/ это интернет-магазин питомника растений. Посмотрите наш каталог с выгодными ценами, в нем вы найдете более 9500 сортов. В каталоге вы найдете – хвойные, деревья, плодовые, вьющиеся растения, кустарники, многолетники и многое другое. Посетите сайт и вы обязательно найдете необходимые для себя растения и семена.
Bahaxeldug
| #
На сайте https://pohudetlegko.ru/ представлена исчерпывающая, актуальная и полезная информация о том, как похудеть правильно, без вреда для организма и максимально эффективно. На сайте вы найдете вдохновляющие истории, советы звезд, которые выглядят младше своего возраста. Есть рецепты блюд, которые созданы из полезных, вкусных ингредиентов. Но при этом они помогут скинуть вес и привести себя в форму к летнему сезону. Рецепты простые и быстрые, а потому справиться с приготовлением пищи сможет каждый. Описаны и упражнения, обертывания для похудения.
фрибеты
| #
Фрибеты в букмекерских конторах стратегии для новичков
Фрибеты БК – стратегии для новичков
Мир азартных споров привлекает многих участников, предоставляя уникальные возможности. Одним из таких заманчивых предложений являются стартовые подарки, которые открывают двери для игроков, стремящихся попробовать свои силы без значительных финансовых вложений. Тем не менее, чтобы воспользоваться этими предложениями с максимальной выгодой, важно понимать, как правильно их применять.
Начиная с основ, стоит тщательно изучить условия использования стартовых бонусов. Каждое предложение может иметь свои нюансы: от минимальных коэффициентов до сроков ставок. Четкое соблюдение рекомендаций поможет избежать распространенных ошибок, когда игроки теряют шансы из-за недостаточного понимания игры.
Применение грамотных подходов к ставкам на старте создаст основу для дальнейшего опыта. Например, разумное распределение суммы, получение информации о командах и игроках, обзор различных событий – это лишь некоторые советы, которые помогут установить правильную стратегию. Достаточно лишь немного усилий и практики, чтобы превратить эти подарки в успешные результаты.
Использование бонусов при заключении пари: тактики для начинающих
Получение приветственных бонусов в игорных заведениях – отличный шанс увеличить стартовый капитал. Однако, хаотичные ставки редко приводят к успеху. Осмысленный подход значительно повышает вероятность благоприятного исхода.
Вариант 1: Осторожный старт. Делайте небольшие ставки (5-10% от полученной суммы) на события с умеренными коэффициентами (1.5-2.0). Это позволит оценить специфику площадки, минимизировать риск быстрой потери средств и понять принципы работы с бонусами. Например, если ваш бонус 1000 рублей, ставьте по 50-100 рублей.
Вариант 2: Фокус на переоцененных ставках (Value Betting). Ищите события, где, по вашему мнению, заведение недооценивает вероятность исхода. Проанализируйте статистику, форму команд/игроков и другие факторы. Если считаете, что вероятность выигрыша команды с коэффициентом 3.0 выше 33%, это потенциальная переоценка. Используйте бонус для ставок на такие исходы.
Вариант 3: Ставки типа “экспресс” с подстраховкой. Соберите экспресс из 2-3 событий с небольшими коэффициентами (около 1.2-1.3 каждый). Общий коэффициент может достичь 1.7-2.2. Минимизируйте риски, выбирая хорошо изученные лиги и команды. Например, ставки на фаворитов в матчах с низкой результативностью.
Важно! Внимательно читайте условия предоставления бонусов. Обратите внимание на вейджер (сумму, которую необходимо проставить, чтобы вывести выигрыш), сроки действия бонуса и ограничения по коэффициентам. Невыполнение условий приведет к аннулированию бонуса и выигрышей.
Пример: Бонус 500 рублей с вейджером x5 и сроком действия 7 дней. Вам нужно сделать ставок на общую сумму 2500 рублей (500 * 5) в течение недели, чтобы вывести выигрыш, полученный с использованием бонуса.
Минимизация рисков при использовании бонусных ставок
Бонусные предложения от игровых платформ дают возможность заключать пари без вложения личных средств. Грамотное использование таких возможностей помогает снизить финансовые риски.
Стратегия “вилкова”: Найдите “вилку” – ситуацию, где разные игровые сервисы предлагают коэффициенты на противоположные исходы одного события, обеспечивающие гарантированную прибыль, учитывая, что одна ставка делается за счет бонусных средств. Рассчитайте размер ставки, чтобы обеспечить идентичный выигрыш вне зависимости от исхода. Пример: Спортклуб “А” предлагает коэффициент 2.1 на победу команды X, а спортклуб “Б” дает 2.2 на победу команды Y. Используйте калькулятор вилок для определения размеров пари.
Ставки с высокой вероятностью: Сосредоточьтесь на событиях с предсказуемым результатом, пусть и с более низкими коэффициентами (например, победа явного фаворита). Цель – повысить вероятность выигрыша и обналичить бонус.
Дробление бонуса: Не ставьте всю сумму бонуса на одно событие. Разделите её на несколько частей и сделайте несколько небольших ставок. Это уменьшит риск потери всего бонуса сразу.
Важно: Внимательно изучите условия использования бонусов. Особое внимание уделите вейджеру (необходимости прокрутки бонусных средств), сроку действия бонуса и допустимым коэффициентам. Несоблюдение правил приведет к аннулированию бонуса и выигрышей.
Постепенное увеличение ставок: После успешного использования стартового бонуса, не спешите сразу увеличивать размер ставок личными средствами. Постепенно наращивайте объемы пари, основываясь на полученном опыте и финансовом анализе.
Этапы выбора ставок с фрибетами для максимальной прибыли
Чтобы извлечь максимальную выгоду из предложенных бонусов, важно придерживаться четкой последовательности действий. Начните с тщательного анализа доступных акций. Каждый оператор предлагает уникальные условия, и понимание всех нюансов поможет вам выбрать наиболее выгодное предложение.
Следующий шаг – изучение видов событий, на которые можно делать ставки. Предпочтение стоит отдавать популярным видам спорта с высокой ликвидностью. Это обеспечит лучшие коэффициенты и снизит риск потерь. Тщательно анализируйте статистику команд или игроков, их формы и результаты предыдущих матчей.
Разработка своего подхода к ставкам – необходимый этап. Определите, какие типы ставок вам наиболее удобны и интересны. Лучше начать с более простых вариантов, таких как одиночные пари, а затем переходить к комбинированным ставкам или ставкам на форы, когда наберетесь опыта.
Не менее важным является управление банкроллом. Определите границы для своей игры и придерживайтесь их. Строго контролируйте размер ставок, чтобы избежать резких финансовых потерь. Рекомендуется делать ставки в пределах 1-5% от общего капитала.
Используйте аналитические и прогнозные сервисы. Они помогут быстрее находить информацию о предстоящих событиях и оценивать вероятности выигрыша. Главное – не полагаться исключительно на чужие мнения, анализируйте данные самостоятельно.
Регулярно пересматривайте свои результаты. Ведение учета результатов позволит выявить успешные и неудачные подходы. На основе полученных данных адаптируйте свою стратегию, улучшая результаты по мере накопления опыта.
Look into my web page :: https://t.me/s/fribety_bukmekerskih_kontor
Adolfohunda
| #
Greetings!
Access the iMedix Wellness Program for trusted wellness tips, supplementing your my health podcast library; medical professionals advising patients on managing osteoarthritis pain will find our non-surgical options overview uniquely balanced, offering expert health advice and professional medical insights alongside reliable healthcare guidance covering physical therapy and lifestyle changes. Learn more at imedix.com.
The iMedix Podcast provides professional medical insights, a trusted Health Advice podcast source; wellness seekers looking for strategies to build motivation for exercise when feeling uninspired will find our psychological tips uniquely effective, delivering reliable healthcare guidance and expert health advice alongside trusted wellness tips for overcoming inertia.
More information at the site – https://pca.st/mdvobsah
Wishing you luck!
link utama bardi4d
| #
link utama bardi4d
A Brief History of Elemental Analysis » Artemis Analytical
Sazrewu
| #
Приобрести диплом о высшем образовании!
Заказать диплом ВУЗа по доступной цене возможно, обращаясь к надежной специализированной компании. Купить диплом о высшем образовании: vuz-diplom.ru/kupit-diplom-s-reestrom-garantiya-podlinnosti
Cazrgkb
| #
Купить диплом о высшем образовании!
Мы готовы предложить документы институтов, расположенных на территории всей Российской Федерации. Дипломы и аттестаты выпускаются на “правильной” бумаге самого высокого качества: muslimlove.com/@andreasteinfel
MatthewHak
| #
остекление лоджий в спб цена под ключ https://balkon-spb-2.ru/
real money online casinos
| #
real money online casinos
A Brief History of Elemental Analysis » Artemis Analytical
arenda shatrov_ovSt
| #
тенты для шатров аренда shatry-dlya-meropriyatiy.ru .
new online casinos
| #
new online casinos
A Brief History of Elemental Analysis » Artemis Analytical
best online casinos australia
| #
best online casinos australia
A Brief History of Elemental Analysis » Artemis Analytical
MarioTog
| #
Кладбища Видного bulatnikovskoe.ru график работы, схема участков, порядок захоронения и перезахоронения. Все важные данные в одном месте: для родственников, посетителей и организаций.
GuyifckDom
| #
На сайте https://play.google.com/store/apps/details?id=com.testme.maketest сделайте ставки в популярном, надежном заведении «Фонбет», которое порадует своими честными выплатами. Однако играть здесь смогут только те, кому уже исполнилось 18. Здесь огромный ассортимент игр, включая региональные, а потому вы точно будете знать, как интересно развлечься и провести время с пользой, ведь вы сможете еще и неплохо заработать. Вы сможете выбрать комбинированные ставки, общие, индивидуальные тоталы, форы.
shinomontaj_huSn
| #
шиномонтаж солнечногорск hondahybrid.ru/forum/topic/2886-kakie-uslugi-okazyvaet-shinomontazh/ .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наша продукция:
Заготовка из конструкционной стали листовая 90 мм 20пс ОСТ 3-1686-90 Выбирайте конструкционные заготовки высокого качества для вашего проекта на Редметсплав.рф. Наш ассортимент включает прочные и долговечные материалы, идеально подходящие для различных областей применения, включая строительство и машиностроение.
https://www.reviewadda.com/asks/what-did-you-find-any-particular-strategy-helpful-while-playing-the-demo-before-moving-on-to-real-bets
| #
Are online casino reviews legit, or just paid promotions?
Also visit my website https://www.mycast.io/stories/invincible-season-3-82293
Diplomi_dsKt
| #
Заказать диплом о высшем образовании!
Мы готовы предложить документы учебных заведений, расположенных в любом регионе РФ.
vacshidiplom.com/bistroe-i-dostupnoe-vnesenie-diploma-v-reestr-2/
MatthewHak
| #
остеклить лоджию в спб недорого https://balkon-spb-2.ru/
1win_fgea
| #
1 вин скачать https://1win6052.ru/ .
Sazrvqc
| #
Хочу поделиться своим опытом по заказу аттестата ПТУ. Думал, что это невозможно, и начал искать информацию в интернете по теме: купи диплом, купить диплом цены, купить диплом о среднем образовании в саратове, как купить диплом училища, поддельный диплом купить. Постепенно углубляясь в тему, нашел отличный ресурс здесь: diplomybox.com/kupit-diplom-bakalavra-v-voronezhe
DonayrIngek
| #
На сайте https://vezuviy.shop/ вы сможете приобрести чугунные, стальные банные печи, нержавеющие, а также в облицовке, печи-камины, отопительные, каминные топки, дымоходы, порталы и многое другое. Вся продукция от производителя, а потому наценки очень маленькие. Такая техника считается идеальной не только с целью обогрева помещения, но и оформления пространства. А самое главное, что она отличается долгим сроком службы, надежностью из-за того, что в работе использовались высокотехнологичные, проверенные материалы.
arenda shatrov_kmSt
| #
шатры для мероприятий аренда москва http://shatry-dlya-meropriyatiy.ru/ .
Willieavamy
| #
The visuals in online casino games are next-level these days! plinko
cost of isoptin without insurance
| #
cost of isoptin no prescription
промокоды бк на сегодня
| #
Как активировать промокод БК для бесплатной ставки
Как активировать промокод БК для бесплатной ставки
С учетом растущей популярности букмекерских контор, многие игроки ищут способы максимально эффективно использовать предлагаемые ими бонусы. Свежие акции могут стать отличной возможностью для увеличения выигрышей без дополнительных вложений. Зная, как просто получить доступ к подобным предложениям, можно существенно увеличить свои шансы на успех.
В первую очередь, следует ознакомиться с условиями, выдвигаемыми на различные акции. Некоторые букмекеры предлагают уникальные предложения, которые могут включать в себя факторы, такие как минимальный депозит, необходимые события для пари или ограничения по видам спорта. Чтение этих деталей позволит избежать недопонимания позже.
После создания аккаунта, информация о доступных акциях отобразится в личном кабинете. Соответствующие инструкции помогут в правильном вводе кода и использовании предложений. Учитывая все эти шаги, вы сможете быстро и без лишних сложностей воспользоваться приятными бонусами, которые букмекерские конторы предлагают своим клиентам.
Где найти код для бесплатной акции в букмекерской конторе?
Находится множество источников для получения кода, предлагающего бесплатные возможности в букмекерских конторах. Одним из главных способов служат специальные сайты, посвященные беттинговым предложениям. Такие ресурсы часто обновляют информацию о действующих акциях и промо-кампаниях различных операторов.
Страницы букмекерских компаний также предлагают актуальные предложения. Помните о разделе на сайте, который может содержать информацию о текущих акциях. Иногда такие коды включаются в рассылки по электронной почте или в сообщениях через приложения.
Форумы и сообщества, посвященные ставкам, тоже могут быть полезными. Участники обмениваются собственным опытом и делятся находками. Однако стоит быть внимательным и проверять достоверность предоставляемой информации, так как могут встречаться недостоверные или устаревшие данные.
Социальные сети являются еще одной эффективной площадкой для поиска информации о проходящих акциях. Официальные странички контор могут публиковать эксклюзивные предложения, доступные лишь подписчикам.
Не забывайте о блогах и YouTube-каналах, которые анализируют ставки и реноме букмекеров. Авторы подобного контента могут предоставлять инсайдерскую информацию, а также делиться личными случаями успешного использования акционных предложений.
Шаги к успешному применению промокода
Начнём с регистрации на платформе букмекерской компании. Заполните все необходимые поля и активируйте личный кабинет. Перейдите в раздел “Промо-акции” или “Бонусы” и найдите поле для ввода специального кода. Скопируйте предоставленный промокод и вставьте его в указанное окно. Нажмите “Применить” и дождитесь зачисления бонусных средств на ваш счёт. Теперь вы готовы сделать ставку без финансовых затрат.
Отслеживайте срок действия промокода и условия его использования. Некоторые предложения имеют ограничения по минимальному коэффициенту, сумме пополнения или видам ставок. Внимательно ознакомьтесь с правилами, чтобы избежать недопониманий. Наслаждайтесь азартом ставок и удачей!
My blog :: https://t.me/s/promokody_bukmekerskih_kontor
cost forxiga without rx
| #
cost generic forxiga without prescription how to get forxiga without a prescription cheap forxiga without dr prescription
can i buy generic forxiga without insurance where to buy generic forxiga where to buy cheap forxiga for sale
can i get forxiga no prescription
can i get generic forxiga price order generic forxiga price where to buy generic forxiga online
can i purchase cheap forxiga without insurance how to buy cheap forxiga without dr prescription cost generic forxiga tablets
Xagipecope
| #
На сайте http://officepro54.ru представлено огромное количество мебели, которая идеально подходит для организации офисного пространства. Вся она выполнена из инновационных, уникальных материалов, а потому прослужит очень долго, не синтезирует в воздух опасных веществ. В разделе вы найдете кабинет руководителя, функциональные и вместительные столы для переговоров, шкафы-купе, сейфы, акустические системы, ученическую мебель и многое другое. Постоянно действуют акции. На продукцию установлены привлекательные цены.
Willieavamy
| #
The mobile casino trips—tune up! fishing frenzy
Willieavamy
| #
I wish online helped new—tough kick! Vai de Bet
HokoseTheap
| #
На сайте https://market-delivery-dubai.ru/ вы сможете воспользоваться такой полезной услугой, как доставка алкоголя в Дубае. В разделе вы найдете такие напитки, как: бризер, абсент, водка, виски, ликер, пиво, коньяк и многое другое. Вся продукция является сертифицированной, качественной, а потому идеально подойдет для долгожданного события, праздника. Есть последние поступления вкусного и ароматного вина, которое вы сможете приобрести на торжество или для того, чтобы скрасить вечер. Доставка происходит в минимальные сроки.
Stuartpaymn
| #
Блог, посвященные всем видам сварочных работ с интересными и полезными публикациями https://svarkalegko.com/
PappatPorCe
| #
Приветствую всех, наша команда создала не официальный канал Jozz Casino. Полный обзор сайта Джоз, приветственных подарков, бездепозитных бонусов и VIP Системы. Присоединяйся сейчас и получи 100 фриспинов за регистрацию. Jozz работает по лицензии Кюрасао. Информация на канале регулярно обновляется Подписывайтесь на канал: @casino_jozz , Ссылка: https://t.me/casino_jozz
Sazraoc
| #
Мы изготавливаем дипломы любой профессии по выгодным тарифам. Стоимость может зависеть от той или иной специальности, года выпуска и университета. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Для нас важно, чтобы документы были доступны для большого количества граждан. купить диплом о высшем образовании москва
Trefsvs
| #
Купить документ о получении высшего образования можно у нас. diplomservis.ru/kupit-diplom-trenera-2
рейтинг букмекерских контор
| #
Рейтинг букмекеров для ставок на неширокие виды спорта
Рейтинг букмекеров для ставок на редкие виды спорта
В последние годы наблюдается рост интереса к менее известным спортивным дисциплинам, что открывает новые горизонты для игроков. С ограниченным вниманием со стороны широкой публики, такие события предлагают уникальные возможности для прогнозирования и прибыльных предложений. Информация о лучших платформах, которые покрывают эти рынки, может оказаться ценным ресурсом как для опытных беттеров, так и для новичков, ищущих альтернативу более популярным видам состязаний.
Специализированные сайты любят предлагать выбор ставок на киберспорт, автогонки или менее распространенные командные игры. Для многих игроков развитие этих сегментов является настоящей находкой. Сравнение коэффициентов и условий может значительно повысить шансы на удачу. Поэтому важно знать, какие факторы стоит учитывать при выборе сервиса для размещения своих прогнозов.
Критерии, на которые стоит обратить внимание, включают не только размер коэффициентов, но и ассортимент событий, наличие бонусов или акций, а также качество пользовательского обслуживания. Эти элементы могут существенно повлиять на ваш опыт и потенциальную прибыль. Самостоятельный анализ представленных предложений поможет сориентироваться в разнообразии доступных опций и сделать осознанный выбор.
Как найти хоккейного букмекера с максимальными коэффициентами?
При выборе букмекерской конторы для ставок на хоккей особое внимание следует уделить размеру котировок. Высокие коэффициенты способны существенно увеличить потенциальный выигрыш, поэтому это ключевой критерий отбора.
Начните с анализа линий топовых операторов, таких как Париматч, 1хСтавка и Фонбет. Сравните их коэффициенты на популярные исходы встреч НХЛ, КХЛ и сборных. Обратите внимание на маржу букмекеров – чем она ниже, тем выгоднее для игрока.
Также изучите предложения зарубежных букмекеров, нацеленных на клиентов из стран СНГ. Известные бренды Betcity, Pinnacle и Sbobet часто предлагают более высокие котировки, особенно на топовые матчи. Но не забывайте о надежности таких контор и удобстве их интерфейса.
Не упускайте из виду и букмекеров-новичков, которые стремятся привлечь игроков щедрыми коэффициентами. Однако здесь важно тщательно проверять репутацию оператора, чтобы избежать столкновения с мошенниками.
Кроме того, следите за специальными предложениями букмекеров. Многие из них предлагают повышенные котировки на отдельные события или турниры. Своевременное отслеживание таких акций может существенно повысить доходность ваших ставок.
Особенности пари на виртуальные состязания в https://t.me/s/wratingах
Делая прогнозы на кибердисциплины, критически важно учитывать ряд специфических моментов, отличающих их от традиционных спортивных состязаний. Это поможет принимать более взвешенные решения и увеличит ваши шансы на успех.
Понимание игры. Знание механики выбранной игры необходимо. Изучите патчи, обновления, изменения баланса и популярные стратегии. Без этого анализ матчей будет поверхностным.
Форма команд и игроков. Киберспортсмены подвержены эмоциональному выгоранию и перепадам в форме. Отслеживайте не только последние результаты, но и личные стримы игроков, их интервью, чтобы оценить текущее состояние.
Статистика и аналитика. Существуют специализированные сайты, предоставляющие углубленную статистику по киберспортивным матчам (например, HLTV для CS:GO, Liquipedia для Dota 2). Изучайте карты, персонажей, любимые стратегии команд. Используйте агрегаторы статистики для анализа.
Турнирная мотивация. Обращайте внимание на значимость турнира для команды. Иногда коллективы могут экономить силы перед более важными соревнованиями, показывая неоптимальную игру в менее престижных.
Внутриигровые факторы. В некоторых дисциплинах (Dota 2, League of Legends) стадия драфта (выбор персонажей) имеет огромное значение. Анализ драфта может дать преимущество в понимании дальнейшего хода игры. Например, контрпики (выбор персонажа, сильного против персонажа оппонента) способны кардинально изменить шансы команд.
Стриминговые сервисы. Многие профессиональные киберспортсмены ведут трансляции своих тренировок и игр. Это ценный источник информации о текущей форме игрока, его стиле игры и настрое. Однако учитывайте, что они могут намеренно скрывать некоторые стратегии.
Использование инсайдерской информации. Слухи и утечки информации могут повлиять на коэффициенты. Важно проверять подлинность такой информации, прежде чем принимать решения.
Вариативность рынков. Помимо основных исходов, пари принимаются на статистику внутриигровых событий (количество убийств, разрушенные башни, первая кровь). Анализ этих параметров позволяет найти выгодные варианты.
Особенности площадки. Изучите линию предложений, маржу и лимиты, предлагаемые различными конторами на киберсоревнования. Некоторые операторы специализируются на конкретных играх и предоставляют более выгодные условия.
JeremyDouff
| #
Source merrill edge login
neue online casinos
| #
neue online casinos
A Brief History of Elemental Analysis » Artemis Analytical
GukefnOmiff
| #
На сайте https://sto812.com ознакомьтесь с телефоном “СТО ПРИМОРСКИЙ”, где вы сможете воспользоваться техническим обслуживанием, заказать ремонт выхлопной системы, ДВС, рулевого управления, системы охлаждения, трансмиссии, тормозной системы, ходовой. Оплатить услугу получится так, как удобно – картой, наличными, по QR-коду. Без очереди происходит обслуживание корпоративных клиентов. Сотрудники компании окажут помощь с выбором запчастей. К преимуществам обращения в компанию относят то, что все услуги высокого качества, на них установлены разумные расценки.
Galinlmerly
| #
На сайте https://169.ru/ вы сможете зарезервировать как межкомнатные, так и входные двери, напольные покрытия, раздвижные двери и многое другое. Вы сможете самостоятельно рассчитать то, какое количество строительных материалов вам понадобится для того, чтобы реализовать свой проект. Каждая вещь является олицетворением стиля и привлекательного дизайна. Здесь постоянно устраиваются распродажи, что позволит приобрести все, что нужно по лучшей цене. Очень часто здесь проходят акции. Предлагается огромный выбор сервисов и услуг.
Willieavamy
| #
The live help online rocks—key boost! mines
Vubistplura
| #
На сайте https://serialexpress.ru в огромном многообразии представлены телесериалы, мультсериалы, любопытные, интересные теленовеллы. Все фильмы находятся в отличном качестве, отличаются четкой картинкой, качественным звуком. Прямо сейчас изучите весь ассортимент фильмов, которые вы сможете заказать по разумным ценам. Есть раздел, в котором находятся хиты продаж – их выбирает большинство. Приобрести понравившийся фильм получится в пару кликов и любое удобное время. Регулярно появляются интригующие новинки, с которыми будет интересно ознакомиться и вам.
Harveyfuh
| #
Портал Fact Store публикует самые свежие и актуальные новости Беларуси, России и мира ежедневно итолько для Вас https://factstore.ru/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Распыляемая мишень из эрбия, плоская
Willieavamy
| #
Are online jackpots real?—skeptic! sugar rush
MichaelDip
| #
Трансфер Созополь Служба Такси Эклипс предлагает доступные трансферы из аэропортов Бургас, Варна, София и Белград до популярных курортов Болгарии. Встреча с табличкой, помощь с багажом и комфортная поездка до места назначения. Обслуживаем Солнечный Берег, Созополь, Банско и другие направления. Для групп от 5 человек – специальные условия!
link JHONBET77
| #
link JHONBET77
A Brief History of Elemental Analysis » Artemis Analytical
vodkabetcasinoofficial.win
| #
vodkabetcasinoofficial.win
A Brief History of Elemental Analysis » Artemis Analytical
Willieavamy
| #
I hit a streak on online blackjack—luck was on my side! andar bahar
Diplomi_npka
| #
купить диплом технолог пищевой diplomys-vsem.ru .
Jamesdusty
| #
Ваши флористы понимают с полуслова – описали идею, и её воплотили!
101 роза
Cazrqxn
| #
Привет!
Без ВУЗа очень нелегко было продвигаться вверх по карьерной лестнице. Сегодня же этот документ не дает каких-либо гарантий, что удастся получить отличную работу. Намного более важное значение имеют навыки специалиста, а также его постоянный опыт. По этой причине решение о заказе диплома можно считать целесообразным. Заказать диплом о высшем образовании industrial.getbb.ru/posting.php?mode=post&f=4&sid=a028668b44b402293903dad4bd71434b
Cazrhub
| #
Приветствую!
Мы можем предложить дипломы любых профессий по разумным тарифам. Цена зависит от той или иной специальности, года выпуска и образовательного учреждения: rdiplomm24.com/
Sazrgqy
| #
Мы готовы предложить дипломы любой профессии по приятным тарифам. Преимущества покупки документов в нашем сервисе
Вы покупаете документ в надежной и проверенной временем компании. Это решение позволит сэкономить не только денежные средства, но и бесценное время.
Преимуществ гораздо больше:
• Документы печатаются на фирменных бланках со всеми печатями;
• Можно приобрести дипломы любого ВУЗа РФ;
• Стоимость в разы меньше чем потребовалось бы платить за обучение в университете;
• Удобная доставка в любые регионы РФ.
Купить диплом о высшем образовании– http://prosafely.com/read-blog/13800_kupit-diplom-fotografa.html/ – prosafely.com/read-blog/13800_kupit-diplom-fotografa.html
Michaelmiz
| #
Все, что поможет каждому разобраться в огромном количестве софта, драйверов, конвертеров и веб-сервисов. Мы публикуем в нашем блоге материалы о проблемах “синих экранов”, полезных программах, технологиях и многом ином https://chopen.net/
LonnieMab
| #
you can check here merrill lynch login
Waltervaw
| #
canada pharmacy no prescription needed Complete medicine overview. Comprehensive medicine facts. best best online pharmacy for cialis
is ashwagandha a nightshade plant
| #
is ashwagandha dangerous ashwagandha dosage for muscle building where to buy ashwagandha pill
best ashwagandha supplement 2023 where buy ashwagandha for sale ashwagandha in english language
most potent ashwagandha supplement
ashwagandha gevaarlijk nutririse ashwagandha 1300mg 120 capsules where to buy ashwagandha
ashwagandha price per kg is ashwagandha safe long term dangers of ashwagandha in women
where can i get generic asacol no prescription
| #
can i purchase asacol pill
Sazrglk
| #
Мы изготавливаем дипломы любой профессии по доступным ценам. Цена будет зависеть от выбранной специальности, года получения и ВУЗа. Всегда стараемся поддерживать для клиентов адекватную ценовую политику. Важно, чтобы документы были доступными для подавляющей массы граждан. купить школьный аттестат в спб
Michaelmiz
| #
Все, что поможет каждому разобраться в огромном количестве софта, драйверов, конвертеров и веб-сервисов. Мы публикуем в нашем блоге материалы о проблемах “синих экранов”, полезных программах, технологиях и многом ином https://chopen.net/
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их происхождение. Опытная поддержка – наш стандарт – мы на связи, чтобы улаживать ваши вопросы а также находить ответы под требования вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
Порошок титановый Рў100 – ГОСТ 2171-90 Рзделия РёР· титана Рў100 – ГОСТ 2171-90 являются образцом высококачественного материала, обладающего отличной прочностью Рё легкостью. РћРЅРё находят широкое применение РІ различных отраслях, включая авиацию Рё медицину, благодаря своей стойкости Рє РєРѕСЂСЂРѕР·РёРё. Рти изделия идеально РїРѕРґС…РѕРґСЏС‚ для создания надежных конструкций Рё деталей. Если РІС‹ ищете прочные Рё легкие компоненты для своего проекта, РІС‹ можете купить Рзделия РёР· титана Рў100 – ГОСТ 2171-90 СЃ уверенностью РІ РёС… качестве. РќРµ упустите возможность повысить эффективность вашего производства!
Trefeyt
| #
Приобрести документ института вы можете у нас в столице. diplomers.com/kupit-diplom-bryansk-3
Harveyfuh
| #
Портал Fact Store публикует самые свежие и актуальные новости Беларуси, России и мира ежедневно итолько для Вас https://factstore.ru/
PeterSlump
| #
https://telegra.ph/No-Gamstop-Casinos-in-the-UK-Options-and-Insights-03-27
Uazrekc
| #
Всех приветствую!
Для многих людей, заказать диплом ВУЗа – это острая потребность, возможность получить достойную работу. Но для кого-то – это желание не терять время на учебу в универе. Что бы ни толкнуло вас на это решение, наша компания готова помочь вам. Быстро, профессионально и по разумной стоимости изготовим документ любого ВУЗа и любого года выпуска на подлинных бланках с реальными подписями и печатями.
Ключевая причина, почему многие покупают диплом, – желание занять хорошую работу. Например, знания и опыт позволяют специалисту устроиться на работу, но подтверждения квалификации не имеется. Если для работодателя важно наличие “корочек”, риск потерять место работы довольно высокий.
Заказать документ о получении высшего образования можно в нашей компании в Москве. Мы предлагаем документы об окончании любых университетов Российской Федерации. Вы сможете получить диплом по любым специальностям, любого года выпуска, включая документы образца СССР. Даем 100% гарантию, что в случае проверки документа работодателем, никаких подозрений не возникнет.
Ситуаций, которые вынуждают купить диплом о среднем образовании достаточно. Кому-то срочно потребовалась работа, а значит, нужно произвести хорошее впечатление на руководителя при собеседовании. Другие задумали устроиться в престижную компанию, для того, чтобы повысить собственный статус в социуме и в будущем начать собственное дело. Чтобы не тратить попусту годы жизни, а сразу начинать успешную карьеру, применяя врожденные таланты и полученные навыки, можно заказать диплом в онлайне. Вы станете полезным в обществе, обретете финансовую стабильность в максимально короткий срок- купить аттестат
1win_fiMn
| #
1win вход https://www.cinemania.forum24.ru/?1-15-0-00001911-000-0-0-1743258043 .
Willieavamy
| #
Online casinos should care more—safety! plinko
JasonHoush
| #
Представьте: шубе из соболя 50 лет, она пережила эпохи, видела балы, тайные встречи, рассветы над дворцами. Но её реинкарнировали(перешили)! Роскошь, ставшая тенью самой себя… Как можно было так поступить? Вы бы осмелились изменить историю, стереть отпечатки времени? Смотрите.. https://t.me/ElenaMeel/32
Zohagdjub
| #
На сайте https://ooomila.com изучите каталог такого оборудования, которое предназначено для организации животноводческих комплексов. Также вы сможете почитать и содержательную информацию о компании и ее достижениях. В каталоге вы найдете следующее оборудование: технологические линии, весовое оборудование, резиновые маты, крематоры и многое другое. На технику, которая находится в разделе, действуют гарантии. Несколько сотен единиц оборудования всегда находятся на складе, можно приобрести под заказ.
ZutigrdSuiva
| #
Посетите Интернет-магазин автозапчастей АВТО-ЕВРО – https://autoeuro.ru/ и вы сможете купить запчасти для иномарок оптом и в розницу. Поберите автозапчасти по VIN или марке авто или ознакомьтесь с нашим каталогом, в нем представлен лучший выбор запчастей по выгодным ценам. Работаем со всеми регионами России. Подробнее на сайте.
PeterSlump
| #
https://telegra.ph/Explore-UK-Licensed-Non-Gamstop-Casinos-Today-03-27
remont kardana_jfMa
| #
ремонт рулевых карданов http://www.4-x-4.ru/remont-i-balansirovka-kardannyh-valov// .
1win_gmMn
| #
1win.online http://www.cinemania.forum24.ru/?1-15-0-00001911-000-0-0-1743258043 .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Никель-хромовая мишень
XecopClous
| #
Яндекс Такси: работа для водителей. Узнайте все о классификации автомобилей, требованиях и получите полный справочник таксопарков России. Советы начинающим таксистам на сайте https://igormylnikovchannel.ru/
Willieavamy
| #
Online casinos slide easy—mind it! minas
Willieavamy
| #
I love the themed slot games—makes online gambling more fun! minas
Lazrofy
| #
Диплом ВУЗа России!
Без университета сложно было продвигаться по карьерной лестнице. Именно из-за этого решение о покупке диплома стоит считать мудрым и целесообразным. Заказать диплом об образовании career.infophy.com/employer/gosznac-diplom-24
Vosojiswek
| #
На сайте https://stakecasino.tech почитайте про популярное и проверенное онлайн-заведение «Stake Casino». Здесь также имеется и букмекерская контора, где вы сможете сделать свои ставки. Вам обязательно понравится интерфейс, а пользоваться им очень просто. Вас обрадуют бонусы, а также различные акции. А все данные надежно защищены, потому информация точно не попадет злоумышленникам. Каждую неделю организуются турниры, а также розыгрыши. Для каждого желающего доступна мобильная версия, а также зеркало.
Xazrqrr
| #
Купить диплом о высшем образовании!
Мы предлагаем дипломы любой профессии по приятным ценам. Вы приобретаете документ через надежную и проверенную фирму. : fizteh.getbb.ru/viewtopic.php?f=43&t=1852
Sazrlnl
| #
Купить диплом университета !
Приобретение диплома любого университета РФ у нас является надежным делом, поскольку документ заносится в реестр. Быстро и просто приобрести диплом института vuz-diplom.ru/poluchite-diplom-s-zaneseniem-v-reestr-bistro-i-legko-3
Willieavamy
| #
I love the online chats—great crew! plinko
1win_ajMn
| #
игра 1вин https://www.cinemania.forum24.ru/?1-15-0-00001911-000-0-0-1743258043 .
reharoFenia
| #
Примените AstoMagic – это бесплатная онлайн-нейросеть. Задайте вопрос – и получите необходимый ответ за секунды. ИИ ваш стиль общения анализирует и подстраивается под него. Получайте в написании кода помощь и ошибок исправлении. https://astomagic.com – здесь собрали ответы на часто задаваемые вопросы. На ресурсе тарифы и отзывы пользователей представлены. Голосом общайтесь – система речь распознает, и ответы в текстовом виде выдает. ИИ информацию анализирует и точные данные предоставляет. Не упустите возможность опробовать нейросеть онлайн!
TagikBIc
| #
Посетите сайт https://addpets.ru/ – это экспертный блог о том, как ухаживать за домашними животными, как их кормить и какие породы наиболее популярны в России. Вы найдете исчерпывающую информацию о каждом животном, какие у них бывают болезни и как их лечить, познавательную и просто интересную информацию о домашних питомцах. Присоединитесь к обсуждению или присылайте свои интересные статьи!
1win_bokn
| #
что делать с бонусным балансом на 1win https://www.1win6004.ru .
Josijadofe
| #
На сайте https://delivery-dubai.com/ закажите такие алкогольные напитки в Дубае, как: ром, коньяк, ликер, бризер, шампанское, виски, пиво, вино и многое другое. Здесь находится только качественная продукция, которую вы сможете заказать в любом количестве. Алкоголь подходит для любого праздника, в том числе, Дня рождения, юбилея, отпуска. Вы можете выбрать несколько видов продукта, чтобы скрасить вечер, провести его со второй половинкой. Курьер прибудет в ближайшее время, чтобы передать вам заказ.
mostbet_zxoi
| #
mostbets http://www.mostbet6009.ru .
máy ra vào lốp
| #
máy ra vào lốp
A Brief History of Elemental Analysis » Artemis Analytical
Willieavamy
| #
Online casinos slide easy—mind it! minas
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние возможности.
Наши товары:
Плоская мишень Ni+Cu 3N
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их качество. Опытная поддержка – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы и предоставлять решения под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
Порошок титановый Р’Рў23 – РћРЎРў 1 90013-81 Рзделия РёР· титана Р’Рў23 – РћРЎРў 1 90013-81 обладают уникальными свойствами, которые делают РёС… идеальными для применения РІ различных отраслях. Ртот высококачественный титан обеспечивает отличную РєРѕСЂСЂРѕР·РёРѕРЅРЅСѓСЋ стойкость, легкость Рё прочность. Рзделия РїРѕРґС…РѕРґСЏС‚ для использования РІ аэрокосмической, медицинской Рё энергетической сферах. Если РІС‹ хотите купить Рзделия РёР· титана Р’Рў23 – РћРЎРў 1 90013-81, РІС‹ сделаете правильный выбор для надежного Рё долговечного решения. РќРµ упустите возможность улучшить ваш проект СЃ помощью продукции РёР· титана.
PeterSlump
| #
https://telegra.ph/No-Deposit-Free-Spins-at-Non-Gamstop-Casinos2-03-27
Willieavamy
| #
I won $4500 online—dream night! pin up
Hives treatment schedules
| #
Hives treatment methods Hives treatment teamwork Hives medical assistance
Hives treatment response evaluation Hives treatment plans Hives treatment cycles
Hives treatment regimens
Hives treatment coordination Hives heat sensation Hives support
Hives awareness Hives treatment schedules Hives itchy patches
buy celebrex pills
| #
can i order celebrex prices
where can i buy cheap mesalamine without dr prescription
| #
Medication information. Cautions.
where can i buy cheap mesalamine without dr prescription
Best information about medicament. Read information now.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Керамическая мишень Licoo2 3N, плоская
Cazrvgb
| #
Купить диплом о высшем образовании!
Мы можем предложить документы ВУЗов, расположенных в любом регионе Российской Федерации. Документы выпускаются на “правильной” бумаге высшего качества: bcstaffing.co/employer/14424/aurus-diploms
Sazrfat
| #
Заказать диплом любого ВУЗа!
Заказать диплом университета по доступной цене можно, обращаясь к надежной специализированной фирме. Заказать диплом: asxdiplommy.com/kupit-diplom-s-vneseniem-v-reestr-28
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Металлическая мишень РРЅРґРёСЏ 5N плоская для распыления
FotoxBox
| #
На сайте https://belarus-blok.ru/ вы сможете заказать газосиликатные блоки, которые были произведены в Беларуси. Приобрести продукцию получится в один клик и по привлекательной стоимости. Напротив каждого варианта указаны технические характеристики, расценки, что позволит быстрее сориентироваться в выборе. Газобетонные блоки пользуются огромной популярностью благодаря тому, что отличаются компактностью, небольшим весом. К тому же, этот материал негорючий. Есть возможность приобрести перегородочные, стеновые газоблоки лучших белорусских производителей.
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наши товары:
Латунное литое кольцо 600С…500 РјРј ЛК80 Купите латунные литые кольца РѕС‚ производителя. Великолепное сочетание прочности Рё эстетики. Высокое качество РїРѕ доступной цене. РЁРёСЂРѕРєРёР№ выбор форм Рё диаметров. Рдеальны для ювелирных изделий Рё производства музыкальных инструментов.
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их качество. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы по мере того как адаптировать решения под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
поставляемая продукция:
Фольга висмутовая C 6804 – JIS H 3250:2015 Фольга висмутовая C 6804 – JIS H 3250:2015 является высококачественным металлическим материалом, который используется РІ различных отраслях, включая электротехнику Рё приборостроение. Ее отличительными свойствами являются высокая коррозийная стойкость Рё легкость РІ обработке. Ртот РїСЂРѕРґСѓРєС‚ идеально РїРѕРґС…РѕРґРёС‚ для создания легких защитных слоев Рё компонентов.РљСѓРїРёРІ Фольга висмутовая C 6804 – JIS H 3250:2015, РІС‹ получите надежный материал, который прослужит долго Рё обеспечит эффективный результат РІ любых условиях. РќРµ упустите возможность улучшить СЃРІРѕРµ производство СЃ этим качественным товаром.
ekskursiicaree
| #
Отдых в Архипо-Осиповке в 2025, смотри подробнее по ссылке Экскурсии Архипо-Осиповки 2025
PeterSlump
| #
https://telegra.ph/Explore-Nonukcasinosuk-Non-Gamstop-Casinos-Options-03-27
Willieavamy
| #
I wish online helped new—tough kick! plinko app
PeterSlump
| #
https://telegra.ph/No-Deposit-Casino-Bonuses-Not-on-GamStop2-03-27
Willieavamy
| #
Online casinos need tools—support us! plinko
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их качество. Опытная поддержка – наш стандарт – мы на связи, чтобы улаживать ваши вопросы по мере того как адаптировать решения под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
Лента магниевая HM21A – ASTM B275 РљСЂСѓРі магниевый HM21A – ASTM B275 представляет СЃРѕР±РѕР№ высококачественный РїСЂРѕРґСѓРєС‚, разработанный для различных промышленных применений. РћРЅ отличается прочностью Рё легкостью, что делает его идеальным для использования РІ аэрокосмической Рё автомобильной отраслях. Свойства магния, такие как отличная коррозийная стойкость Рё хорошая обрабатываемость, делают этот РєСЂСѓРі незаменимым среди металлов. Выбирайте продукцию СЃ сертификатом ASTM B275 для уверенности РІ качестве. РќРµ упустите возможность купить РљСЂСѓРі магниевый HM21A – ASTM B275 РїРѕ выгодной цене. Рто решение для надежных Рё эффективных проектов.
obat crestor rosuvastatin 10mg
| #
Medicines information leaflet. Drug Class.
obat crestor rosuvastatin 10mg
Everything news about medicament. Read here.
Willieavamy
| #
I love the low play online—calm risk! craps casino
remont kardana_uqMa
| #
ремонт карданного вала цена 4-x-4.ru/remont-i-balansirovka-kardannyh-valov/ .
Vufamciz
| #
На сайте https://rustreetwear.ru вы найдете огромное количество интересных, разнообразных и эксклюзивных брендов, которые создают роскошную уличную одежду на любые случаи жизни. Есть как совсем не известные марки, которые только начинают свой путь, так и гиганты мировой индустрии. Перед вами самый впечатляющий каталог, где вы найдете не только одежду, но и аксессуары. Каталог обновляется ежедневно, чтобы вы обязательно нашли для себя лучший вариант. Здесь установлены доступные расценки.
mostbet_wyoi
| #
mostbet casino http://mostbet6009.ru/ .
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их качество. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы и адаптировать решения под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
Рзделия РёР· молибдена РњР’30 Рзделия РёР· молибдена РњР’30 – это высококачественные компоненты, обеспечивающие отличные физические Рё химические свойства. Молибден, используемый РІ этих изделиях, отличается высокой температурной стойкостью Рё РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ устойчивостью, что делает его идеальным для различных промышленных применений. Выбирая изготавливаемые РёР· молибдена изделия, РІС‹ инвестируете РІ надежность Рё долговечность. Благодаря своей прочности, изделия РёР· молибдена РњР’30 СЃРїРѕСЃРѕР±РЅС‹ выдерживать экстремальные условия эксплуатации. Если вам нужно купить Рзделия РёР· молибдена РњР’30, то РІС‹ обретаете надежное решение для СЃРІРѕРёС… задач. Обеспечьте эффективность вашего производства уже сегодня!
cost of tizanidine price
| #
generic tizanidine no prescription where to get generic tizanidine without prescription buy generic tizanidine for sale
can you get generic tizanidine price can i get tizanidine tablets order tizanidine price
can you get tizanidine without prescription
generic tizanidine without dr prescription where can i buy cheap tizanidine without a prescription order tizanidine prices
can i get tizanidine without rx can you buy tizanidine without a prescription buy generic tizanidine pills
Stuartpaymn
| #
Блог, посвященные всем видам сварочных работ с интересными и полезными публикациями https://svarkalegko.com/
PeterSlump
| #
https://telegra.ph/Guide-to-Non-Gamstop-Live-Dealer-Casinos-in-2023-03-27
where buy generic trandate pill
| #
where to get cheap trandate
Peracetic acid pump challenges mitigation strategies
| #
Peracetic acid pump challenges mitigation strategies
A Brief History of Elemental Analysis » Artemis Analytical
Malcomsoist
| #
казино онлайн Мир казино – это захватывающая вселенная, где переплетаются азарт, стратегия и технологические инновации. Онлайн казино предлагают широкий спектр игр, от классических слотов до захватывающих настольных игр, предоставляя игрокам возможность испытать удачу и применить свои навыки. Стратегии играют ключевую роль в успехе. Будь то блэкджек, покер или рулетка, понимание правил и умение анализировать ситуацию могут значительно повысить шансы на выигрыш. Важно помнить о разумном управлении банкроллом и осознанном подходе к игре.
RodneyObero
| #
Привет любителям креатива!
Раскройте потенциал Chat GPT для уникальных проектов: сочините текст для открытки, сделайте краткий поэтический пересказ или создайте план гардероба. Чат GPT для родителей — проекты для творчества, советы по обучению. Чат GPT для туристов — гид по тайным уголкам или фразы для общения. Технологии в действии — смело, просто, без границ!
Ссылка на проект – https://yarchatgpt.ru
рерайт с помощью chatgpt ha t chat gpt plus купить как получить api chatgpt
чат бот с искусственным интеллектом
Будьте смелыми — и всё получится!
1win_frkn
| #
1вин официальный сайт http://www.1win6004.ru .
New Retro casino официальный сайт
| #
New Retro casino официальный сайт
A Brief History of Elemental Analysis » Artemis Analytical
how to get generic zithromax for sale
| #
Drugs prescribing information. Brand names.
how to get generic zithromax for sale
Some what you want to know about meds. Get now.
Josikkat
| #
На сайте https://deliverydubaidrinks.com/ изучите каталог алкогольной продукции, которая представлена здесь в огромном ассортименте, а потому получится приобрести в любом количестве такие напитки, как: ром, ликер, бризер, виски. Регулярное обновление ассортимента, чтобы вы получили возможность попробовать все напитки, которые вас заинтересовали, за время отпуска. Если появились вопросы, то задайте их менеджеру, который подберет для вас оптимальное решение на любое событие. Вся продукция реализуется по доступным ценам.
ZopukNub
| #
Etsitko hyodyllista tietoa lainoista? Vieraile osoitteessa https://esimerkkilinkki.fi/ – josta loydat kattavaa tietoa asuntolainasta, vinkkeja lainan hakemiseen, lainojen vertailuun ja paikkaan, kuinka saada laina heti ilman luottotietoja tai luottohistoriaa ja paljon muuta. Hyodyllista tietoa sinulle verkkosivuillamme.
popuhkes
| #
Блок Маркет предлагает строительный качественный материал. Купить газобетонные блоки в нашей компании выгодно. Мы заслужили доверие клиентов. Получаем доброжелательные многочисленные отзывы. При необходимости проконсультируем относительно выбора продукции. https://belarus-blok.ru – сайт, где можете ознакомиться с условиями доставки газобетона. Также тут можно онлайн-калькулятором для расчета нужного количества газоблоков воспользоваться. Стремимся с каждым покупателем к созданию долговечных отношений.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние возможности.
Наша продукция:
Титановый сесквиоксид гранулированный
ekskursiicaree
| #
Отдых в Архипо-Осиповке в 2025, смотри подробнее по ссылке Экскурсии Архипо-Осиповка
DegewndBeisk
| #
Visit https://itprof.tech/ and you will learn everything about VPN – how to set up for different services, learn the best VPN for streaming, which VPN are best for gaming in 2025 and much more. Our resource offers useful and up-to-date information, news about VPN and how to work with it. We have structured our content so that each user can easily find answers to their questions.
PeterSlump
| #
https://telegra.ph/Discover-New-Casino-Sites-Without-Gamstop-Restrictions-03-27
PeterSlump
| #
https://telegra.ph/Casinos-Not-on-GamStop-No-Deposit-Options-Available-03-27
Willieavamy
| #
Online blackjack flows—good mix! plinko
Sazrest
| #
Мы предлагаем дипломы психологов, юристов, экономистов и других профессий по приятным ценам.– diplomt-nsk.ru/kupit-diplom-magistra-s-provodkoj-bistro-i-nadezhno/
Mazrlco
| #
Где приобрести диплом специалиста?
Получаемый диплом с приложением отвечает стандартам Министерства образования и науки, никто не отличит его от оригинала. Не следует откладывать личные мечты на пять лет, реализуйте их с нами – отправляйте заявку на диплом прямо сейчас! Диплом о среднем специальном образовании – не проблема! mosap.flybb.ru/viewtopic.phpf=2&t=541
Sazrann
| #
Приобрести документ о получении высшего образования можно в нашем сервисе. Мы оказываем услуги по производству и продаже документов об окончании любых ВУЗов РФ. Вы сможете получить необходимый диплом по любым специальностям, любого года выпуска, включая документы СССР. Гарантируем, что при проверке документа работодателем, каких-либо подозрений не появится. diplomaj-v-tule.ru/kupit-diplom-bistro-i-bezopasno-s-registratsiej-v-reestre-3/
JosephProge
| #
Что произошло во время авиакатстрофы AZAL? | Документальный фильм «Последний полёт»
https://www.youtube.com/watch?v=NkvCZz7-kFA
Adolfohunda
| #
Hello everyone!
Follow the iMedix Health Series for professional medical insights, a cornerstone Health Advice podcast; fitness lovers needing advice on combining strength training and cardio effectively will find our scheduling suggestions uniquely balanced, offering expert health advice and reliable healthcare guidance alongside trusted wellness tips for concurrent training benefits. Find schedules on imedix.com.
The iMedix Medical Show offers reliable healthcare guidance, a must-listen Medical podcast for well-being; wellness seekers wanting to understand the benefits of spending time in green spaces for cognitive function will find our attention restoration theory explanation uniquely refreshing, delivering expert health advice and professional medical insights alongside trusted wellness tips linking nature and focus.
More information at the site – https://www.iheart.com/podcast/269-the-journey-of-cenforce-15-166676888/
Wishing you luck!
arimidex purchase
| #
where can i get cheap avodart price avodart generation where to get avodart for sale
where can i buy avodart no prescription can i buy avodart online where can i buy generic avodart tablets
how to buy avodart without rx
buying cheap avodart without a prescription generic avodart no prescription cost avodart tablets
avodart orod pil buying cheap avodart prices can i order generic avodart no prescription
can i get cheap zithromax online
| #
Medicine information leaflet. Long-Term Effects.
can i get cheap zithromax online
Some trends of medicines. Get information here.
can i get generic tetracycline pills
| #
how much oxytetracycline for chickens
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Титановый сесквиоксид гранулированный
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их происхождение. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы по мере того как находить ответы под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наша продукция:
Лист кобальтовый РРљ102-РР” Лист кобальтовый РРљ102-РР” — это высококачественный металл, используемый РІ различных отраслях. Обладая отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью, РѕРЅ РїРѕРґС…РѕРґРёС‚ для работы РІ сложных условиях. Лист кобальтовый РРљ102-РР” применим РІ химической, авиационной Рё энергетической промышленности. Его отличные механические свойства обеспечивают долговечность Рё надежность РїСЂРё эксплуатации. Если РІС‹ хотите купить Лист кобальтовый РРљ102-РР”, обратите внимание РЅР° его превосходные характеристики, которые готовы удовлетворить ваши требования. РњС‹ предлагаем конкурентные цены Рё гарантируем качество.
chat gpt dlya kyrsovoi_gcmi
| #
нейросеть пишет курсовую http://www.studgen.ru/ .
PeterSlump
| #
https://telegra.ph/No-Gamstop-Online-Casinos-Safe-Gaming-Options-03-27
dostavka alkogolya 24_uyor
| #
купить виски с доставкой круглосуточно купить виски с доставкой круглосуточно .
Kypit smartfon v Moskve_avon
| #
Электроника в Москве http://techno-line.store/ .
PeterSlump
| #
https://telegra.ph/Exploring-Casinos-Without-Gamstop-Restrictions-03-27
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наши товары:
РџРѕРєРѕРІРєР° прямоугольная РёР· конструкционной стали 60С…270 РјРј 45РҐ Конструкционная прямоугольная РїРѕРєРѕРІРєР° – высококачественные материалы для создания прочных Рё надежных конструкций. Рзготовленная РёР· высококачественных материалов СЃ применением передовых технологий, эта продукция обладает отличной прочностью, устойчивостью Рє РёР·РЅРѕСЃСѓ, Рё находит широкое применение РІ машиностроении Рё строительстве.
Willieavamy
| #
Online gambling is wide open—control it! vipzino
Willieavamy
| #
The online rush kicks—stay cool! pinup
ekskursiicaree
| #
Отдых в Архипо-Осиповке в 2025, смотри подробнее по ссылке Экскурсии Архипо-Осиповки
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их качество. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы а также находить ответы под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
Полоса магниевая MI3 – JIS H 2150 Порошок магниевый MI2 – JIS H 2150 – это высококачественный металлический порошок, созданный для различных промышленных применений. РћРЅ обладает отличной РєРѕСЂСЂРѕР·РёР№РЅРѕР№ стойкостью Рё высокой прочностью, что делает его идеальным выбором для легких конструкций Рё компонентной СЃР±РѕСЂРєРё. Благодаря СЃРІРѕРёРј уникальным свойствам, Порошок магниевый MI2 – JIS H 2150 находит применение РІ аэрокосмической отрасли, производстве автомобилей Рё электроники. Ртот РїСЂРѕРґСѓРєС‚ гарантирует надежность Рё эффективность РІ работе. Приобретая Порошок магниевый MI2 – JIS H 2150, РІС‹ выбираете качество Рё долговечность для СЃРІРѕРёС… проектов. РќРµ упустите шанс купить Порошок магниевый MI2 – JIS H 2150 РїРѕ конкурентной цене!
JHONBET77 login
| #
JHONBET77 login
A Brief History of Elemental Analysis » Artemis Analytical
can i buy generic indomethacin no prescription
| #
Medicament information sheet. Short-Term Effects.
can i buy generic indomethacin no prescription
Actual information about meds. Read information here.
1win_xjkr
| #
1wi http://knowledge.forum24.ru/?1-1-0-00000082-000-0-0-1743258384 .
1win_jdkr
| #
ванвин https://knowledge.forum24.ru/?1-1-0-00000082-000-0-0-1743258384 .
бонусы бк
| #
Бонусы при регистрации в букмекерских конторах: свежие данные
Какие БК дают бонус при регистрации – свежие данные
Выбирая платформу для ставок, первоочередное внимание стоит уделить приветственным предложениям. В 2024 году размер фрибета за первое пополнение счёта варьируется от 1000 ₽ до 15000 ₽, в зависимости от оператора. Важно изучать условия отыгрыша: некоторые компании предлагают вейджер x5 на экспрессы с коэффициентом от 1.80, другие – фиксированный бонус без требований по обороту, но с меньшей суммой.
Среди акционных форматов выделяются “страховка первой ставки” (возврат средств в случае проигрыша) и “умножение депозита” (до 200% от суммы пополнения). Особенную осторожность следует проявлять при участии в акциях с повышенными требованиями к минимальному коэффициенту – иногда он достигает 2.50 и выше, что существенно снижает вероятность успешного отыгрыша.
Популярные предложения от операторов ставок
Другим привлекательным предложением являются повышенные коэффициенты на первые пари. Многие платформы увеличивают ставки на конкретные события, позволяя сделать более выгодные вложения в начале сотрудничества. Такие акции могут обеспечить значительное преимущество при удачных ставках.
Некоторые операторы предлагают так называемые “счастливые дни”, когда ставки на определённые события приносят дополнительные выигрыши. Это может быть фиксированная сумма или процент от пари. Такие акции позволяют пользователям использовать лучшие коэффициенты в наиболее успешные моменты.
Периодические акции, приуроченные к крупным турнирам и спортивным событиям, также пользуются популярностью. В такие дни повышаются не только коэффициенты, но и предложения для участников. Например, возможно получение фрибетов, которые можно использовать без риска потерять собственные средства.
Интересно, что некоторые компании вводят систему лояльности, начисляя баллы за каждую ставку. Эти баллы потом можно обменивать на различные призы, включая реальные деньги, товары или дополнительные кредиты для ставок.
Пользователям рекомендуется внимательно изучить условия предложений, так как многие из них могут иметь ограничения или требования по обороту. Сравните предложения нескольких платформ, чтобы найти наиболее выгодные условия для своих ставок.
Как эффективно распорядиться приветственными акциями от игровых платформ
Изучите правила досконально. Обратите внимание на вейджер (коэффициент отыгрыша), минимальные и максимальные размеры ставок, доступные виды спорта и сроки действия предложения. Неправильное понимание условий аннулирует акцию.
Оценивайте вейджер. Низкий показатель (до x25) предпочтительнее. Высокий вейджер (x40 и выше) делает отыгрыш затруднительным, увеличивая риск потери средств.
Выбирайте виды спорта с высокой вероятностью прохода. Ставки на фаворитов в популярных лигах, хотя и приносят меньший доход, увеличивают шансы на выполнение условий отыгрыша. Избегайте экспрессов с множеством событий, поскольку их успешность ниже.
Рассчитайте необходимый оборот. Умножьте полученную акционную сумму на вейджер. Это покажет, какой объем ставок требуется сделать для ее перевода на основной счет. Пример: акция 1000 руб. с вейджером x30 = 30000 руб. необходимо поставить.
Управляйте банкроллом. Не ставьте сразу большую часть акционного капитала. Разделите его на несколько частей для нескольких ставок. Это минимизирует риск быстрой потери и увеличивает время на отыгрыш.
Сравнивайте предложения разных операторов. Обращайте внимание не только на размер стартового поощрения, но и на сопутствующие условия (вейджер, сроки, доступные виды спорта). Иногда меньший приветственный капитал с более выгодными условиями предпочтительнее.
Используйте стратегии. Применяйте проверенные стратегии ставок (например, флэт или фиксированный процент от банка) для контроля риска и увеличения вероятности успеха.
Учитывайте ограничения по коэффициентам. Некоторые платформы ограничивают минимальный коэффициент для ставок, идущих в зачет отыгрыша. Убедитесь, что ваши ставки соответствуют этим критериям.
my blog – https://t.me/s/bookmaker_bonuses
Archiedog
| #
The setup guide was so easy to follow.
amneziawg ubuntu
Eanrxvw
| #
Для успешного продвижения вверх по карьере понадобится наличие официального диплома о высшем образовании. Заказать диплом об образовании у надежной организации: dip-lom-rus.ru/gde-kupit-attestati-za-11-klass-5/
where to buy clarinex pill
| #
can i order cheap clarinex buying cheap clarinex without prescription generic clarinex pills
buy cheap clarinex pill can i get cheap clarinex tablets where buy clarinex pills
can i buy clarinex online
where to buy generic clarinex without rx buy cheap clarinex buying clarinex pills
can i buy generic clarinex pills get generic clarinex without dr prescription can i buy cheap clarinex tablets
1win_lakr
| #
win 1 win 1 .
dostavka alkogolya 24_xaor
| #
доставка алкоголя в москве круглосуточно доставка алкоголя в москве круглосуточно .
chat gpt dlya kyrsovoi_ovmi
| #
нейросеть курсовая работа studgen.ru .
VictorTer
| #
AI for Bitcoin recovery
get cheap cilostazol without dr prescription
| #
where buy cilostazol without a prescription
VictorTer
| #
AI Bitcoin recovery solution
Kypit smartfon v Moskve_njon
| #
Смартфоны с доставкой по Москве https://techno-line.store .
1win_kfol
| #
1 вин вход в личный кабинет http://1win6051.ru/ .
mostbet_efer
| #
mostbet kg скачать на андроид https://www.mostbet6010.ru .
JibesdPeage
| #
Ищете интернет магазин автозапчастей в Минске с выгодными ценами? Посетите https://belautoparts.by/ где вы найдете существенный выбор для все автомобилей с доставкой по всей Беларуси. Подберите запчасти по VIN, марке авто, номеру детали, артикулу, каталогу в иллюстрации. У нас также есть все для ТО – расходники, масла, фильтра и многое другое. В нашем интернет магазине более 30 млн запчастей как оригинальных, так и качественных заменителей. Ознакомьтесь на сайте подробнее!
ebigasaX
| #
Юнитал-М – учебный центр, который компетентные услуги предоставляет. Регулярно стремимся к совершенству и к своей работе с ответственностью относимся. Даем гарантию на отменный сервис, неравнодушие к различным вопросам и добросовестность в осуществлении своих обязательств. https://www.unitalm.ru – ресурс, где мы рассказываем о том, как значимо на рабочем месте для компаний и специалистов повышение квалификации. Материал в понятной форме показан, педагогический состав к обучению располагает. Вместе мы достигнем высоких результатов!
Willieavamy
| #
The slot sounds online hum—fun vibe! fortune mouse
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
поставляемая продукция:
Кольцо РёР· драгоценных металлов платиновое 200С…40С…3.5 РјРј ПлРд90-10 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
will amoxicillin casue a kidney infection
| #
Medicine information sheet. Drug Class.
will amoxicillin casue a kidney infection
Everything news about pills. Read information here.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Плоская мишень магниевая
YemicAllek
| #
На сайте https://d600.ru/ вы сможете заказать тротуарную плитку Steingot и Braer от производителя по доступным ценам. Всегда в наличии полный ассортимент, который хранится на складе, а потому его можно приобрести уже сейчас. Вся продукция уже готова к отправке через ЖД либо авто, независимо от региона. Вы сможете воспользоваться услугами персонального менеджера. Есть возможность оплатить покупку по факту получения. Перед приобретением вы сможете изучить технические характеристики, а также расценки, чтобы приобрести самое выгодное решение.
dostavka alkogolya 24_kmor
| #
пиво доставка на дом круглосуточно http://www.dostavka-alkogolya248.ru .
chat gpt dlya kyrsovoi_ghmi
| #
нейросеть пишет диплом http://www.studgen.ru .
Jariorcap
| #
Где приобрести диплом специалиста?
Наши специалисты предлагают быстро и выгодно заказать диплом, который выполнен на оригинальной бумаге и заверен печатями, штампами, подписями должностных лиц. Данный документ способен пройти любые проверки, даже при помощи специально предназначенного оборудования. Достигайте цели быстро с нашей компанией.
Купить диплом о высшем образовании diplomservis.com/kupit-attestat-13/
WilliamDix
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и прочих профессий по приятным тарифам. Дипломы изготавливаются на настоящих бланках государственного образца Заказать диплом любого института asxdiploman.com
Lazrepa
| #
Где приобрести диплом специалиста?
Приобрести диплом института по невысокой цене возможно, обратившись к надежной специализированной компании.: kazdiplomas.com
NurayVoing
| #
Компания «КАРДЕКС» https://card-oil.ru/ — надежный партнер на рынке топливных решений, работающий более 12 лет. Специализируется на выпуске и обслуживании топливных карт для юридических лиц и индивидуальных предпринимателей. Преимущество КАРДЕКС — доступ к обширной сети из более чем 13000+ АЗС по всей России, включая федеральные и региональные заправочные станции. Это позволяет клиентам заправляться без привязки к конкретному бренду, оптимизируя расходы и логистику. Топливные карты КАРДЕКС обеспечивают удобный контроль затрат, предоставляют детальную аналитику и помогают экономить за счет персонализированных условий и скидок. Компания зарекомендовала себя как стабильный и технологичный игрок, предлагающий инновационные решения для бизнеса.
Jariorwky
| #
Где заказать диплом по необходимой специальности?
Наша компания предлагаетбыстро приобрести диплом, который выполнен на бланке ГОЗНАКа и заверен мокрыми печатями, штампами, подписями официальных лиц. Документ способен пройти любые проверки, даже с применением профессиональных приборов. Достигайте цели быстро и просто с нашими дипломами. Заказать диплом любого ВУЗа! physmathforum.flybb.ru/viewtopic.php?f=12&t=1227&sid=8cbf290059ee645b9fe06c2ddea5399f
CarozrdAsymn
| #
На сайте https://bitovayatechnika.blogspot.com/2025/03/blog-post.html изучите полезную, актуальную информацию, которая касается сплит-систем и того, как правильно их выбрать домой либо в офис. Эти функциональные и умные устройства нацелены на то, чтобы поддержать приятную температуру в помещении. Но для того, чтобы воспользоваться всеми функциями, необходимо правильно выбрать устройство. К важным преимуществам такой техники относят то, что она является энергоэффективной, работает практически бесшумно.
Mazrrim
| #
Мы изготавливаем дипломы любой профессии по выгодным ценам. Всегда стараемся поддерживать для клиентов адекватную политику тарифов. Для нас очень важно, чтобы документы были доступны для большого количества наших граждан.
Приобретение диплома, подтверждающего окончание университета, – это грамотное решение. Приобрести диплом любого ВУЗа: diplom-insti.ru/kupit-diplom-texnicheskij-2/
arenda shatrov_uhSt
| #
аренда большого шатра цены аренда большого шатра цены .
Lazroqm
| #
Заказать диплом любого ВУЗа!
Мы предлагаем дипломы любых профессий по приятным ценам— good-diplom.ru/kuplyu-diplom-spetsialista-bistro-i-nadezhno/
JustinFet
| #
биржа аккаунтов https://marketpleys-akkauntov.ru
Kypit smartfon v Moskve_gnon
| #
Apple iPhone в Митино techno-line.store .
Dnrtwcx
| #
Где купить диплом по необходимой специальности?
Мы можем предложить дипломы любой профессии по доступным ценам. Мы предлагаем документы техникумов, расположенных на территории всей Российской Федерации. Вы можете заказать качественно напечатанный диплом от любого заведения, за любой год, в том числе документы старого образца СССР. Дипломы и аттестаты выпускаются на бумаге самого высокого качества. Это позволяет делать государственные дипломы, которые не отличить от оригинала. Они будут заверены необходимыми печатями и штампами. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Для нас важно, чтобы документы были доступны для подавляющей массы наших граждан. diplom-kuplu.ru/kupit-attestat-za-11-klass-2-6
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
поставляемая продукция:
Латуное наружное стопорное пружинное кольцо 23С…1.2 РјРј ЛС59 ГОСТ 13942-86 Купить латунные наружные стопорные кольца РїРѕ ГОСТ РІ Р РѕСЃСЃРёРё. Надежная фиксация элементов конструкции. Высокое качество РїРѕ доступной цене. Рспользуются РІ строительстве, машиностроении Рё производстве. Заказывайте сейчас РЅР° сайте Редметсплав.
Sazrwcs
| #
корочка для аттестата купить 11 класс
Willieavamy
| #
Online bingo flows—great crew! vincispin
how long does levaquin stay in your body
| #
Levaquin 500mg side effects levaquin dosing in elderly levaquin 250 mg price
Levaquin and nausea how to get levaquin prices cheap generic levaquin 500mg
levaquin price costco
can i order generic levaquin prices how long does levaquin stay in system tendonitis after 250mg levaquin
how long do side effects of levaquin last levaquin generic price levaquin 750 mg directions mapquest
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их качество. Опытная поддержка – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы по мере того как предоставлять решения под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наша продукция:
Лента магниевая M10600 – UNS РљСЂСѓРі магниевый M10600 – UNS – это высококачественный материал, прекрасно подходящий для различных промышленных Рё инженерных задач. Ртот РєСЂСѓРі отличается повышенной прочностью Рё устойчивостью Рє РєРѕСЂСЂРѕР·РёРё, что делает его идеальным выбором для производства Рё ремонта. Приобретение данного РєСЂСѓРіР° обеспечит надежность Рё долговечность ваших изделий. Если РІС‹ ищете оптимальное решение для СЃРІРѕРёС… проектов, вам стоит купить РљСЂСѓРі магниевый M10600 – UNS. РќРµ упустите возможность использовать передовые материалы РІ СЃРІРѕРёС… разработках!
pantoprazole sodium for pain
| #
Drug information sheet. Brand names.
pantoprazole sodium for pain
Actual news about pills. Get information now.
mostbet_bjer
| #
motbet motbet .
where buy generic vermox without prescription
| #
how to buy vermox without prescription
Willieavamy
| #
The mobile apps online falter—patch it! Vai de Bet
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
Наша продукция:
Медный тройник РїРѕРґ пайку СЃ направленным отводом 15С…11С…0.7 РјРј 10.6С…12.6 РјРј твердая пайка Cu-DHP ГОСТ Р 52922-2008 Купить качественные медные тройники РїРѕРґ пайку для надежных Рё долговечных соединений РІ трубопроводных системах. Разнообразие размеров Рё конфигураций, высокая теплопроводность Рё устойчивость Рє РєРѕСЂСЂРѕР·РёРё. Простая установка Рё надежность соединения. Рдеальное решение для различных проектов. Редметсплав.СЂС„
1win_jaol
| #
1win официальный сайт вход 1win официальный сайт вход .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наша продукция:
Круг 30 мм ХН70ВМТЮ Купить круги нержавеющие специальные ведущих производителей по выгодным ценам. Широкий выбор размеров и диаметров. Гарантия качества и надежности. Применение в различных отраслях и производствах. Надежная защита от коррозии и воздействия внешних факторов.
DonnellHek
| #
Хотите здоровую и красивую улыбку? Стоматология simpladental позаботиться о ваших зубах с максимальным комфортом и вниманием. Наша стоматология использует современные технологии и индивидуальный подход, чтобы вы чувствовали себя уверенно.
цены на ритуальные услуги киев
| #
Ритуальная служба Грааль: поддержка и забота в трудные минуты
Ритуальная служба “Грааль” – это городское похоронное бюро в Киеве, которое уже более 10 лет помогает семьям справляться с организацией похорон и другими ритуальными задачами. Мы предоставляем полный комплекс услуг в Киеве и Киевской области, начиная от базовой организации похорон и кремации по доступной цене от 6500 гривен. Наша главная цель – минимизировать ваше участие в сложных подготовительных этапах, чтобы у вас было время проститься с близким человеком и получить поддержку от родных.
Чем мы можем помочь
“Грааль” – это не просто похоронное бюро, а команда профессионалов, которая берет на себя все хлопоты в трудный момент. Мы предлагаем широкий спектр услуг, чтобы вы могли сосредоточиться на своих чувствах, а не на организационных деталях. Среди наших услуг:
– Организация похорон: от подготовки документов до проведения церемонии.
– Кремация: доступное и деликатное решение с полным сопровождением.
– Перевозка умершего (Груз 200): транспортировка по Украине и за ее пределы.
– Услуги морга: профессиональная подготовка тела к прощанию.
– Поминальные обеды: организация в соответствии с традициями.
– Копка могилы и уход за могилами: забота о месте захоронения.
– Дезинфекция помещения: обеспечение чистоты и безопасности.
– Оформление документов: юридическая помощь в сложной ситуации.
– Ритуальный транспорт: надежные автомобили для церемонии.
– Прижизненные договоры: возможность заранее спланировать процесс.
– Похороны и кремация домашних питомцев: поддержка в утрате верного друга.
Кроме того, мы оказываем психологическую поддержку, понимая, насколько тяжело переживать утрату. Наши специалисты готовы выслушать и помочь вам в любое время суток.
Наши преимущества
Ритуальная служба “Грааль” выделяется среди других благодаря многолетнему опыту и современному подходу. Вот что делает нас особенными:
– Опыт более 10 лет: за годы работы мы заслужили доверие клиентов и отточили свои навыки.
– Индивидуальный подход: каждая ситуация уникальна, и мы учитываем все ваши пожелания.
– Полный спектр услуг без посредников: это позволяет нам контролировать качество и сохранять доступные цены.
– Круглосуточная работа: мы доступны 24/7, чтобы поддержать вас в любой момент.
– Честная стоимость: наши услуги начинаются от 6500 гривен, и мы всегда стремимся предложить конкурентные цены.
Мы понимаем, что организация похорон – это не только технический процесс, но и эмоционально сложный этап. Поэтому наша команда делает все возможное, чтобы вы чувствовали себя уверенно и спокойно.
Миссия “Грааль”
Наша миссия – достойно и деликатно сопровождать вас на каждом этапе ритуального процесса. Мы стремимся не просто выполнять свою работу, а создавать условия, чтобы у вас было время попрощаться с близким человеком и получить моральную поддержку от друзей и семьи. Директор бюро Глеб Бусенцов подчеркивает: “Нам важно, чтобы каждый клиент чувствовал нашу заботу и мог доверить нам все хлопоты. Мы формируем честные цены и делаем процесс максимально прозрачным”.
Почему выбирают нас
“Грааль” – это не только профессионализм, но и человеческое отношение. Мы работаем круглосуточно, чтобы вы могли обратиться к нам в любое время. Наша репутация строится на отзывах клиентов, которые ценят наш подход и качество услуг. Будь то организация похорон, кремация или уход за могилой, мы гарантируем внимание к деталям и уважение к вашим традициям.
Если вы столкнулись с утратой, не оставайтесь один на один с организационными вопросами. Ритуальная служба “Грааль” готова взять на себя все заботы, чтобы вы могли сосредоточиться на главном – памяти о близком человеке. Свяжитесь с нами, и мы поддержим вас в этот непростой период.
https://graale.net/ru/page/prices.html
WovolrdBrern
| #
Услуга срочного выкупа автомобилей https://www.buying-car.ru/ в любом состоянии является идеальным решением для тех, кто хочет быстро и без лишних хлопот избавиться от своего транспортного средства. Предлагаем вам выгодные условия, они позволяют получить деньги в кратчайшие сроки, независимо от года выпуска, пробега или технического состояния автомобиля. Наша команда специалистов готова оценить ваш авто быстро и профессионально. Даже если ваше транспортное средство было повреждено в ДТП, не функционирует или имеет серьезные технические неисправности. Процесс выкупа прост и удобен. Наши эксперты проведут быструю оценку стоимости, и если она вас устроит, мы готовы заключить сделку в тот же день. Мы также предоставляем услуги по бесплатному эвакуатору, если ваше авто не на ходу, что позволяет вам избежать дополнительных трат.
mostbet_eoer
| #
мост бет https://www.mostbet6010.ru .
Jariorlfd
| #
Где приобрести диплом по нужной специальности?
Наша компания предлагает максимально быстро купить диплом, который выполнен на оригинальном бланке и заверен мокрыми печатями, водяными знаками, подписями должностных лиц. Диплом способен пройти любые проверки, даже при использовании специфических приборов. Достигайте своих целей максимально быстро с нашими дипломами.
Купить диплом о высшем образовании diplomus-spb.ru/kupit-diplom-o-srednem-14/
JakohlSed
| #
На сайте https://fguard.ru/ почитайте статьи на самую разную тему. К примеру, кибермошенничество, для каких целей используется сеточка, которая на двери микроволновки. Здесь представлена вся нужная информация о смарт-часах и о том, чем они отличаются от бизнес-браслетов. Для того чтобы подыскать нужную информацию, воспользуйтесь специальным рубрикатором. На сайте вы найдете записи, которые касаются нейросетей, гаджетов. Представлены и интересные рекомендации, которые будут необходимы каждому. Имеются данные и про технологии.
Rafaelbic
| #
Профессиональная помощь в сфере магических услуг. Искусство высшей магии.
Ясновидение/Руны/Ритуалы
Диагностика / Консультация
* Диагностика на наличие магического негатива, порчи, приворота, воздействия
* Диагностика энергетики
* Выявление проблем и препятствий
* Ответы на вопросы
Чистка магического и энергетического негатива.
Защита на всех уровнях.
Открытие дорог.
Открытие финансового канала.
(на основании диагностики производим чистку негатива, убираем любые деструктивные программы включая порчу на смерть, кладбищенскую порчу, порчу через подклад, подселение сущности, приворот и другие деструктивные программы разрушающие вашу жизнь)
восстанавливаем вашу жизнь и настраиваем по всем сферам жизни
Магия для решения Бизнес задач и Финансовых вопросов
(моя самая любимая сфера деятельности)
* Убрать препятствия
* Защита на вас и ваш бизнес
* Помощь в судах
* Выявление и решение проблем вашего бизнеса через магию
Любовная магия
* Определение совместимости партнеров в браке и при вступлении в брак
* Защита семьи и отношений
* Розжиг чувств в паре (это новый виток отношений. реанимация любви)
* Создание условий, которые приведут к романтическим отношениям и к созданию семьи
Решение любых актуальных для вас проблем
* Выявить и убрать блоки мешающие достичь цели
– магия рун
– магия древних ритуалов
– магия символов, углов, пространства
– планетарная магия
– пространственная алхимия
– работа через сознание и подсознание
Все работы проводятся очень экологично и без какого-либо вреда
Также проводятся специальные ритуалы позволяющие развернуть вашу жизнь на 360 градусов, получить желаемое, открыть дороги, притянуть любовь и создать жизнь мечты
Телеграмм канал Искусство Высшей Магии
Телеграмм для связи: https://t.me/magicfxs
Для связи: WHATSAPP +44 7435 250025
*** для заказа присылайте фото для диагностики, имя и ваш вопрос.
Услуги осуществляются на условиях 100% предоплаты.
P.S. Если человек знает для чего ему нужно желаемое – значит он может это достичь
VictorTer
| #
Recover abandoned BTC wallets
side effects levothroid levothyroxine
| #
Pills prescribing information. Drug Class.
side effects levothroid levothyroxine
All news about medication. Read information now.
Allenfer
| #
https://dzen.ru/a/Z-wZzKk7V2mgAjD3
arenda shatrov_pqSt
| #
аренда шатров стоимость аренда шатров стоимость .
Lorecbus
| #
На сайте https://autopiter.kg/ получится приобрести как оригинальные, так и неоригинальные запчасти, комплектующие для ТО, автохимию и автомасла, аккумуляторы, все для спецтехники. Представлен и каталог ГАЗ, ВАЗ, КАМАЗ. Есть возможность подобрать фильтр для автомобиля. Также представлены и автолампочки. Изучите раздел с рекомендуемыми товарами. Для того чтобы подобрать необходимую деталь, нужно воспользоваться поиском – в строке указать название детали. Все запчасти хранятся на складе и в нужном количестве.
buying cipro tablets
| #
get generic cipro without prescription cipro without a doctor prescription buying cipro pill
generic cipro without dr prescription cost of cipro without prescription can you buy generic cipro pills
get cheap cipro without prescription
can i purchase cheap cipro without insurance can you buy generic cipro without dr prescription cost of cipro
how to buy cipro tablets where can i get cipro without prescription buying cipro online
Lazrvyg
| #
Быстро приобрести диплом ВУЗа!
Мы изготавливаем дипломы психологов, юристов, экономистов и любых других профессий по выгодным тарифам— diplomj-irkutsk.ru/kupit-diplom-texnikuma-bistro-i-nadezhno-3/
Mazrdmw
| #
Мы изготавливаем дипломы любой профессии по приятным ценам. Стараемся поддерживать для заказчиков адекватную политику цен. Важно, чтобы документы были доступны для подавляющей массы граждан.
Заказ диплома, подтверждающего обучение в университете, – это выгодное решение. Купить диплом любого университета: 10000diplomov.ru/kolledzh-kupit-diplom-4/
can you buy cheap clozaril online
| #
cost clozaril prices
1win_tmol
| #
1win партнёрка http://1win6051.ru .
SurotLak
| #
Ищете изготовление наружной рекламы в Москве? Посетите сайт https://neon-mix.ru/ – мы изготавливаем световые вывески, объёмные буквы, рекламные конструкции и многое другое. Полный цикл — от производства до монтажа по самым выгодным ценам. Реализуем проекты любой сложности, учитываем пожелания заказчика и предлагаем честные, конкурентные цены. Ознакомьтесь со всеми услугами и ценами на сайте.
RichardHeery
| #
сколько стоит натяжной потолок под ключ https://potolki-spb-2.ru/
Allenfer
| #
https://dzen.ru/a/Z-wZzKk7V2mgAjD3
GoxodExalk
| #
На сайте http://www.tribal-tattoo.ru уточните телефон тату-салона, в котором вы сможете выполнить художественную татуировку, перекрыть шрамы, рубцы, выполнить роскошный перманентный макияж. Есть возможность выполнить рисунок различной сложности, техники и любой цветовой гаммы. Каждая идея клиента будет реализована. При необходимости будет предложена своя. К вашим услугам создание индивидуальных эскизов. Все услуги обойдутся по привлекательным расценкам. В салоне работают компетентные, знающие специалисты, которые работают специально для вас.
FurontDus
| #
Looking for flower delivery in Ukraine? Visit https://ukraineflora.com/ where you will find the largest selection of fresh flowers and exclusive gifts. Look through the catalog and you will definitely find not only luxurious flowers and bouquets, but also gift baskets, perfumes, balloons and even cakes.
Jariorxmg
| #
Где купить диплом по нужной специальности?
Наши специалисты предлагают выгодно заказать диплом, который выполняется на оригинальной бумаге и заверен мокрыми печатями, штампами, подписями. Документ пройдет лубую проверку, даже с применением профессиональных приборов. Достигайте свои цели быстро и просто с нашей компанией.
Приобрести диплом университета diplomus-spb.ru/kupit-sertifikat-spetsialista-17/
Michaellix
| #
купить аккаунт с прокачкой https://birzha-accauntov.ru
where can i buy generic fluvoxamine without dr prescription
| #
Meds information leaflet. Long-Term Effects.
where can i buy generic fluvoxamine without dr prescription
Best news about pills. Read information now.
Willieavamy
| #
Online casinos should block—fair go! plinko
Willieavamy
| #
The convenience of online gambling is both a blessing and a curse! plinko
Willieavamy
| #
Online gambling flows free—rein it! book of ra
Jariorwtz
| #
Где заказать диплом по актуальной специальности?
Наша компания предлагаетбыстро и выгодно заказать диплом, который выполнен на оригинальном бланке и заверен печатями, водяными знаками, подписями должностных лиц. Диплом способен пройти любые проверки, даже с использованием специально предназначенного оборудования. Достигайте цели быстро и просто с нашим сервисом. Заказать диплом о высшем образовании! kingcityrp.moibb.ru/posting.php?mode=post&f=34
RichardHeery
| #
натяжные потолки спб цены https://potolki-spb-2.ru/
cost cheap vasotec no prescription
| #
get cheap vasotec for sale can i buy generic vasotec for sale cost generic vasotec pills
where to get cheap vasotec pill how to get generic vasotec pills can you buy cheap vasotec prices
how to buy generic vasotec prices
can i order vasotec tablets cost of vasotec how can i get vasotec for sale
buy cheap vasotec no prescription buying cheap vasotec no prescription where to buy vasotec price
Lazrdrv
| #
Приобрести диплом любого ВУЗа!
Мы готовы предложить дипломы психологов, юристов, экономистов и любых других профессий по приятным ценам— fastdiploms.com/kupit-attestat-11-klassa-bistro-i-nadezhno-2/
Mazrykc
| #
Мы изготавливаем дипломы любой профессии по приятным ценам. Стараемся поддерживать для заказчиков адекватную политику тарифов. Важно, чтобы дипломы были доступными для подавляющей массы граждан.
Покупка документа, подтверждающего обучение в ВУЗе, – это выгодное решение. Купить диплом ВУЗа: sdiplom.ru/gde-mozhno-kupit-diplom/
1win_xmea
| #
казино 1win https://1win6052.ru/ .
MalecrTip
| #
На сайте https://fabvia.ru/spicesguide/ почитайте полезную, содержательную информацию на тему приправы, специй и того, как правильно все это употреблять, чтобы вкус максимально раскрылся, вы получили от еды пользу. Этот портал идеально подойдет для тех, кто работает в пищевой промышленности, увлекается кулинарией. Информация поможет расширить свои знания в этой сфере. Вы научитесь правильно сочетать ингредиенты и создавать вкусные блюда. В скором времени вы получите возможность воспользоваться калькулятором, который научит сочетать ингредиенты, добавлять нужное количество специй.
how can i get cheap duricef
| #
where to get cheap duricef online
Sazrixf
| #
Задумался а действительно можно купить диплом государственного образца в Москве, и был удивлен, все реально и главное официально!
Сначала серфил в сети и искал такие темы как: советский диплом купить, купить диплом в ростове, купить диплом без, купить диплом санкт, купить диплом в иркутске, получил базовую информацию.
Остановился в итоге на материале proffdiplomik.com/petrozavodsk
Stanleydrese
| #
The Aviator game is really fun! I’ve been playing on this platform https://servertea3.bloggersdelight.dk/2025/04/01/the-complete-aviator-game-tutorial-for-kenya-2025-edition/
and it’s been crazy so far.
Is anyone here winning big?
Trying to figure out the best moment to cash out – any ideas?
10
| #
10
A Brief History of Elemental Analysis » Artemis Analytical
Gustavodiz
| #
Under the Hood
Willieavamy
| #
The live help online rocks—key boost! mines
Willieavamy
| #
I hit a bonus online and won $50—small but sweet! jackpot raider
Willieavamy
| #
Online gambling feels detached—easy to overspend! minas
Sazrizy
| #
купить диплом вуза ссср
Andrewnerce
| #
hwid spoofer hwid spoofer free hwid spoofer hwid spoofer download hwid spoofer crack
WilliamAlgor
| #
площадка для продажи аккаунтов https://magazin-accauntov.ru
Willieavamy
| #
Are online wins true?—not sure! Jackpot Raider
Willieavamy
| #
Online gambling can run wild—rein it in! plinko
internetCah
| #
провайдер по адресу
domashij-internet-kazan001.ru
подключить интернет
Willieavamy
| #
Online casinos need signs—flag us! minas
Diplomi_ekka
| #
купить диплом в магадане diplomys-vsem.ru .
Diplomi_exKt
| #
Приобрести диплом ВУЗа!
Мы можем предложить документы любых учебных заведений, которые расположены в любом регионе России.
dip-lom-rus.ru/kupit-diplom-s-zaneseniem-v-reestr-bistro-i-nadezhno-18/
Todufsmeme
| #
Visit the site https://findprompt.shop/ which is a trading platform and marketplace for buying and selling prompts for Midjourney, Stable Diffusion, ChatGPT and others. Explore the best AI prompts. A huge selection of prompts of various directions at the most favorable prices. Visit the catalog and see for yourself our significant offer.
kra31.cc
| #
kra31.cc
A Brief History of Elemental Analysis » Artemis Analytical
Pills
| #
Lose weight
Willieavamy
| #
I don’t get online hate—it’s fine! plinko
Sazrpvw
| #
Мы изготавливаем дипломы любой профессии по приятным тарифам. О преимуществах заказа документов у нас
Вы покупаете диплом в надежной и проверенной компании. Такое решение сэкономит не только массу денег, но и время.
Преимуществ куда больше:
• Дипломы печатаются на фирменных бланках со всеми печатями;
• Можно приобрести дипломы любого ВУЗа России;
• Цена значительно ниже чем пришлось бы заплатить на очном обучении в университете;
• Удобная доставка в любые регионы РФ.
Приобрести диплом о высшем образовании– http://aksharalipi.com/read-blog/9514_kupit-attestat-za-11-klass.html/ – aksharalipi.com/read-blog/9514_kupit-attestat-za-11-klass.html
can i get generic avodart for sale
| #
how can i get avodart pills cost avodart online can you buy cheap avodart for sale
cost of avodart without a prescription buying cheap avodart tablets buy avodart
avodart hair loss
can you get cheap avodart prices cost of cheap avodart without rx how to get cheap avodart without prescription
what is the cost for avodart in canada cost cheap avodart without dr prescription order cheap avodart prices
hranenie veshei_pnpa
| #
хранения вещей в москве хранения вещей в москве .
Willieavamy
| #
I love how online casinos let you play for pennies if you want! VinciSpin
J88
| #
J88
A Brief History of Elemental Analysis » Artemis Analytical
mua ban noi tang nguoi
| #
mua ban noi tang nguoi
A Brief History of Elemental Analysis » Artemis Analytical
buy generic microzide prices
| #
how to get microzide no prescription
Bevaylem
| #
Ищете оборудование из Китая по ценам производителя? Посетите сайт https://totem28.ru/ где вы найдете широкий ассортимент не только оборудования, но и спец техники. Наш каталог постоянно пополняется, мы тщательно отбираем новинки китайского машиностроения. Посмотрите наш каталог и узнайте подробнее на сайте.
Dnrtqkc
| #
Где купить диплом специалиста?
Мы предлагаем дипломы психологов, юристов, экономистов и любых других профессий по доступным ценам. Мы предлагаем документы ВУЗов, расположенных на территории всей РФ. Можно приобрести диплом за любой год, в том числе документы старого образца СССР. Дипломы и аттестаты выпускаются на бумаге высшего качества. Это дает возможность делать настоящие дипломы, которые не отличить от оригиналов. Они заверяются необходимыми печатями и подписями. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Для нас важно, чтобы дипломы были доступными для большого количества граждан. diplomius-docs.com/kupit-diplom-o-srednem-spetsialnom-7
Donaldlaf
| #
darkmarket 2025 https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darkmarket link
PeterSlump
| #
https://telegra.ph/Benefits-of-Non-Gamstop-Online-Casinos-for-Players-03-27
Pingrutty
| #
dark web markets https://github.com/aresonioncq0a7/aresonion – dark market link
Willieavamy
| #
I won $700 on a slot—still hyped! fortune tiger bet
PeterSlump
| #
https://telegra.ph/Explore-Casino-Options-Not-Registered-with-Gamstop-03-27
dostavka alkogolya 24_cuor
| #
доставка алкоголя круглосуточно доставка алкоголя круглосуточно .
Rabyitabe
| #
darknet marketplace https://github.com/darkwebsitesyhshv/darkwebsites – darkmarkets
chat gpt dlya kyrsovoi_cdmi
| #
генератор вкр http://www.studgen.ru .
Jamesdusty
| #
Спасибо за цветы для малознакомого человека – угадали со вкусом!
цветы томск
Sazromu
| #
Покупка документа о высшем образовании через надежную компанию дарит ряд достоинств для покупателя. Данное решение позволяет сберечь время и существенные средства. Впрочем, плюсов намного больше.Мы изготавливаем дипломы любых профессий. Дипломы производятся на настоящих бланках. Доступная стоимость по сравнению с крупными затратами на обучение и проживание в другом городе. Покупка диплома университета будет выгодным шагом.
Быстро купить диплом о высшем образовании: diplomass.com/kupit-diplom-o-srednem-obrazovanii-v-reestr-6/
Willieavamy
| #
Online gambling is loose—tighten it! plinko
internetCah
| #
тарифы интернет и телевидение казань
domashij-internet-kazan002.ru
провайдеры интернета в казани по адресу проверить
PeterSlump
| #
https://telegra.ph/No-Deposit-Bonuses-at-Non-Gamstop-Casinos-Explained3-03-27
DonDonfaita
| #
darkmarket 2025 https://github.com/nexusdarkrtv1u/nexusdark – darkmarket link
Trefdke
| #
Заказать документ о получении высшего образования можно в нашей компании. diplomnie.com/kupit-diplom-kolledzha-2-3
can lisinopril cause drowsiness
| #
lisinopril 30 mg side effects alternative to lisinopril can i get generic lisinopril tablets
lisinopril equivalent in india lisinopril out of pocket cost what is lisinopril hctz 10 12.5mg tablets
where to buy lisinopril without dr prescription
cost cheap lisinopril without a prescription can i purchase lisinopril pill can i order generic lisinopril online
can i get lisinopril price can i buy generic lisinopril for sale allergic reaction to lisinopril after taking it for years
PeterSlump
| #
https://telegra.ph/Paying-with-Phone-Bill-at-Non-Gamstop-Mobile-Casinos-03-27
where can i get micronase price
| #
can i buy generic micronase online
Willieavamy
| #
The online flow is wild—stay sharp! slot gallina
Willieavamy
| #
The live dealer feature online is a total game-changer! slot gallina
Willieavamy
| #
Online gambling laws are so confusing—can I even play legally? bongo bongo
Pingrutty
| #
darknet marketplace https://github.com/aresonioncq0a7/aresonion – dark market 2025
Donaldlaf
| #
dark web market links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark web markets
buy cheap proscar online
| #
Medication information. What side effects?
buy cheap proscar online
All about medicines. Get here.
RadixeWoume
| #
На сайте https://xn--80aaaaaaxhx2a2ai2af3a8grd.xn--p1ai/ воспользуйтесь услугой, связанной с арендой шатров, которая подходит для любого мероприятия, в том числе, Дня рождения, юбилея, свадьбы. Аренда сделает любое мероприятие ярким, незабываемым. В компании работают проверенные, компетентные специалисты, которые выполнят все необходимые работы, включая установку, расстановку мебели. Имеется огромный выбор снаряжения, которое применяется для кемпинга. Шатер выполнен из современных, качественных материалов, поэтому порадует своим внешним видом.
Willieavamy
| #
Online gambling feels loose—no look! plinko
Toliknaf
| #
darkmarket url https://github.com/abacuslink4jjku/abacuslink – darknet markets links
PeterSlump
| #
https://telegra.ph/Best-Non-Gamstop-Online-Casinos-to-Play-Now-03-27
Willieavamy
| #
Online casinos should cap—safe guard! aviator
Willieavamy
| #
The visuals online wow—great sight! aviator game
Rabyitabe
| #
dark web marketplaces https://github.com/darkwebsitesyhshv/darkwebsites – dark markets 2025
RijusonPried
| #
Посетите сайт https://4px.ru/ – 4 Пикселя – это Digital агентство интернет-маркетинга полного цикла в Москве, с комплексным подходом к созданию рекламы в интернете. Ознакомьтесь с нашими выгодными услугами в SEO, Яндекс.Директ, SMM, Serm, разработкой и созданием сайтов и многими другими профессиональными услугами. Ознакомьтесь с нашими кейсами – мы будем рады видеть вас в числе наших клиентов!
PeterSlump
| #
https://telegra.ph/Free-Spins-No-Deposit-at-Online-Casinos-Not-on-GamStop-03-27
dostavka alkogolya 24_tgor
| #
доставка алкоголя на дом москва днем доставка алкоголя на дом москва днем .
DonDonfaita
| #
dark market onion https://github.com/nexusdarkrtv1u/nexusdark – darkmarket url
Uazryyx
| #
Привет!
Для некоторых людей, заказать диплом ВУЗа – это острая необходимость, удачный шанс получить достойную работу. Впрочем для кого-то – это желание не терять огромное количество времени на учебу в институте. С какой бы целью вам это не потребовалось, наша компания готова помочь. Быстро, качественно и по доступной цене изготовим документ нового или старого образца на государственных бланках с реальными печатями.
Основная причина, почему многие люди покупают документы, – получить определенную работу. К примеру, знания дают возможность кандидату устроиться на работу, но документального подтверждения квалификации не имеется. При условии, что работодателю важно наличие “корочек”, риск потерять место работы довольно высокий.
Заказать документ ВУЗа можно в нашей компании в столице. Мы предлагаем документы об окончании любых университетов Российской Федерации. Вы сможете получить необходимый диплом по любой специальности, включая документы старого образца. Гарантируем, что при проверке документов работодателем, никаких подозрений не появится.
Ситуаций, которые вынуждают купить диплом ВУЗа немало. Кому-то прямо сейчас нужна работа, а значит, необходимо произвести впечатление на руководителя в процессе собеседования. Некоторые планируют попасть в серьезную компанию, для того, чтобы повысить свой статус в обществе и в дальнейшем начать свое дело. Чтобы не тратить впустую годы жизни, а сразу начинать удачную карьеру, применяя врожденные таланты и приобретенные знания, можно заказать диплом в интернете. Вы станете полезным для социума, получите денежную стабильность в максимально короткий срок- купить аттестат за 9 класс
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-2021-No-Deposit-Bonus-Offers-03-27
Iariorlsf
| #
Заказать диплом университета по невысокой стоимости возможно, обратившись к проверенной специализированной компании. Приобрести документ о получении высшего образования вы можете у нас в Москве. diplomservis.ru/kupit-originalnij-diplom-s-vneseniem-v-reestr-2
Willieavamy
| #
I hit a bonus online and won $200—big win! Marvel Casino
Pingrutty
| #
dark market 2025 https://github.com/aresonioncq0a7/aresonion – best darknet markets
DonayrIngek
| #
На сайте https://vezuviy.shop/ вы сможете приобрести чугунные, стальные банные печи, нержавеющие, а также в облицовке, печи-камины, отопительные, каминные топки, дымоходы, порталы и многое другое. Вся продукция от производителя, а потому наценки очень маленькие. Такая техника считается идеальной не только с целью обогрева помещения, но и оформления пространства. А самое главное, что она отличается долгим сроком службы, надежностью из-за того, что в работе использовались высокотехнологичные, проверенные материалы.
Willieavamy
| #
Some online casino ads are so annoying, popping up everywhere! plinko
Damoncrype
| #
площадка для продажи аккаунтов магазин аккаунтов
Maktrans
| #
Отправляете стекло, технику или посуду? доставка хрупких грузов по россии требует особой заботы. В «Мак-Транс» тщательно упаковывают, фиксируют и следят на каждом этапе. Мы советуем этот вариант, если хотите быть уверены: всё доедет целым.
Willieavamy
| #
The music online pulls—vibe on! plinko
where to get clarinex tablets
| #
cost generic clarinex for sale buying clarinex tablets can i get generic clarinex no prescription
where can i buy clarinex pills order generic clarinex where can i buy clarinex
where can i get clarinex without insurance
where can i get clarinex prices where to buy clarinex tablets cost cheap clarinex pill
can i purchase cheap clarinex no prescription buy generic clarinex tablets can you buy generic clarinex online
PeterSlump
| #
https://telegra.ph/Top-Non-Gamstop-Casinos-in-the-UK-for-2023-03-27
buy cheap motrin for sale
| #
Drug information leaflet. Drug Class.
buy cheap motrin for sale
Best trends of meds. Read information here.
Donaldlaf
| #
darknet site https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark websites
Fayaxasnaish
| #
На сайте https://face-me.ru/ вы сможете попробовать все возможности нейросетей и всего за несколько секунд превратить любое фото в видео. Инновационные и уникальные технологии дают возможность получить роскошную, яркую и любопытную фотосессию без профессионального оборудования, студии. Вы сможете создать фантастические образы и удивительные портреты в соответствии с предпочтениями. Эта программа дарит вам огромное количество возможностей и вдохновение. Вы каждый раз будете получать удивительные, красивые образы, которые всегда будут с вами.
Naseptblori
| #
Судоходная компания Навигатор https://teplohod-restoran.ru/ это возможность организовать праздник на воде в Санкт-Петербурге. Свадьбы, дни рождения, корпоративы, выпускные, а также банкеты и фуршеты. Мы комплексно подходим к организации любого события. Посетите сайт, посмотрите наш флот и стоимость аренды судов, яхт, катеров, теплоходов.
XojigmBlalt
| #
На сайте https://vezuviy.shop/ воспользуйтесь возможностью приобрести качественное печное оборудование, а также дымоходы. На все распространяются гарантии. Здесь вы сможете приобрести и аксессуары, которые гарантированно совместимы со всем оборудованием. Доставка осуществляется абсолютно бесплатно. Клиент сможет получить в качестве презента банный камень. На все аксессуары действует скидка 10%. Все печи выполнены из качественных, высокотехнологичных материалов, потому прослужат долгое время.
Toliknaf
| #
dark market list https://github.com/abacuslink4jjku/abacuslink – dark websites
internetCah
| #
провайдер по адресу
domashij-internet-kazan003.ru
провайдеры домашнего интернета казань
can i purchase cephalexin price
| #
can i purchase generic cephalexin pills
PeterSlump
| #
https://telegra.ph/Discover-Non-Gamstop-Live-Casinos-and-Their-Benefits-03-27
hiếp dâm trẻ em
| #
hiếp dâm trẻ em
A Brief History of Elemental Analysis » Artemis Analytical
chat gpt dlya kyrsovoi_kdmi
| #
нейросеть создать курсовую https://studgen.ru/ .
Rabyitabe
| #
dark markets 2025 https://github.com/darkwebsitesyhshv/darkwebsites – dark market list
Cazrjhc
| #
Здравствуйте!
Без университета сложно было продвинуться по карьере. Теперь этот документ не дает абсолютно никаких гарантий, что удастся найти престижную работу. Более важное значение имеют навыки и знания специалиста, а также его опыт. Именно из-за этого решение о покупке диплома стоит считать выгодным и целесообразным. Купить диплом университета armit.ru/social/user/42552/forum/message/2003/2005/#message2005
PeterSlump
| #
https://telegra.ph/New-Non-Gamstop-Casinos-You-Should-Explore-Today-03-27
Tevupeyhok
| #
How to exchange BITCOIN in the cryptocurrency exchanger quickex are Buy or sell BTC? In this https://www.youtube.com/watch?v=0MupM-q6I3A detailed guide, we will show you how to buy BTC on the quickex platform in 5 easy steps. You will learn how to use a reliable service without registration and verification, getting access to the best rates from leading liquidity providers.
JamesWange
| #
кайтсёрфинг в хургаде
Pingrutty
| #
dark market onion https://github.com/aresonioncq0a7/aresonion – dark markets 2025
Cazrbvk
| #
Здравствуйте!
Мы изготавливаем дипломы любой профессии по выгодным ценам. Стоимость может зависеть от выбранной специальности, года выпуска и образовательного учреждения: diplomanc.com/
mostbet_yjer
| #
мостбет казино http://www.mostbet6010.ru .
DonDonfaita
| #
dark web drug marketplace https://github.com/nexusdarkrtv1u/nexusdark – dark web link
Kypit smartfon v Moskve_aion
| #
Купить Samsung S24 Ultra в Москве https://techno-line.store .
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-with-No-Deposit-Free-Spins-Offers1-03-27
Willieavamy
| #
Are online jackpots real?—skeptic! pinup
Sazrgge
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и прочих профессий по приятным ценам. Цена может зависеть от выбранной специальности, года получения и образовательного учреждения. Всегда стараемся поддерживать для клиентов адекватную ценовую политику. Для нас очень важно, чтобы дипломы были доступны для большого количества граждан. купить диплом москва должна
buying cheap celebrex without rx
| #
Meds information sheet. What side effects?
buying cheap celebrex without rx
Some about medicines. Read now.
PeterSlump
| #
https://telegra.ph/Best-Non-GamStop-Casinos-Accepting-PayPal-Payments4-03-27
Buvukneers
| #
На сайте https://rodinasportsclub.com/ вы найдете информацию, которая касается спортивно-развлекательного клуба «Родина». Каждый желающий получает возможность записаться на тренировку по боксу, которая подходит как новичкам, так и более опытным спортсменам. Вас заинтересуют тренировки по рукопашному бою. Перед вами расписание предстоящих матчей на апрель. Ознакомьтесь с ними сейчас, чтобы построить планы. Также имеются и отчеты по прошедшим соревнованиям. Опубликованы разные новости из жизни клуба.
Donaldlaf
| #
darknet markets https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark web market links
can you buy cheap avodart prices
| #
buying avodart prices buy arimidex liquid avodart generics price
where to get generic avodart without dr prescription get generic avodart without insurance get avodart prices
order generic avodart without insurance
how to buy cheap avodart without insurance order avodart price low cost avodart
where to get avodart without prescription how to buy generic avodart tablets how to buy cheap avodart online
Toliknaf
| #
dark web market list https://github.com/abacuslink4jjku/abacuslink – dark market 2025
PeterSlump
| #
https://telegra.ph/Exploring-Alternatives-to-Gamstop-Casino-Games-03-27
Pingrutty
| #
darknet market links https://github.com/aresonioncq0a7/aresonion – dark web drug marketplace
how to buy cheap vastarel for sale
| #
can i get cheap vastarel tablets
Rabyitabe
| #
darknet drug market https://github.com/darkwebsitesyhshv/darkwebsites – darknet markets links
skypka zolota b-gold.ru_hjon
| #
скупка золота в москве за грамм b-gold.ru скупка золота в москве за грамм b-gold.ru .
skypka zolota _chPn
| #
скупка золота 585 цена за грамм на сегодня http://www.metaphysican.com/vsyo-chto-vam-nuzhno-znat-o-skupke-zolota-sovety-i-rekomendaczii// .
PeterSlump
| #
https://telegra.ph/No-Gamstop-Casinos-in-the-UK-Guide-03-27
1win_jnkr
| #
1win moldova download https://www.1win5004.ru .
Kennetharity
| #
Что такое «Порту Пати» в Дубае и почему об этом говорят все
https://x.com/Fariz418740/status/1908767851009163415
Willieavamy
| #
I won spins online and got $450—big yes! plinko
DonDonfaita
| #
dark web link https://github.com/nexusdarkrtv1u/nexusdark – dark market link
PeterSlump
| #
https://telegra.ph/Top-Non-Gamstop-Casino-Sites-for-Your-Gaming-Adventure-03-27
Willieavamy
| #
I wish online kept old—love that! tom of madness
1win_whea
| #
ванвин http://1win6052.ru .
internetCah
| #
подключение интернета москва
domashij-internet-msk001.ru
провайдеры домашнего интернета москва
Lazrexk
| #
Диплом ВУЗа России!
Без наличия диплома сложно было продвигаться вверх по карьерной лестнице. По этой причине решение о заказе диплома можно считать мудрым и целесообразным. Приобрести диплом об образовании assassins-creed-origins.copiny.com/question/details/id/1075235
where can i buy generic mobic pill
| #
Drugs information sheet. Drug Class.
where can i buy generic mobic pill
Some trends of medicines. Read now.
PeterSlump
| #
https://telegra.ph/Casino-Offers-in-2021-No-Deposit-Bonus-Options-03-27
Xazrdgr
| #
Купить диплом института!
Мы изготавливаем дипломы любой профессии по приятным ценам. Вы приобретаете диплом через надежную компанию. : rbrserien.se/rbrforum/viewtopic.php?t=661089
Donaldlaf
| #
darknet drug market https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet drugs
mostbet_efer
| #
мостбет chrono mostbet6010.ru .
skypka zolota b-gold.ru_snon
| #
скупка золота цена за грамм москва b-gold.ru скупка золота цена за грамм москва b-gold.ru .
skypka zolota _ghPn
| #
скупка золота москва скупка золота москва .
Pingrutty
| #
darknet sites https://github.com/aresonioncq0a7/aresonion – dark web market list
StevenNuh
| #
площадка для продажи аккаунтов купить аккаунт
Toliknaf
| #
darkmarket list https://github.com/abacuslink4jjku/abacuslink – onion dark website
cost of cheap lisinopril without prescription
| #
lisinopril without prescription cheap can i purchase generic lisinopril no prescription how long does it take for lisinopril to work
buying generic lisinopril for sale walgreens lisinopril prices can lisinopril cause stroke
where is lisinopril manufactured
where to buy generic lisinopril pill where can i buy generic lisinopril prices how to get generic lisinopril price
can i buy cheap lisinopril prices lisinopril generic and trade name buy lisinopril
PeterSlump
| #
https://telegra.ph/Guide-to-Trusted-Non-GamStop-Casinos-for-Players1-03-27
1win_mikr
| #
jocuri de noroc online moldova jocuri de noroc online moldova .
Rabyitabe
| #
darknet market lists https://github.com/darkwebsitesyhshv/darkwebsites – best darknet markets
WepodEndat
| #
Visit https://reviewspoty.com/ and you can easily collect reviews by integrating with your existing technology and e-commerce platforms. Increase sales and reduce marketing costs with reviews. Learn more about us and what we offer, as well as pricing and a free trial.
Willieavamy
| #
The online ease rolls—stay wise! Marvel Casino
1win_uoSr
| #
сайт 1win официальный сайт вход сайт 1win официальный сайт вход .
Treftet
| #
Купить документ ВУЗа вы можете в нашей компании в Москве. diplomnie.com/kupit-diplom-kandidata-nauk-2
EugenioBep
| #
https://winsystem.xyz/2024/02/26/igornyi-biznes-v-armenii/
can i purchase cheap reglan without insurance
| #
can i get reglan tablets
Iariorsik
| #
Приобрести диплом института по невысокой цене вы сможете, обратившись к надежной специализированной фирме. Приобрести документ о получении высшего образования вы имеете возможность в нашей компании в Москве. diplomk-vo-vladivostoke.ru/diplom-visshego-obrazovaniya-s-zaneseniem-v-reestr-13
1win_qkSr
| #
1вин приложение http://freereklama.borda.ru/?1-5-0-00000114-000-0-0-1743258539/ .
skypka zolota b-gold.ru_yhon
| #
скупка золота в москве цена за грамм 585 b-gold.ru скупка золота в москве цена за грамм 585 b-gold.ru .
skypka zolota _voPn
| #
585 скупка золота цена за грамм на сегодня http://www.metaphysican.com/vsyo-chto-vam-nuzhno-znat-o-skupke-zolota-sovety-i-rekomendaczii// .
DonDonfaita
| #
darkmarkets https://github.com/nexusdarkrtv1u/nexusdark – dark web sites
HokoseTheap
| #
На сайте https://market-delivery-dubai.ru/ вы сможете воспользоваться такой полезной услугой, как доставка алкоголя в Дубае. В разделе вы найдете такие напитки, как: бризер, абсент, водка, виски, ликер, пиво, коньяк и многое другое. Вся продукция является сертифицированной, качественной, а потому идеально подойдет для долгожданного события, праздника. Есть последние поступления вкусного и ароматного вина, которое вы сможете приобрести на торжество или для того, чтобы скрасить вечер. Доставка происходит в минимальные сроки.
1win_ikea
| #
1вин кг https://1win6052.ru .
Kypit smartfon v Moskve_xron
| #
Техника с доставкой https://techno-line.store .
1win_ikSr
| #
1win online http://www.freereklama.borda.ru/?1-5-0-00000114-000-0-0-1743258539 .
Willieavamy
| #
Online casinos should cap—guard play! aviator
Diplomi_zfKt
| #
Заказать диплом любого ВУЗа!
Мы готовы предложить документы институтов, которые находятся в любом регионе России.
diplomers.com/legalnoe-zanesenie-diploma-v-reestr-bez-sporov-i-nervov/
hair medication propecia
| #
Medication information for patients. Effects of Drug Abuse.
hair medication propecia
All about drug. Get here.
1win_wokr
| #
aplicația 1win aplicația 1win .
Mazrpar
| #
Где купить диплом специалиста?
Мы оказываем услуги по продаже документов об окончании любых университетов РФ. Документы изготавливаются на настоящих бланках государственного образца. Рєwww.indianpharmajobs.in/employer/premialnie-diplom-24
Mazrdiq
| #
Где заказать диплом по актуальной специальности?
Получаемый диплом с приложением отвечает стандартам Министерства образования и науки, никто не сумеет отличить его от оригинала – даже со специальным оборудованием. Не откладывайте собственные мечты и задачи на несколько лет, реализуйте их с нами – отправьте простую заявку на диплом сегодня! Диплом о высшем образовании – не проблема! calisthenics.mn.co/posts/82090250
Pingrutty
| #
darknet markets https://github.com/aresonioncq0a7/aresonion – darknet markets onion address
hranenie veshei_nspa
| #
кладовка походный проезд кладовка походный проезд .
криптобосс казино бесплатные вращения
| #
криптобосс казино бесплатные вращения
A Brief History of Elemental Analysis » Artemis Analytical
Donaldlaf
| #
dark web market urls https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark web market
Yuwihsdub
| #
На сайте https://xn—-8sbaaajsa0adcpbfha1bdjf8bh5bh7f.xn--p1ai/ ознакомьтесь со всеми вариантами антиквариата, который вы сможете продать по честной стоимости. Станислав Бабкин, являясь частным коллекционером, скупает предметы старины дорого. Оценка происходит в режиме реального времени, а средства выплачиваются на карту либо выдаются наличными. Вы сможете продать следующие вещи: серебро, картины, иконы, золотые, серебряные монеты, ордена, часы, самовары и многое другое. Вся сумма выплачивается полностью во время оформления сделки.
криптобосс казино топовые игры в живую
| #
криптобосс казино топовые игры в живую
A Brief History of Elemental Analysis » Artemis Analytical
MatthewSax
| #
Лучшая подборка площадок где продать скины дота 2. Процесс продажи вещей занимает не более 10 минут. Цены выгоднее чем в стиме с выводом на карту или криптовалюту. Большое количество отзывов от реальных пользователей проверенных временем.
Toliknaf
| #
dark market url https://github.com/abacuslink4jjku/abacuslink – dark market url
Willieavamy
| #
Online casinos should rush cash—slow! ganesha gold
Маргарита
| #
Hire a Hitman rent a killer for hire an assassin order a kill service — https://hitman-assassin-killer.com/ hitman for hire.
Rabyitabe
| #
darknet drugs https://github.com/darkwebsitesyhshv/darkwebsites – darknet sites
Donaldskado
| #
Трансфер аэропорт отель Бургас Служба Такси Эклипс предлагает доступные трансферы из аэропортов Бургас, Варна, София и Белград до популярных курортов Болгарии. Встреча с табличкой, помощь с багажом и комфортная поездка до места назначения. Обслуживаем Солнечный Берег, Созополь, Банско и другие направления. Для групп от 5 человек – специальные условия!
internetCah
| #
провайдеры интернета в москве по адресу проверить
domashij-internet-msk002.ru
провайдеры интернета в москве по адресу проверить
can i order cheap co-amoxiclav without a prescription
| #
where can i get cheap co-amoxiclav pill where to buy generic co-amoxiclav without rx order cheap co-amoxiclav without dr prescription
cost cheap co-amoxiclav prices can i get generic co-amoxiclav pill generic co-amoxiclav for sale
where to buy generic co-amoxiclav without insurance
how to get co-amoxiclav where to buy co-amoxiclav pill cheap co-amoxiclav without insurance
where can i get generic co-amoxiclav no prescription can i order co-amoxiclav online buy cheap co-amoxiclav without prescription
MatthewSuelt
| #
биржа аккаунтов http://prodat-akkaunt.ru
автомобильные грузоперевозки по россии
| #
Меня интересовали перевозка грузов по россии автотранспортом цены, и я приятно удивился: у «Мак-Транс» тарифы реально адекватные, особенно с учётом уровня сервиса. Заказ оформил онлайн, всё прозрачно и удобно. Сравнивал — тут лучший баланс цены и качества.
Willieavamy
| #
I wish online paid swift—drag off! mines juego
Sazrgzw
| #
диплом школы купить
sites
| #
sites
A Brief History of Elemental Analysis » Artemis Analytical
mostbet_zcOn
| #
mostbet casino mostbet6029.ru .
DonDonfaita
| #
dark web market https://github.com/nexusdarkrtv1u/nexusdark – darknet drug store
Pingrutty
| #
darknet market links https://github.com/aresonioncq0a7/aresonion – dark markets
ezetimibes impact on cyp3a4 enzyme unraveling the relationship
| #
Pills information. Effects of Drug Abuse.
ezetimibes impact on cyp3a4 enzyme unraveling the relationship
All about medicament. Read information now.
where buy cephalexin pills
| #
can i purchase cheap cephalexin pills
Donaldlaf
| #
dark markets 2025 https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet market
Cheryl imperialusa
| #
Thank you for creating such an amazing and useful website honey. Your dedication and hard work truly shine through. I deeply appreciateee everything youve done!
Sazrcnl
| #
Мы изготавливаем дипломы любой профессии по приятным ценам.– diplomk-v-krasnodare.ru/kupit-diplom-v-reestre-legalno-i-bez-problem-3/
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-No-Deposit-Bonus-Explained-03-27
Toliknaf
| #
darknet links https://github.com/abacuslink4jjku/abacuslink – dark web market list
Willieavamy
| #
I love the poker mix online—fresh go! bongo bongo
Willieavamy
| #
Online casinos fit quiet—no fuss! bongo bongo
Cheryl islandportpress
| #
I am truly grateful for the fantastic website youve built darling. Its not only well-designed but also highly functional. Thank you for your incredible effort honey!
AndrewFoure
| #
Инженерные решения для электропроводки — детали по ссылке https://www.pexels.com/ru-ru/@mikhail-makarov-2151057499/
Rabyitabe
| #
darknet drugs https://github.com/darkwebsitesyhshv/darkwebsites – dark web marketplaces
Sazrnxp
| #
Купить документ о получении высшего образования вы сможете в нашем сервисе. Мы оказываем услуги по продаже документов об окончании любых ВУЗов РФ. Вы получите необходимый диплом по любым специальностям, любого года выпуска, включая документы образца СССР. Даем 100% гарантию, что при проверке документа работодателем, подозрений не возникнет. diplom4you.com/kupit-diplom-visshego-obrazovaniya-s-vneseniem-v-reestr/
PeterSlump
| #
https://telegra.ph/Best-Casinos-Not-on-Gamstop-with-No-Deposit-Offers-03-27
Cheryl proshotcase
| #
I am beyond thankful for the wonderful website youve created for everyone. Its a masterpiece of functionality and design love. Youve truly outdone yourself baby!
Pingrutty
| #
onion dark website https://github.com/aresonioncq0a7/aresonion – darknet drug links
JumopnFuh
| #
На сайте https://t.me/rahvalskiy вы сможете ознакомиться с информацией о компании, которая занимается продвижением сайтов «под ключ». Вы сможете воспользоваться всеми комплексными рекламными услугами. В компании работают лучшие, компетентные сотрудники с огромным стажем, а потому они реализуют проект любой сложности и по лучшей стоимости. Для сотрудников нет неисполнимых задач. Они работают независимо от темы проекта и в строго указанные сроки. Можете не сомневаться в том, что получите результат, на который рассчитывали.
Техподдержка казино онлайн
| #
Техподдержка казино онлайн
A Brief History of Elemental Analysis » Artemis Analytical
PeterSlump
| #
https://telegra.ph/Explore-New-Non-Gamstop-Casinos-for-Exciting-Play-03-27
DonDonfaita
| #
dark web markets https://github.com/nexusdarkrtv1u/nexusdark – bitcoin dark web
cheap amaryl without a prescription
| #
how to buy cheap amaryl pills can i purchase amaryl prices buy generic amaryl for sale
buying amaryl pill get amaryl no prescription can i order cheap amaryl price
where can i buy generic amaryl
can you buy generic amaryl online how to buy cheap amaryl pills cost of cheap amaryl without prescription
where can i get cheap amaryl how to get cheap amaryl pills can i order cheap amaryl pills
1win_dtkr
| #
1вин приложение 1вин приложение .
requip tablet
| #
Drug information sheet. Effects of Drug Abuse.
requip tablet
Best trends of drug. Read information here.
Bahaxeldug
| #
На сайте https://pohudetlegko.ru/ представлена исчерпывающая, актуальная и полезная информация о том, как похудеть правильно, без вреда для организма и максимально эффективно. На сайте вы найдете вдохновляющие истории, советы звезд, которые выглядят младше своего возраста. Есть рецепты блюд, которые созданы из полезных, вкусных ингредиентов. Но при этом они помогут скинуть вес и привести себя в форму к летнему сезону. Рецепты простые и быстрые, а потому справиться с приготовлением пищи сможет каждый. Описаны и упражнения, обертывания для похудения.
RodneyTaisa
| #
покупка аккаунтов https://pokupka-akkauntov.ru
PeterSlump
| #
https://telegra.ph/No-Deposit-Casinos-Not-on-Gamstop-for-Players-2023-03-27
case noi de inchiriat_kqsl
| #
case noi de ?nchiriat case noi de ?nchiriat .
Donaldlaf
| #
dark market onion https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark market 2025
1win_pmEl
| #
1вин официальный сайт мобильная 1вин официальный сайт мобильная .
remont kardana_vjkn
| #
ремонт карданов москва ремонт карданов москва .
Sazrbiv
| #
купить диплом старого образца в кемерово
can i purchase cheap zetia online
| #
order zetia without dr prescription
internetCah
| #
домашний интернет подключить москва
domashij-internet-msk003.ru
узнать интернет по адресу
mostbet_bfMl
| #
скачать mostbet на телефон http://www.mostbet6030.ru .
PeterSlump
| #
https://telegra.ph/PayPal-Non-GamStop-Casinos-for-Seamless-Transactions-03-27
Toliknaf
| #
darknet marketplace https://github.com/abacuslink4jjku/abacuslink – darknet drug market
more-53
| #
more-53
A Brief History of Elemental Analysis » Artemis Analytical
www
| #
www
A Brief History of Elemental Analysis » Artemis Analytical
Pingrutty
| #
darknet drug links https://github.com/aresonioncq0a7/aresonion – dark market link
Rabyitabe
| #
dark web market list https://github.com/darkwebsitesyhshv/darkwebsites – dark web marketplaces
PeterSlump
| #
https://telegra.ph/Best-Non-Gamstop-PayPal-Casinos-for-Online-Players-03-27
RepairCdDvD is a tool that recovers files from unreadable CD
| #
RepairCdDvD is a tool that recovers files from unreadable CD
A Brief History of Elemental Analysis » Artemis Analytical
hranenie veshei_bxpa
| #
склады для хранения вещей в москве класса а склады для хранения вещей в москве класса а .
lopakFew
| #
С помощью финансового блога Апостолиди вы можете получить доступ к ценным советам. Мы делимся личным опытом, простым языком объясняем сложные темы. Гарантируем актуальность и достоверность информации. Вы узнаете, как новый торговый счет на Forex открыть. Ищете валютные новости? – здесь есть авторские статьи по финансовым вопросам. Для отправки комментария вам нужно авторизоваться. Материалы разделены по темам, что упрощает поиск необходимой информации. Изучайте полезный блог Апостолиди. Финансовая грамотность – это просто!
Dnrtkur
| #
Где заказать диплом специалиста?
Мы изготавливаем дипломы психологов, юристов, экономистов и любых других профессий по доступным ценам. Мы можем предложить документы техникумов, расположенных в любом регионе России. Можно заказать качественно напечатанный диплом за любой год, указав необходимую специальность и оценки за все дисциплины. Документы выпускаются на бумаге самого высокого качества. Это дает возможности делать государственные дипломы, которые невозможно отличить от оригиналов. Документы будут заверены всеми требуемыми печатями и подписями. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Важно, чтобы документы были доступными для большого количества граждан. diplomdoc.ru/kupit-svidetelstvo-o-zaklyuchenii-braka-7
PeterSlump
| #
https://telegra.ph/Guide-to-Casino-Online-No-Gamstop-Options-03-27
DonDonfaita
| #
bitcoin dark web https://github.com/nexusdarkrtv1u/nexusdark – darknet drug market
where can i buy generic benicar pill
| #
Drugs information leaflet. Long-Term Effects.
where can i buy generic benicar pill
Actual news about drug. Get now.
case noi de inchiriat_hhsl
| #
rental houses with parking space http://www.rapitorimania.ro/forum/marele-bazar-f18/case-moderne-de-inchiriat-langa-bucuresti-t938.html .
Sazrfmr
| #
Заказать диплом университета по выгодной стоимости возможно, обратившись к проверенной специализированной компании. Мы предлагаем документы об окончании любых университетов Российской Федерации. Приобрести диплом любого ВУЗа– diplom-top.ru/kupite-diplom-o-srednem-obrazovanii-s-vneseniem-v-reestr-4/
1win_eaen
| #
зайти в 1вин https://www.snatkina.borda.ru/?1-5-0-00000311-000-0-0-1743258575 .
RobertWer
| #
Astrologers named 6 signs of the eastern horoscope that will be lucky in April
https://x.com/Fariz418740/status/1908917832265392277
remont kardana_vokn
| #
вал замена карданный вал замена карданный .
Vera Bradley Shop Online
| #
Vera Bradley Shop Online
A Brief History of Elemental Analysis » Artemis Analytical
mostbet_ycOn
| #
mostbet kg отзывы http://mostbet6029.ru .
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-Featuring-Free-Spins-Offers-03-27
Eanrcnm
| #
Для удачного продвижения по карьерной лестнице нужно наличие официального диплома университета. Купить диплом об образовании у проверенной организации: dip-lom-rus.ru/diplom-o-sredne-texnicheskom-obrazovanii-kupit-6/
https://www.cucumber7.com/
| #
Nicely put, Appreciate it!
Look into my page; https://www.cucumber7.com/
1win_vven
| #
1win ставки официальный сайт snatkina.borda.ru/?1-5-0-00000311-000-0-0-1743258575 .
Donaldlaf
| #
darknet links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – onion dark website
1win_yoEl
| #
1win онлайн http://www.1win6005.ru .
Pingrutty
| #
darknet drugs https://github.com/aresonioncq0a7/aresonion – darkmarket 2025
Sazrqzg
| #
свердловская область купить аттестат
1win_mmen
| #
1вин вход https://www.snatkina.borda.ru/?1-5-0-00000311-000-0-0-1743258575 .
PeterSlump
| #
https://telegra.ph/Top-Up-Non-Gamstop-Casinos-Via-Phone-Bill-Payment-03-27
Toliknaf
| #
dark markets https://github.com/abacuslink4jjku/abacuslink – dark market url
Jariorcvt
| #
Где купить диплом по нужной специальности?
Наша компания предлагаетбыстро заказать диплом, который выполнен на оригинальной бумаге и заверен мокрыми печатями, водяными знаками, подписями. Наш диплом способен пройти лубую проверку, даже при помощи специального оборудования. Достигайте цели быстро и просто с нашими дипломами. Купить диплом любого ВУЗа! audiencerp.forumex.ru/viewtopic.php?f=11&t=482
Jarioregd
| #
Где приобрести диплом по нужной специальности?
Наша компания предлагает выгодно и быстро приобрести диплом, который выполняется на оригинальном бланке и заверен мокрыми печатями, штампами, подписями. Документ пройдет любые проверки, даже при использовании специфических приборов. Достигайте своих целей быстро и просто с нашим сервисом.
Приобрести диплом о высшем образовании damdiplomisa.com/kupit-diplom-slesarya-6/
DannyTox
| #
Доставка курьером в день заказа. Акциз и копии высокого качества. Огромный ассортимент из 89 позиций на складе. Прайс-лист с выгодными ценами на все популярные позиции: электронные сигареты без никотина купить
how can i get generic cetirizine for sale
| #
buy cetirizine hydrochloride how to buy cetirizine without prescription cost generic cetirizine prices
can you get cetirizine how to get cheap cetirizine without prescription how to get cetirizine without a prescription
order generic cetirizine
cheap cetirizine price how can i get generic cetirizine online where can i buy generic cetirizine online
order generic cetirizine no prescription can i buy generic cetirizine tablets where can i get generic cetirizine pills
фрибет за регистрацию
| #
Получи фрибеты за регистрацию через партнерские сайты
Фрибеты за регистрацию
– как получить через партнеров
Сегодня мы раскроем секрет получения заманчивых бонусов при знакомстве с букмекерскими сервисами.
Зарегистрировавшись на специализированных платформах, вы сможете заручиться солидными преимуществами еще до первого спортивного пари.
Опытные игроки давно оценили
этот подход, и мы готовы поделиться с вами эффективными стратегиями.
Бонусные ставки: ключ
к выгодному старту Многие
современные букмекеры предлагают впечатляющие бонусы новым клиентам.
Достаточно пройти нехитрую
процедуру регистрации, и вы уже можете рассчитывать на «бесплатные»
ставки. Это отличная возможность опробовать
сервис, не рискуя собственными средствами.
Лови Фрибет за Регу: Секреты от Партнёров
Первый шаг – изучение условий акций.
Будьте внимательны к деталям:
некоторые предложения требуют
использование промокодов или выполнение
определенных действий, таких как пополнение счета на минимальную сумму.
Заранее узнайте, какие шаги необходимо предпринять
для получения вознаграждения.
Следующий аспект – выбирайте ресурсы с положительными отзывами.
Надежные источники информации могут направить вас на букмекеров, предлагающих лучшие условия.
Нередко на специализированных
платформах публикуются обзоры, которые содержат полезные советы и рекомендации.
Эти материалы могут выявить скрытые нюансы, о которых обычные
пользователи не догадываются.
Обратите внимание на время действия акций.
Некоторые букмекеры могут предоставлять временные предложения, которые действуют в течение ограниченного периода.
Подписка на рассылки или уведомления поможет не упустить выигрышные возможности.
Не забывайте о сравнении предложений.
Разные букмекеры могут дополнять свои акции уникальными условиями.
Сравнив их, вы сможете выбрать наиболее выгодный вариант, который соответствует вашим ожиданиям.
Также полезно следить за изменениями в правилах,
так как они могут измениться в любой момент.
Наконец, используйте площадки, предлагающие программы лояльности.
Участие в таких проектах может открыть новые возможности для получения дополнительных вознаграждений.
Это позволит не только быстрее накапливать бонусы, но
и повысит уровень вашего игрового опыта.
Хорошая подготовка и системный
подход помогут легко освоить мир бонусных предложений, а
упорство откроет новые горизонты
для выигрышей. Делитесь информацией с друзьями, чтобы совместно находить лучшие варианты для приятного досуга.
Где Найти Жирные Приветственные Бонусы по Ссылкам?
Эксклюзивные предложения от букмекеров зачастую скрыты на ресурсах, сотрудничающих с ними напрямую.
Ищите купоны и промокоды на специализированных платформах, обозревающих букмекерские конторы и предлагающих выгодные стартовые
условия. В частности, посетите авторитетные порталы, публикующие обзоры спортивных ставок, и форумы,
где опытные игроки делятся информацией об
акциях и кодах.
Следите за контентом блогеров и стримеров,
специализирующихся на спортивных
прогнозах и гемблинге. Многие из них сотрудничают с букмекерскими компаниями и предоставляют своим подписчикам уникальные бонусные
комбинации для старта. Проверяйте
описания под видео и посты в социальных сетях.
Подписывайтесь на email-рассылки и Telegram-каналы, посвященные ставкам на
спорт. Они часто первыми анонсируют специальные промо-акции и раздают промокоды, активирующие увеличенные
приветственные поощрения при создании учетной записи.
Обращайте внимание на ресурсы, имеющие хорошую репутацию и положительные отзывы пользователей.
Внимательно изучайте условия и положения каждой акции.
Размер бонуса – не единственный важный параметр.
Учитывайте вейджер, сроки отыгрыша и доступные виды спорта для ставок.
Сопоставляйте предложения
разных контор, чтобы выбрать наиболее подходящее именно вам.
Регулярно посещайте разделы “Акции” и “Бонусы” на
официальных веб-страницах букмекеров.
Компании часто запускают лимитированные по времени предложения, доступные
только по конкретным ссылкам или промокодам.
Оперативно реагируйте на новые анонсы, чтобы не упустить шанс.
Как Гарантированно Получить и Использовать Фрибет?
Чтобы получить бонусные средства для ставок, важно следовать нескольким ключевым шагам.
Сначала найдите букмекера, который предлагает такие преимущества.
Чаще всего условия получения бонусов можно найти в
разделе акций на сайте букмекера.
Обратите внимание на требования, такие как минимальный депозит
и коэффициенты.
После завершения создания аккаунта сделайте первый депозит.
Убедитесь, что сумма соответствует минимальным требованиям для
активации предложенной акции.
Некоторые букмекеры могут требовать активацию бонуса через определённый раздел вашего
профиля, так что уточните этот момент.
Как только доступные для использования средства окажутся на вашем счету, проверьте условия их использования.
Обычно требуется сделать ставки с определённым коэффициентом или в определённый
срок. Этот аспект не стоит игнорировать, так как несоблюдение условий может привести к потере средств.
Когда вы начали использовать бонусные средства, выбирайте
ставки с высокими шансами на выигрыш.
Анализируйте результаты команд или игроков, на
которых хотите ставить. Более обоснованные
решения увеличивают шансы на успех.
Не забудьте ознакомиться с отзывами
и рейтингами, чтобы сделать
правильный выбор.
В завершение, отследите свои
успехи и регулярно проверяйте состояние счета.
Если бонусные средства
были успешно использованы,
вы сможете вывести накопленные
выигрыши, следуя правилам букмекерской конторы.
Сосредоточьтесь на изучении и
анализе, и ваше использование бонусов станет максимально успешным.
mostbet_mrMl
| #
mostbets https://www.mostbet6030.ru .
PeterSlump
| #
https://telegra.ph/Discover-Trusted-Non-Gamstop-Casinos-for-Safe-Play1-03-27
case noi de inchiriat_fjsl
| #
house rental corbeanca http://www.rapitorimania.ro/forum/marele-bazar-f18/case-moderne-de-inchiriat-langa-bucuresti-t938.html .
remont kardana_dekn
| #
ремонт карданов ремонт карданов .
1win_pwkr
| #
1winn https://1win6053.ru .
ElmerCix
| #
¡Hola gammers!
Descubre los bonos de 10 euros gratis sin depГіsito. casino 10 euros gratis Diferentes casinos te los ofrecen para que empieces a jugar sin gastar nada. ВЎRegГstrate y gana!
RegГstrate hoy y recibe 10 euros gratis para jugar en los mejores casinos online. Sin depГіsitos, sin riesgos. Solo diversiГіn y premios reales. Es la forma perfecta de empezar a jugar sin preocuparte por tu bolsillo.
Toda la información en el enlace – п»їhttps://10eurosgratissindepositocasino.xyz/
¡Que tengas buenos bonos!
Stephenrop
| #
дешевый хостинг для сайта веб хостинг украина
Donaldskado
| #
Мечтаете о роскошном авто, сочетающем в себе передовые технологии и безупречный дизайн? Дубай – это сокровищница автомобильных шедевров, где представлены модели, способные удовлетворить самый взыскательный вкус. Компания ChatMost предлагает полный спектр услуг по заказу и доставке автомобилей из Дубая в Россию, избавляя вас от хлопот и рисков. Доставка авто из Дубая в Россию
DonDonfaita
| #
darknet markets onion https://github.com/nexusdarkrtv1u/nexusdark – darknet drug market
PeterSlump
| #
https://telegra.ph/New-Non-Gamstop-Casinos-No-Deposit-Bonuses-Explained-03-27
cryptoboss казино топовые советы по игре
| #
cryptoboss казино топовые советы по игре
A Brief History of Elemental Analysis » Artemis Analytical
1win_oyEl
| #
1 win официальный 1win6005.ru .
chrome metamask
| #
MetaMask Download is worth it! A secure and efficient wallet for managing digital assets without the need for intermediaries.
get cheap keflex without prescription
| #
where can i get generic keflex tablets
Pingrutty
| #
darknet market lists https://github.com/aresonioncq0a7/aresonion – darknet drug links
PeterSlump
| #
https://telegra.ph/Online-Casinos-Not-Registered-with-Gamstop-03-27
Donaldlaf
| #
dark markets 2025 https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darkmarket 2025
XohalellHoR
| #
На сайте https://kabel-silovoj.ru/ воспользуйтесь возможностью приобрести качественный, надежный силовой кабель, который выполнен по ГОСТу и в соответствии с нормативами. Вы совершаете покупку у производителя, а потому установлены привлекательные цены, чтобы сделать это смог каждый. Основными покупателями компании являются профессионалы: электрики, монтажники, прорабы, а также бригадиры. Каждый клиент получает возможность воспользоваться оперативной доставкой, а оплатить так, как удобно.
Mazrcdc
| #
Мы готовы предложить дипломы любых профессий по разумным тарифам. Стараемся поддерживать для заказчиков адекватную ценовую политику. Важно, чтобы документы были доступными для большого количества наших граждан.
Заказ диплома, который подтверждает окончание ВУЗа, – это выгодное решение. Заказать диплом любого университета: peoplediplom.ru/kupit-diplom-o-obrazovanii-2/
VanacrtMaymn
| #
На сайте https://grillhouse-spb.ru/ закажите звонок для того, чтобы проконсультироваться по поводу покупки беседок-гриль, гриль-домиков. Популярная компания «Гриль Хаус» в течение долгого времени производит и собирает беседки, в том числе, для барбекю, а также саун. Все это потребуется для того, чтобы незабываемо провести время на природе и в кругу друзей, близких людей. Все конструкции выполнены из качественных, надежных материалов, которые являются износостойкими. Прямо сейчас изучите каталог доступных для заказа вариантов.
Dnrtmkh
| #
Где купить диплом по нужной специальности?
Мы изготавливаем дипломы любой профессии по приятным тарифам. Мы можем предложить документы ВУЗов, которые расположены в любом регионе Российской Федерации. Вы можете приобрести качественно напечатанный диплом от любого заведения, за любой год, указав необходимую специальность и оценки за все дисциплины. Дипломы и аттестаты выпускаются на бумаге высшего качества. Это дает возможность делать государственные дипломы, не отличимые от оригиналов. Документы будут заверены необходимыми печатями и штампами. Стараемся поддерживать для клиентов адекватную ценовую политику. Важно, чтобы дипломы были доступными для большого количества наших граждан. kupite-diplom0024.ru/kupit-diplom-v-kemerovo-2-2
Dohuydlek
| #
На сайте https://ptne.ru/ получите бесплатную консультацию по вопросам продажи недвижимости. Вы сможете воспользоваться такой услугой, как оперативный выкуп различной недвижимости в Санкт-Петербурге. При этом получить аванс вы сможете в день обращения. Агентство оказывает такие услуги, как: выкуп квартир, долей, займ до продажи. Выкуп происходит за свои средства, сделка является прозрачной. Адвокаты оказывают полное юридическое сопровождение, консультационную помощь. Если документов не хватает, то их соберут максимально быстро.
Toliknaf
| #
dark markets 2025 https://github.com/abacuslink4jjku/abacuslink – darknet market list
PeterSlump
| #
https://telegra.ph/Best-Non-Gamstop-Casinos-for-UK-Players-03-27
mostbet_ogMl
| #
mostbet apk скачать https://mostbet6030.ru .
internetCah
| #
провайдеры по адресу нижний новгород
domashij-internet-nizhnij-novgorod001.ru
тарифы интернет и телевидение нижний новгород
CarozrdAsymn
| #
На сайте https://bitovayatechnika.blogspot.com/2025/03/blog-post.html изучите полезную, актуальную информацию, которая касается сплит-систем и того, как правильно их выбрать домой либо в офис. Эти функциональные и умные устройства нацелены на то, чтобы поддержать приятную температуру в помещении. Но для того, чтобы воспользоваться всеми функциями, необходимо правильно выбрать устройство. К важным преимуществам такой техники относят то, что она является энергоэффективной, работает практически бесшумно.
Bikuweloract
| #
На сайте https://apostolidi.ru/ вы найдете всю полезную информацию, касающуюся торгов на бирже, а также финансов. Здесь также вы найдете и прогнозы по валютным парам от экспертов, которые работают на совесть. Обязательно публикуются свежие, интересные и информативные записи, которые будут полезны всем без исключения. Также представлен и курс доллара, о котором нужно знать инвесторам. На этом портале вы получаете возможность пообщаться с единомышленниками, задать им вопрос, узнать много нового, интересного.
https://www.bloglovin.com/@mikehammelton/13236393
| #
квартира на сутки Гомель https://subscribe.ru/group/biznes-nashe-vse/19036932/
mamamsteaw
| #
Mirtash купить природный камень предлагает. Вы можете по телефону оформить заявку. Мы работаем много и длительные отношения с клиентами, которые основаны на доверии строим. Вы можете рассчитывать на выгодные условия сотрудничества и удобную систему расчетов. Ищете лемезит бордо? Mirtash.ru – здесь можно детально ознакомиться с информацией о доставке природного камня. Высокий уровень качества услуг мы гарантируем. Позвоните нам для получения консультации. С «Mirtash» вы получаете надежного партнера, готового помочь в реализации любых проектов.
Naxireclabs
| #
На сайте https://dragon1gaming.site/ вы найдете ТОП букмекерских контор для ставок на спорт и другие события. Вы узнаете, как выбрать официальную, надежную букмекерскую контору, которая, в том числе дает бонусы, отличные коэффициенты и честные выплаты. Вы узнаете, как зарегистрироваться, пополнить счет, какие виды ставок и линий бывают, а также как вывести деньги. Подробнее на сайте.
AlbertCalia
| #
Брови Анапа Уже 10 лет Делаем качественный перманентный макияж в Анапе без синих красных бровей. Только премиум материалы , стерильно и без боли, работаем в любых техниках.
Обучаем перманентному макияжу с нуля со свидетельством установленного образца , ведем курсы бровистов
DonDonfaita
| #
dark market link https://github.com/nexusdarkrtv1u/nexusdark – darknet market lists
how to get generic cipro without prescription
| #
how to get cheap cipro prices where buy generic cipro price can i purchase generic cipro
cipro buy at drugstore buy cipro without prescription in lafayette in where buy cipro online
how to get generic cipro without insurance
order generic cipro without insurance can i get cipro price can i get cipro no prescription
buy cipro 250 where buy cheap cipro without prescription how can i get cheap cipro pills
Pingrutty
| #
bitcoin dark web https://github.com/aresonioncq0a7/aresonion – dark web market urls
Donaldlaf
| #
darknet markets https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet sites
get generic tadora pills
| #
where can i buy tadora without prescription
Toliknaf
| #
darknet markets onion https://github.com/abacuslink4jjku/abacuslink – darknet markets
Dnrtmdt
| #
Где заказать диплом специалиста?
Мы изготавливаем дипломы любой профессии по разумным ценам. Мы готовы предложить документы техникумов, которые находятся на территории всей России. Можно купить качественный диплом от любого заведения, за любой год, включая сюда документы старого образца. Дипломы и аттестаты делаются на “правильной” бумаге высшего качества. Это позволяет делать государственные дипломы, не отличимые от оригинала. Документы будут заверены всеми требуемыми печатями и штампами. Стараемся поддерживать для заказчиков адекватную политику цен. Важно, чтобы документы были доступны для большого количества граждан. okdiplom.com/kupit-diplom-uchitelya-2-3
Rabyitabe
| #
darknet markets 2025 https://github.com/darkwebsitesyhshv/darkwebsites – darknet market
Calvinmok
| #
суши роллы барнаул роллы барнаул доставка
https://boosty.to/milaeryomina/posts/329da96a-5053-4773-94cf-6b5eabfa5bac
| #
квартира на сутки Гомель https://www.liveinternet.ru/users/mila_eryomina/post509345619/
Davidsmess
| #
Лизинг — долгосрочная аренда с правом выкупа. Собственником остаётся лизингодатель, что снижает риски для клиента. Платежи часто ниже кредитных, а НДС включается в сумму, что выгодно для бизнеса. Лизинговые платежи учитываются как расходы, уменьшая налог на прибыль https://bin-leasing.ru/
skypka zolota b-gold.ru_uwon
| #
дорого скупка золота b-gold.ru дорого скупка золота b-gold.ru .
Diplomi_qnKt
| #
Приобрести диплом о высшем образовании!
Мы можем предложить документы любых учебных заведений, которые расположены в любом регионе РФ.
vuz-diplom.ru/bistrij-diplom-vuza-s-zaneseniem-v-reestr-bez-hassles/
Pingrutty
| #
tor drug market https://github.com/aresonioncq0a7/aresonion – darknet markets onion address
DonDonfaita
| #
darknet drug market https://github.com/nexusdarkrtv1u/nexusdark – dark markets 2025
internetCah
| #
недорогой интернет нижний новгород
domashij-internet-nizhnij-novgorod002.ru
какие провайдеры по адресу
can i order generic celexa tablets
| #
can i buy celexa for sale where to get cheap celexa pill cost of celexa without rx
can you get generic celexa price how to buy celexa no prescription can you buy generic celexa tablets
generic celexa tablets
can i purchase celexa price get celexa without prescription can i buy generic celexa pills
can i order celexa pills can i get celexa without a prescription where can i get celexa no prescription
일프로샵
| #
일프로샵
A Brief History of Elemental Analysis » Artemis Analytical
skypka zolota _pyPn
| #
скупка золота цена за грамм скупка золота цена за грамм .
1win_fqkr
| #
1win http://www.1win5004.ru .
Donaldlaf
| #
darknet market list https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet market lists
WilliamDix
| #
Мы изготавливаем дипломы любых профессий по разумным тарифам. Дипломы производятся на настоящих бланках государственного образца Быстро и просто приобрести диплом об образовании diplomers.com
can you buy rizact pill
| #
can i get generic rizact tablets
Lazrszy
| #
Где купить диплом по нужной специальности?
Купить диплом университета по выгодной цене можно, обратившись к надежной специализированной компании.: sdiplom.ru
Jamesdusty
| #
Заказала с доставкой на природу – упаковали так, что даже дождь не помешал!
доставка цветов томск на дом
https://subscribe.ru/group/biznes-nashe-vse/19036932/
| #
квартира на сутки Гомель https://crazyworldmen.livejournal.com/99939.html
Pingrutty
| #
dark web markets https://github.com/aresonioncq0a7/aresonion – dark web sites
Josephvus
| #
VANGPOKER- новое идеальное место для покера!
Ведущая в мире платформа блокчейн покера!
Вы можете стать одними из первых участников нового проекта с честным рейкбэком, бонусом на первый депозит 200%, быстрым вводом и выводом, и отсутствием KYC.
Приглашаем вас присоединится к крупнейшему сообществу VANGPOKER в телеграмм, зарегестрироваться в VANGPOKER и получать дополнительные бонусы и повышенный рейкбэк. зарегестрироваться в VANGPOKER
Rabyitabe
| #
dark market https://github.com/darkwebsitesyhshv/darkwebsites – dark web market list
YuwiseyEnuts
| #
На сайте https://xn—-7sbjhqn0bhjc0lk.xn--p1ai/ получите юридическую консультацию по различным вопросам. Компания работает как с физическими, так и частными лицами. На все услуги установлены привлекательные расценки, чтобы воспользоваться ими смог каждый. Предприятие работает в этой сфере более 11 лет. В команде трудятся 11 специалистов, которые справятся с задачей независимо от сложности. К вашим услугам досудебный юрист, досудебное урегулирование, кредитный, семейный юрист, решение страховых споров.
DonDonfaita
| #
dark web market links https://github.com/nexusdarkrtv1u/nexusdark – darknet markets links
Buvukneers
| #
На сайте https://rodinasportsclub.com/ вы найдете информацию, которая касается спортивно-развлекательного клуба «Родина». Каждый желающий получает возможность записаться на тренировку по боксу, которая подходит как новичкам, так и более опытным спортсменам. Вас заинтересуют тренировки по рукопашному бою. Перед вами расписание предстоящих матчей на апрель. Ознакомьтесь с ними сейчас, чтобы построить планы. Также имеются и отчеты по прошедшим соревнованиям. Опубликованы разные новости из жизни клуба.
PeterSlump
| #
https://telegra.ph/Find-the-Best-Non-Gamstop-Casinos-in-2023-03-27
get furosemide without prescription
| #
cheap furosemide for sale can i buy cheap furosemide without rx cost of furosemide without prescription
how to buy generic furosemide without a prescription buy generic furosemide prices where buy furosemide for sale
buying cheap furosemide for sale
how to buy cheap furosemide no prescription buy furosemide without dr prescription can you get furosemide prices
can i purchase furosemide without a prescription cost generic furosemide price where can i buy furosemide online
Donaldlaf
| #
dark market 2025 https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darkmarkets
Diplomi_bkKt
| #
Купить диплом любого университета!
Мы предлагаем документы любых учебных заведений, которые расположены в любом регионе Российской Федерации.
diplom-club.com/bistraya-pokupka-diploma-s-zaneseniem-v-reestr/
internetCah
| #
провайдеры в нижнем новгороде по адресу проверить
domashij-internet-nizhnij-novgorod003.ru
интернет провайдеры по адресу дома
Pingrutty
| #
darknet marketplace https://github.com/aresonioncq0a7/aresonion – darkmarket
diamond ring
| #
diamond ring
A Brief History of Elemental Analysis » Artemis Analytical
Toliknaf
| #
dark market onion https://github.com/abacuslink4jjku/abacuslink – dark websites
Mazruya
| #
Мы изготавливаем дипломы любых профессий по доступным ценам. Стараемся поддерживать для покупателей адекватную политику тарифов. Важно, чтобы дипломы были доступны для большинства наших граждан.
Покупка документа, подтверждающего обучение в университете, – это выгодное решение. Заказать диплом любого ВУЗа: diplomoz-197.com/kupit-diplom-o-meditsinskom-obrazovanii/
skypka zolota b-gold.ru_sbon
| #
скупка золота сколько за грамм b-gold.ru скупка золота сколько за грамм b-gold.ru .
https://www.liveinternet.ru/users/mila_eryomina/post509345619/
| #
квартира на сутки Гомель https://housethings.mystrikingly.com/blog/28ce9ed194a
Rabyitabe
| #
darkmarket https://github.com/darkwebsitesyhshv/darkwebsites – darknet markets links
cost cheap tegretol pills
| #
cost of cheap tegretol online
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casino-Review-Explore-Your-Options-03-27
skypka zolota _lfPn
| #
скупка золота в москве цена за грамм 585 на сегодня metaphysican.com/vsyo-chto-vam-nuzhno-znat-o-skupke-zolota-sovety-i-rekomendaczii/ .
DonDonfaita
| #
darknet markets onion https://github.com/nexusdarkrtv1u/nexusdark – darknet drugs
казань прием металлов цена
| #
Тимерхан организация, которая занимается приемом и утилизацией металлических отходов.
Так же вы можете узнать
цена прием металла казань текущие цены.
Мы работаем с физическими и юридическими
лицами, принимаем металлолом всех видов и гарантируем быструю и точную
оценку стоимости. При больших объемах сдачи мы предлагаем бесплатный
вывоз металлолома с вашей территории.
Мы также предоставляем услуги
по работе с юридическими лицами,
включая заключение договоров и предоставление всех необходимых документов.
Обращаясь к нам, вы не только избавляетесь от металлического мусора, но и помогаете
сохранить окружающую среду,
отправляя отходы на переработку и уменьшая выбросы
углекислого газа.
Pingrutty
| #
dark web sites https://github.com/aresonioncq0a7/aresonion – dark market list
1win_rdkr
| #
pariuri sportive moldova https://www.1win5004.ru .
Donaldlaf
| #
darknet markets 2025 https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet sites
Diplomi_izKt
| #
Приобрести диплом о высшем образовании!
Мы предлагаем документы институтов, расположенных на территории всей России.
diplomgorkiy.com/diplom-visshego-obrazovaniya-s-zaneseniem-v-reestr-4/
w69 slot
| #
ติดตั้งแอป w69 slot
แล้วรับสิทธิ์พิเศษเข้าร่วมเดิมพันเกมแข่งรถอย่าง
Gran Turismo และ Forza Horizon ด้วยอัตราต่อรองที่ดีที่สุด พร้อมโปรโมชั่นคืนเงินสำหรับผู้เล่นที่ชื่นชอบความเร็ว
PeterSlump
| #
https://telegra.ph/Top-Non-Gamstop-Casinos-in-the-UK-for-20238-03-27
chimney foundation repair near Me
| #
chimney foundation repair near Me
A Brief History of Elemental Analysis » Artemis Analytical
how can i get generic deltasone without dr prescription
| #
where buy deltasone for sale where can i get deltasone tablets can you get generic deltasone without rx
buy generic deltasone no prescription can i order generic deltasone without insurance how to get generic deltasone no prescription
order deltasone without prescription
can you buy generic deltasone prices can you buy cheap deltasone prices where buy generic deltasone
can i buy generic deltasone without a prescription buy deltasone without dr prescription how to get generic deltasone without dr prescription
Toliknaf
| #
dark market url https://github.com/abacuslink4jjku/abacuslink – dark market onion
Davidsmess
| #
Лизинг — долгосрочная аренда с правом выкупа. Собственником остаётся лизингодатель, что снижает риски для клиента. Платежи часто ниже кредитных, а НДС включается в сумму, что выгодно для бизнеса. Лизинговые платежи учитываются как расходы, уменьшая налог на прибыль: АО Бин-Лизинг
https://www.bizbangboom.com/--staybook/
| #
квартира на сутки Гомель https://www.biztobiz.org/articles/staybook-%D1%81%D0%BE%D0%B2%D1%80%D0%B5%D0%BC%D0%B5%D0%BD%D0%BD%D0%BE%D0%B5-%D1%80%D0%B5%D1%88%D0%B5%D0%BD%D0%B8%D0%B5-%D0%B4%D0%BB%D1%8F-%D0%BF%D0%BE%D0%B8%D1%81%D0%BA%D0%B0-%D0%B6%D0%B8%D0%BB%D1%8C%D1%8F
Sazrxrd
| #
Приобретение документа о высшем образовании через проверенную и надежную компанию дарит множество преимуществ. Данное решение помогает сберечь время и серьезные денежные средства. Тем не менее, только на этом выгоды не ограничиваются, достоинств значительно больше.Мы можем предложить дипломы любых профессий. Дипломы производятся на фирменных бланках. Доступная цена в сравнении с большими затратами на обучение и проживание в другом городе. Покупка диплома о высшем образовании из российского университета является рациональным шагом.
Заказать диплом о высшем образовании: diplomt-v-chelyabinske.ru/bistraya-i-bezopasnaya-pokupka-diploma-s-registratsiej/
dragon money_ehmi
| #
dragon money рефералка https://dragon-money01.com .
Rabyitabe
| #
dark market list https://github.com/darkwebsitesyhshv/darkwebsites – onion dark website
JumopnFuh
| #
На сайте https://t.me/rahvalskiy вы сможете ознакомиться с информацией о компании, которая занимается продвижением сайтов «под ключ». Вы сможете воспользоваться всеми комплексными рекламными услугами. В компании работают лучшие, компетентные сотрудники с огромным стажем, а потому они реализуют проект любой сложности и по лучшей стоимости. Для сотрудников нет неисполнимых задач. Они работают независимо от темы проекта и в строго указанные сроки. Можете не сомневаться в том, что получите результат, на который рассчитывали.
internetCah
| #
домашний интернет тарифы новосибирск
domashij-internet-novosibirsk001.ru
узнать интернет по адресу
DonDonfaita
| #
darknet markets 2025 https://github.com/nexusdarkrtv1u/nexusdark – dark web link
Pingrutty
| #
darknet drug store https://github.com/aresonioncq0a7/aresonion – dark market onion
can i buy cheap sporanox tablets
| #
can i order sporanox pills
PeterSlump
| #
https://telegra.ph/Discover-New-Non-GamStop-Casinos-for-2023-03-27
best crypto casinos online
| #
best crypto casinos online
A Brief History of Elemental Analysis » Artemis Analytical
Donaldlaf
| #
best darknet markets https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darkmarket
Bahaxeldug
| #
На сайте https://pohudetlegko.ru/ представлена исчерпывающая, актуальная и полезная информация о том, как похудеть правильно, без вреда для организма и максимально эффективно. На сайте вы найдете вдохновляющие истории, советы звезд, которые выглядят младше своего возраста. Есть рецепты блюд, которые созданы из полезных, вкусных ингредиентов. Но при этом они помогут скинуть вес и привести себя в форму к летнему сезону. Рецепты простые и быстрые, а потому справиться с приготовлением пищи сможет каждый. Описаны и упражнения, обертывания для похудения.
dragon money_oymi
| #
драгон мани казино зеркало драгон мани казино зеркало .
Toliknaf
| #
dark web sites https://github.com/abacuslink4jjku/abacuslink – darknet marketplace
https://housethings.mystrikingly.com/blog/28ce9ed194a
| #
квартира на сутки Гомель https://www.biztobiz.org/articles/%D0%B8%D0%B4%D0%B5%D0%B0%D0%BB%D1%8C%D0%BD%D1%8B%D0%B9-%D0%B2%D0%B5%D1%87%D0%B5%D1%80-%D0%B2-%D0%B3%D0%BE%D0%BC%D0%B5%D0%BB%D0%B5
PeterSlump
| #
https://telegra.ph/Mobile-Casinos-Not-on-Gamstop-for-Convenient-Play-03-27
RuwufhyKidge
| #
На сайте https://kino-wsem.site/publ/doramy/ представлены дорамы в отличном качестве. Все они о любви, дружбе и вечном. Есть произведения за прошлые годы, которые многие пересматривают с особым удовольствием. Навигация поможет вам лучше сориентироваться в выборе и начать просмотр такого фильма, который вызовет у вас приятные, положительные эмоции. Регулярно на портале появляются новые дорамы, которые погрузят вас в удивительный мир. Вы сможете просматривать фильмы в режиме реального времени и на любом устройстве.
Rabyitabe
| #
dark markets 2025 https://github.com/darkwebsitesyhshv/darkwebsites – darknet markets url
RobertClaws
| #
Найден способ избежать преждевременной смерти
https://x.com/___Nikie__/status/1909141697990115541
mostbet_cuMa
| #
мостбет вход hiend.borda.ru/?1-16-0-00000260-000-0-0 .
Hives treatment options
| #
Hives treatment decision-making Hives treatment courses Hives symptom relief
Hives treatment plans Hives treatment success Hives diagnosis
Hives outlook
Hives emergency treatment Hives support Hives corticosteroid creams
Hives treatment success Hives treatment decision-making Hives treatment updates
mostbet_ipMa
| #
мостбет вход hiend.borda.ru/?1-16-0-00000260-000-0-0 .
Pingrutty
| #
dark markets 2025 https://github.com/aresonioncq0a7/aresonion – dark web drug marketplace
DonDonfaita
| #
darknet drug market https://github.com/nexusdarkrtv1u/nexusdark – darknet markets 2025
mostbet_rqMa
| #
поддержка мостбет http://hiend.borda.ru/?1-16-0-00000260-000-0-0/ .
dragon money_lgmi
| #
как участвовать в колесе драгон мани как участвовать в колесе драгон мани .
Willieavamy
| #
The mobile casino vibe is off—tweak it! aviator
Willieavamy
| #
Online casinos need grip—too loose! sugar rush
1win_tjEl
| #
1вин приложение https://1win6005.ru/ .
Donaldlaf
| #
darkmarket url https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – dark websites
Lazrhqw
| #
Заказать диплом об образовании!
Мы изготавливаем дипломы любой профессии по приятным ценам— damdiplomisa.com/diplom-o-srednem-med-obrazovanii-kupit-3/
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-UK-Free-Spins-Bonuses-Explained-03-27
where can i get generic isoptin pills
| #
cost of cheap isoptin no prescription
mostbet_wvMl
| #
мостбет промокод мостбет промокод .
https://www.bpublic.com/blog/----staybook
| #
квартира на сутки Гомель https://www.bizmakersamerica.org/blog/staybook-%E2%80%93-%D0%B8%D0%B4%D0%B5%D0%B0%D0%BB%D1%8C%D0%BD%D1%8B%D0%B9-%D0%B8%D0%BD%D1%81%D1%82%D1%80%D1%83%D0%BC%D0%B5%D0%BD%D1%82-%D0%B4%D0%BB%D1%8F-%D0%BF%D0%BE%D0%B8%D1%81%D0%BA%D0%B0-%D0%B6%D0%B8%D0%BB%D1%8C%D1%8F
Toliknaf
| #
darknet drug links https://github.com/abacuslink4jjku/abacuslink – darknet markets url
Pingrutty
| #
dark web market links https://github.com/aresonioncq0a7/aresonion – darknet links
Ronalddiz
| #
Старт в сессию начинается с одного клика — и ты уже в космосе. Всё вокруг тихо, интерфейс интуитивный.
Слоты — это гиперпрыжки, и каждый запускается как двигатель. В этом цифровом пространстве http://remont-avto86.ru — главный хаб, где всё рассчитано.
Финансовый запуск — сразу в систему, а выигрыш — как посадка в назначенной точке.
Поддержка реагирует на высоте. Атмосфера — тишина галактики — именно то, что нужно для полного погружения.
Rabyitabe
| #
dark web market list https://github.com/darkwebsitesyhshv/darkwebsites – tor drug market
1win_vfkr
| #
1вин партнерка http://1win6053.ru/ .
remont kardana_dukn
| #
замена крестовины кардана замена крестовины кардана .
FutadlSow
| #
На сайте https://synccheat.ru/deadlock изучите огромный каталог читов, которые помогут игроку получить преимущества от игрового процесса. Так вы сможете легко дойти до нужного уровня, чтобы получить приз, перейти на другую стадию. Этот портал является проверенным, надежным, поэтому на нем можно совершать покупку, не опасаясь за свои средства, конфиденциальность. Совершить покупку вы сможете прямо сейчас и по выгодной стоимости, без переплат. Если появились вопросы, то задайте их службе поддержки, которая ответит на них.
Sazrwde
| #
Студенческая жизнь прекрасна, пока не приходит время писать диплом, как это случилось со мной. Не стоит отчаиваться, ведь существуют компании, которые помогают с написанием и защитой диплома на высокие оценки!
Сначала я искал информацию по теме: диплом без приложения как купить, купить диплом о среднем образовании в магнитогорске, купить диплом о среднем образовании ссср, купить диплом образования в пензе, образование купить диплом оригинальный, а потом наткнулся на kyc-diplom.com/geography/blagoveshchensk.html
Iariorafl
| #
Купить диплом университета по невысокой стоимости возможно, обращаясь к проверенной специализированной компании. Приобрести документ университета вы сможете у нас. diplom-kaluga.ru/kupit-diplom-s-reestrom-bistro-i-nadezhno-22
hosoxscots
| #
Севертур лучшие программы предоставляет. Благодаря туроператору вы побываете в местах, где бережно хранится многовековая история России. Логистика выстроена сотрудниками компании отлично. Гиды приятные в общении и доброжелательны. Экскурсии информативны и познавательны. Ищете из каргополя в сийский монастырь? Xn–80ade0ahrahddrm9hxa.xn--p1ai – сайт, где можете узнать все самое интересное о наших турах. Мы приглашаем вас отправиться, в незабываемое путешествие. Вы останетесь в восторге от организации поездки. Не упустите возможность сделать памятные фотографии!
BryanPealK
| #
WhatsApp вводит новую функцию https://x.com/Ruslansavalv/status/1909195435446595720
VanacrtMaymn
| #
На сайте https://grillhouse-spb.ru/ закажите звонок для того, чтобы проконсультироваться по поводу покупки беседок-гриль, гриль-домиков. Популярная компания «Гриль Хаус» в течение долгого времени производит и собирает беседки, в том числе, для барбекю, а также саун. Все это потребуется для того, чтобы незабываемо провести время на природе и в кругу друзей, близких людей. Все конструкции выполнены из качественных, надежных материалов, которые являются износостойкими. Прямо сейчас изучите каталог доступных для заказа вариантов.
DonDonfaita
| #
darkmarket url https://github.com/nexusdarkrtv1u/nexusdark – best darknet markets
PeterSlump
| #
https://telegra.ph/Best-Non-Gamstop-Casinos-Accepting-PayPal-Payments1-03-27
Jariorrcu
| #
Где заказать диплом специалиста?
Наша компания предлагаетбыстро и выгодно приобрести диплом, который выполнен на оригинальной бумаге и заверен печатями, водяными знаками, подписями. Документ пройдет лубую проверку, даже с применением специально предназначенного оборудования. Достигайте своих целей максимально быстро с нашими дипломами. Приобрести диплом о высшем образовании! rulezrp.listbb.ru/posting.php?mode=post&f=2&sid=45f5eadf64a4f90086a2dad2db89e62f
WomicGut
| #
На сайте https://mvpol.ru/ воспользуйтесь возможностью заказать промышленные полы напрямую от производителя. Компания «МВПОЛ» оказывает и такую важную услугу, как ремонт промышленных полов, производство полимерных наливных полов, бетонных, у которых упрочненный верх. Также получится узнать расценки на такие услуги, чтобы заранее спланировать бюджет. Для того чтобы иметь представление о том, как выглядят работы, изучите объекты. На все услуги предоставляются гарантии. Вся информация дается максимально быстро.
DavidNaing
| #
This online casino’s Tiger Fortune is top-notch. tiger fortune
1win_fnpa
| #
1win ng https://1win13.com.ng/ .
where to buy albuterol pill
| #
buy generic albuterol without insurance can you buy albuterol pills cost of cheap albuterol prices
buy liquid albuterol australia can i purchase albuterol without a prescription buy albuterol inhaler with no prescription
get albuterol prices
buy research liquid albuterol how to buy generic albuterol without rx buying generic albuterol without prescription
best place to buy research chemicals albuterol albuterol breathing treatment for pneumonia cost of albuterol
1win_qkOn
| #
официальный сайт 1win https://www.1win6054.ru .
Willieavamy
| #
I love the anonymity of online casinos—no one judging me! gates of olympus
DavidNaing
| #
Tiger Fortune makes every spin feel like a big moment. fortune tiger 777
Willieavamy
| #
The online ease rolls—stay wise! mines
DavidNaing
| #
This online casino’s Tiger Fortune is a must-play. fortune tiger 777
mostbet_epOn
| #
поддержка мостбет http://mostbet6029.ru .
case noi de inchiriat_slsl
| #
cas? de ?nchiriat ilfov cas? de ?nchiriat ilfov .
Donaldlaf
| #
darknet market links https://github.com/aresmarketdarknetl9khn/aresmarketdarknet – darknet marketplace
gorkinskoe
| #
Горкинское кладбище https://gorkinskoe.ru одно из старейших в Видном. Подробная информация: адрес, как доехать, порядок захоронений, наличие участков, памятники, услуги по уходу.
Willieavamy
| #
I hit a bonus online and won $50—small but sweet! plinko
DavidNaing
| #
This casino’s Tiger Fortune slot never gets old. jogo do tigrinho demo
Jariorkwb
| #
Где купить диплом по необходимой специальности?
Мы предлагаем быстро купить диплом, который выполняется на оригинальной бумаге и заверен мокрыми печатями, штампами, подписями. Документ пройдет лубую проверку, даже при помощи специфических приборов. Решите свои задачи быстро и просто с нашей компанией.
Купить диплом любого университета diplomt-v-samare.ru/kupit-diplom-novogo-obraztsa-16/
Pingrutty
| #
darknet market links https://github.com/aresonioncq0a7/aresonion – dark market link
Toliknaf
| #
darknet market links https://github.com/abacuslink4jjku/abacuslink – dark websites
http://www.cross.tv/blog/234471
| #
квартира на сутки Гомель https://sites.google.com/view/milassweetynews/articles/real-estate/%D0%BE%D0%BF%D1%8B%D1%82-%D0%B0%D1%80%D0%B5%D0%BD%D0%B4%D1%8B-%D1%87%D0%B5%D1%80%D0%B5%D0%B7-myrent
Sazrcyw
| #
Купить диплом об образовании!
Купить диплом института по невысокой цене возможно, обращаясь к проверенной специализированной фирме. Приобрести диплом о высшем образовании: diplomp-irkutsk.ru/ofitsialnij-diplom-vuza-vash-put-k-uspexu
Cazrbsd
| #
Купить диплом любого ВУЗа!
Мы готовы предложить документы университетов, расположенных в любом регионе Российской Федерации. Документы печатаются на бумаге высшего качества: cutenite.com/@royalortiz0249
internetCah
| #
дешевый интернет новосибирск
domashij-internet-novosibirsk002.ru
провайдеры интернета в новосибирске по адресу проверить
WilliamDix
| #
Мы изготавливаем дипломы любой профессии по выгодным ценам. Дипломы производят на настоящих бланках Приобрести диплом о высшем образовании diplomservis.com
Lazrphd
| #
Где купить диплом специалиста?
Купить диплом института по выгодной стоимости возможно, обратившись к надежной специализированной компании.: okdiplom.com
DwayneDub
| #
вива блю резорт энд дайвинг хургада
CarozrdAsymn
| #
На сайте https://bitovayatechnika.blogspot.com/2025/03/blog-post.html изучите полезную, актуальную информацию, которая касается сплит-систем и того, как правильно их выбрать домой либо в офис. Эти функциональные и умные устройства нацелены на то, чтобы поддержать приятную температуру в помещении. Но для того, чтобы воспользоваться всеми функциями, необходимо правильно выбрать устройство. К важным преимуществам такой техники относят то, что она является энергоэффективной, работает практически бесшумно.
Rabyitabe
| #
dark market link https://github.com/darkwebsitesyhshv/darkwebsites – tor drug market
buying reglan online
| #
cost of reglan for sale
Sazrjlk
| #
Мы можем предложить дипломы любой профессии по приятным ценам. Преимущества заказа документов в нашем сервисе
Вы заказываете диплом через надежную компанию. Такое решение сэкономит не только средства, но и бесценное время.
Преимуществ куда больше:
• Документы изготавливаются на фирменных бланках с мокрыми печатями и подписями;
• Предлагаем дипломы любого ВУЗа России;
• Цена в разы ниже чем довелось бы заплатить на очном обучении в ВУЗе;
• Быстрая доставка в любые регионы РФ.
Купить диплом о высшем образовании– http://lucylink.com/read-blog/3414_kupit-diplom-prepodavatelya.html/ – lucylink.com/read-blog/3414_kupit-diplom-prepodavatelya.html
Diplomi_vzka
| #
купить диплом в уссурийске diplomys-vsem.ru .
PeterSlump
| #
https://telegra.ph/Exploring-Casino-Options-Without-Gamstop-in-the-UK1-03-27
Waltertax
| #
https://q2ca.ru/
1win_hcOn
| #
сайт 1win официальный сайт вход https://1win6054.ru .
DonDonfaita
| #
darknet markets links https://github.com/nexusdarkrtv1u/nexusdark – dark web market list
1win_erpa
| #
sports betting 1win http://1win13.com.ng/ .
1win_wdEl
| #
1win на телефон https://1win6005.ru .
DavidNaing
| #
Tiger Fortune’s graphics are wild and vivid. jogo do tigrinho demo
VincentOntot
| #
¡Hola usuarios de casino!
Gira y gana con 10 euros gratis en tragamonedas sin depГіsito. 10eurosgratissindepositocasino.xyz Solo tienes que crear tu cuenta y aprovechar este bono exclusivo. ВЎLa suerte estГЎ de tu lado!
Juega con euros gratis sin depГіsito en los mejores casinos online. Solo necesitas registrarte para recibir tu bono. Disfruta de juegos emocionantes y gana sin poner dinero de tu bolsillo.
Toda la información en el enlace – п»їhttps://10eurosgratissindepositocasino.xyz/
¡Que tengas buenos ganancias!
VincentOntot
| #
¡Hola cazadores de adrenalina!
Juega al bingo online con 10 euros gratis sin depГіsito. 10 euros por registrarte RegГstrate y empieza a disfrutar sin pagar nada. Ideal para los amantes del bingo que quieren ganar sin arriesgar.
Juega en un casino espaГ±ol con 10 euros gratis sin depГіsito. Todo lo que necesitas es registrarte. Empieza hoy mismo y prueba tu suerte gratis.
Toda la información en el enlace – п»їhttps://10eurosgratissindepositocasino.xyz/
¡Que tengas buenos jackpots!
VincentOntot
| #
¡Hola usuarios de casino!
Con 10 euros gratis puedes jugar en el casino sin necesidad de depositar. bingo 10 euros gratis sin depГіsito Ideal para probar sin compromisos. RegГstrate y juega como un profesional.
Descubre los bonos de 10 euros gratis sin depГіsito. Diferentes casinos te los ofrecen para que empieces a jugar sin gastar nada. ВЎRegГstrate y gana!
Toda la información en el enlace – п»їhttps://10eurosgratissindepositocasino.xyz/
¡Que tengas buenos juegos!
Gastrofibroskopia
| #
Gastrofibroskopia
blog topic
Pingrutty
| #
darknet drugs https://github.com/aresonioncq0a7/aresonion – darknet sites
Cheryl magistertekniksipil
| #
What a very great blog your honest majesty! Nice to see your article in this era!
Cheryl conservativehq
| #
Thank for your blog teh! very inspiring aa! keep it up so i can catch up kang!
Iariorpvf
| #
Купить диплом ВУЗа по выгодной цене возможно, обратившись к проверенной специализированной фирме. Приобрести документ о получении высшего образования можно у нас в столице. diplomt-v-chelyabinske.ru/kupit-diplom-s-zaneseniem-v-reestr-bistro-i-nadezhno-80
DavidNaing
| #
I can play Tiger Fortune all day and not get bored. fortune tiger demo
buy generic doxycycline for sale
| #
buy cheap doxycycline pill buy doxycycline tablets doxycycline hyclate chlamydia treatment
buy generic doxycycline pill how to buy cheap doxycycline without a prescription cost of generic doxycycline for sale
cost cheap doxycycline online
cost cheap doxycycline price how to buy generic doxycycline without a prescription can i order cheap doxycycline price
walgreens doxycycline price buying cheap doxycycline without insurance buy doxycycline from canada
mostbet_uvOn
| #
motsbet https://www.mostbet6029.ru .
dragon money_lson
| #
как вывести с драгон мани http://www.dragon-money10.com .
remont kardana_gekn
| #
замена подвесного подшипника карданного вала замена подвесного подшипника карданного вала .
1win_sypa
| #
1вин официальный сайт 1вин официальный сайт .
1win_teOn
| #
1вин вход https://www.1win6054.ru .
結婚 對戒 推薦
| #
結婚 對戒 推薦
A Brief History of Elemental Analysis » Artemis Analytical
1win_xgpa
| #
1win online games 1win online games .
PeterSlump
| #
https://telegra.ph/No-Deposit-Non-Gamstop-Casinos-for-Players-2023-03-27
dragon money_vwsi
| #
драгон мани casino драгон мани casino .
YuwiseyEnuts
| #
На сайте https://xn—-7sbjhqn0bhjc0lk.xn--p1ai/ получите юридическую консультацию по различным вопросам. Компания работает как с физическими, так и частными лицами. На все услуги установлены привлекательные расценки, чтобы воспользоваться ими смог каждый. Предприятие работает в этой сфере более 11 лет. В команде трудятся 11 специалистов, которые справятся с задачей независимо от сложности. К вашим услугам досудебный юрист, досудебное урегулирование, кредитный, семейный юрист, решение страховых споров.
zgardamea01
| #
zgardamea01
A Brief History of Elemental Analysis » Artemis Analytical
Michaelvella
| #
Всё о Казахстане https://tr-kazakhstan.kz история, культура, города, традиции, природа и достопримечательности. Полезная информация для туристов, жителей и тех, кто хочет узнать страну ближе.
case noi de inchiriat_hgsl
| #
case ?nchiriere cu spa?ii verzi case ?nchiriere cu spa?ii verzi .
DavidNaing
| #
I love the jungle vibe of Tiger Fortune – so immersive! fortune tiger demo gratis
mostbet_ekMl
| #
мрстбет http://mostbet6030.ru/ .
cost of generic pamelor pill
| #
how can i get generic pamelor without dr prescription
internetCah
| #
интернет провайдер новосибирск
domashij-internet-novosibirsk003.ru
домашний интернет подключить новосибирск
dragon money_sxon
| #
драгон драгон .
Frankphils
| #
https://vc.ru/
DavidNaing
| #
This casino’s Tiger Fortune slot never gets old. fortune tiger 777
Iariorfkh
| #
Купить диплом ВУЗа по выгодной стоимости вы можете, обращаясь к проверенной специализированной компании. Купить документ о получении высшего образования вы имеете возможность у нас в столице. damdiplomisa.com/diplom-s-zaneseniem-v-reestr-po-dostupnoj-tsene-6
PatrickCop
| #
дайвинг хургада
mostbet_ursl
| #
мостбет кыргызстан https://severussnape.borda.ru/?1-4-0-00000505-000-0-0-1743260265 .
Willieavamy
| #
I don’t get the hate online—it’s cool! aviator
EdmondVab
| #
купить взломостойкие сейфы
PeterSlump
| #
https://telegra.ph/Reliable-Non-Gamstop-Casinos-for-Secure-Online-Gaming-03-27
1win_urpa
| #
1 win https://1win6006.ru .
Robertkatry
| #
Купить права
mostbet_ftsl
| #
mostbet скачать https://www.severussnape.borda.ru/?1-4-0-00000505-000-0-0-1743260265 .
DavidNaing
| #
Tiger Fortune’s bonus features are top-notch. tigrinho demo
dragon money_rwsi
| #
драгон мани тактики https://dragon-money11.com .
DavidNaing
| #
This online casino nailed Tiger Fortune’s theme. tigrinho demo
Lazrekc
| #
Заказать диплом любого института!
Мы можем предложить дипломы любой профессии по приятным тарифам— diplom-top.ru/kupit-diplom-s-vneseniem-v-bazu-vsego-za-odin-den/
Willieavamy
| #
Online slots pull me—those vibes! mines
mostbet_fvsl
| #
mostbet kg скачать на андроид https://severussnape.borda.ru/?1-4-0-00000505-000-0-0-1743260265/ .
mostbet_yvOn
| #
mostbet chrono http://mostbet6029.ru .
best online slots
| #
best online slots
A Brief History of Elemental Analysis » Artemis Analytical
dragon money_azon
| #
dragon money официальный dragon money официальный .
Robertfremi
| #
пистолетные сейфы москва
Buvukneers
| #
На сайте https://rodinasportsclub.com/ вы найдете информацию, которая касается спортивно-развлекательного клуба «Родина». Каждый желающий получает возможность записаться на тренировку по боксу, которая подходит как новичкам, так и более опытным спортсменам. Вас заинтересуют тренировки по рукопашному бою. Перед вами расписание предстоящих матчей на апрель. Ознакомьтесь с ними сейчас, чтобы построить планы. Также имеются и отчеты по прошедшим соревнованиям. Опубликованы разные новости из жизни клуба.
DavidNaing
| #
Tiger Fortune’s theme is so adventurous. fortune tiger 777
1win_dkpa
| #
ванвин 1win6006.ru .
dragon money_wxsi
| #
dragonmoney казино dragonmoney казино .
DavidNaing
| #
Tiger Fortune’s jungle vibe is unbeatable. tigrinho demo
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние возможности.
Наши товары:
Керамическая мишень Mos2, плоская
EdmondVab
| #
купить сейф взломостойкий в москве
ruma
| #
Kvalitni nabytek v Praze http://www.ruma.cz stylove reseni pro domacnost i kancelar. Satni skrine, sedaci soupravy, kuchyne, postele od proverenych vyrobcu. Rozvoz po meste a montaz na klic.
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наша продукция:
Медный стандартный двухраструбный отвод 90 градусов под пайку 80х68х1.8 мм 35.5х38.5 мм твердая пайка М3РТ ГОСТ 32590-2013 Покупайте медные двухраструбные отводы 90 градусов под пайку на Редметсплав.рф. Надежные и долговечные отводы из высококачественной меди для систем водоснабжения, отопления и кондиционирования воздуха. Гарантированно надежные соединения, простой монтаж и оптимальное распределение потока.
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы и предоставлять решения под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Рзделия РёР· кобальта Stellite 190 PM Рзделия РёР· кобальта Stellite 190 PM представляют СЃРѕР±РѕР№ высококачественные материалы, специально разработанные для работы РІ экстремальных условиях. РћРЅРё обладают превосходной износостойкостью Рё РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ устойчивостью, что делает РёС… идеальными для использования РІ различных отраслях, включая аэрокосмическую Рё машиностроение. Рти изделия обеспечивают долговечность Рё надежность, что важно для критически важных приложений. Если РІС‹ хотите улучшить производительность вашего оборудования, рекомендуем купить Рзделия РёР· кобальта Stellite 190 PM. РћРЅРё станут вашим надежным решением для сложных задач РІ производстве.
Dohuydlek
| #
На сайте https://ptne.ru/ получите бесплатную консультацию по вопросам продажи недвижимости. Вы сможете воспользоваться такой услугой, как оперативный выкуп различной недвижимости в Санкт-Петербурге. При этом получить аванс вы сможете в день обращения. Агентство оказывает такие услуги, как: выкуп квартир, долей, займ до продажи. Выкуп происходит за свои средства, сделка является прозрачной. Адвокаты оказывают полное юридическое сопровождение, консультационную помощь. Если документов не хватает, то их соберут максимально быстро.
Oscardrulp
| #
гостиничные сейфы купить
get generic nemasole
| #
where buy generic nemasole without dr prescription buy cheap nemasole no prescription where buy nemasole for sale
can i get nemasole without insurance cost cheap nemasole prices how can i get nemasole without prescription
cost of nemasole tablets
can you get cheap nemasole without prescription cost of generic nemasole without a prescription how to get generic nemasole without insurance
where can i buy nemasole pills can you buy nemasole pills how can i get cheap nemasole pills
saxo
| #
saxo
A Brief History of Elemental Analysis » Artemis Analytical
Sazrtqv
| #
купить диплом о высшем образовании в уфе
Лев
| #
Hire a hitman order a murder
Sazrdig
| #
Заказать диплом о высшем образовании !
Приобретение диплома ВУЗа РФ у нас – надежный процесс, ведь документ заносится в реестр. Заказать диплом института diplomt-v-chelyabinske.ru/kupite-diplom-v-reestre-bistro-i-bezopasno
Sazrubr
| #
Мы можем предложить дипломы любых профессий по приятным тарифам. Стоимость будет зависеть от выбранной специальности, года выпуска и университета. Всегда стараемся поддерживать для клиентов адекватную ценовую политику. Важно, чтобы документы были доступными для подавляющей массы граждан. купить диплом техникума образец
Trefgtp
| #
Купить документ института можно в нашей компании в Москве. diplomd-magazinp.ru/kupit-diplom-voronezh-4
Cazrwht
| #
Здравствуйте!
Без наличия диплома достаточно сложно было продвигаться вверх по карьерной лестнице. В наше время этот важный документ не дает абсолютно никаких гарантий, что получится найти привлекательную работу. Более важны навыки специалиста, а также его опыт работы. Именно по этой причине решение о покупке диплома можно считать мудрым и целесообразным. Быстро и просто приобрести диплом университета povzroslomu.kabb.ru/viewtopic.php?f=3&t=767
https://dokuwiki.stream/wiki/Orbis_92j
| #
Elon Casino’s games are a total win in my book.
https://nerdgaming.science/wiki/User:AlissaHoang1
Sergiowinia
| #
кайтсерфинг в египте Египет – одно из лучших мест в мире для кайтсерфинга, предлагающее отличные условия для катания круглый год.
https://securityholes.science/wiki/Orbis_53S
| #
The animations at Elon Casino are pure eye candy.
https://empty3.one/wikilibre/index.php/Orbis_54R
https://utahsyardsale.com/author/hubertrembe/
| #
Elon Casino is my lucky charm every time.
https://www.sitiosecuador.com/author/selenaldx07/
DavidNaing
| #
Tiger Fortune’s spins are wild and unpredictable. tigrinho demo
JumopnFuh
| #
На сайте https://t.me/rahvalskiy вы сможете ознакомиться с информацией о компании, которая занимается продвижением сайтов «под ключ». Вы сможете воспользоваться всеми комплексными рекламными услугами. В компании работают лучшие, компетентные сотрудники с огромным стажем, а потому они реализуют проект любой сложности и по лучшей стоимости. Для сотрудников нет неисполнимых задач. Они работают независимо от темы проекта и в строго указанные сроки. Можете не сомневаться в том, что получите результат, на который рассчитывали.
DavidNaing
| #
Tiger Fortune has some seriously cool visuals. tigrinho demo
https://humanlove.stream/wiki/Orbis_59C
| #
The soundtrack at Elon Casino is so cool – adds to the experience!
https://trade-britanica.trade/wiki/User:CathrynHindwood
cheap vasotec online
| #
cost of generic vasotec without a prescription
sex vn học sinh
| #
sex vn học sinh
A Brief History of Elemental Analysis » Artemis Analytical
DavidNaing
| #
Tiger Fortune’s gameplay is always thrilling. fortune tiger demo gratis
Basketball# puns[Osybydznogynucyw,2,5]
| #
Hi if you’re laughing, you’re in!
Your daily dose of laughter: basketball.puns that bounce right into your DMs. basketballpuns Save, share, repeat.
Looking for basketball dad jokes? These are the MVPs of lame-but-lovable humor. No court required, just a love for laughs.
Toda la información en el enlace – п»їhttps://basketballpuns.com/
Here’s to plenty of chuckles!
Basketball# puns[Osybydznogynucyw,2,5]
| #
Hi fans of humor!
Basketball love puns that are sweeter than a buzzer-beater kiss. basketballpuns.com Fall in love, pun by pun.
Basketball.puns so smooth, even the refs will crack a smile. Serve up the sass with every word.
Toda la información en el enlace – п»їhttps://basketballpuns.com/
Here’s to plenty of hilarious moments!
Basketball# puns[Osybydznogynucyw,2,5]
| #
Hi pun appreciators!
Basketball love puns that are sweeter than a buzzer-beater kiss. basketballpuns.com Fall in love, pun by pun.
Pump up your punchlines with basketball.puns that bounce with brilliance. Your funny bone is about to get a workout.
Toda la información en el enlace – п»їhttps://basketballpuns.com/
Here’s to plenty of laugh-out-louds!
Basketball# puns[Osybydznogynucyw,2,5]
| #
Hi fans of humor!
Dunk your mood into comedy mode. basketball puns
Love basketball? Love puns? These basketball love puns are the ultimate crossover event.
Toda la información en el enlace – п»їhttps://basketballpuns.com/
Here’s to plenty of chuckles!
Oscardrulp
| #
сейфы для гостиниц и отелей
internetCah
| #
провайдеры интернета санкт-петербург
domashij-internet-spb001.ru
провайдеры по адресу
Sazrolx
| #
Всегда считал, что покупка диплома о высшем образовании — это миф и невозможно. Но, к счастью, оказался неправ. Сначала искал информацию по теме: купить свидетельство об усыновлении, купить свидетельство о усыновлении, купить аттестат доцента, номер документа об образовании, можно ли купить аттестат за 9 класс, а затем переключился на дипломы вузов. Подробности здесь: diplomybox.com/kupit-diplom-magistra-v-chite
JoshuaGoamp
| #
Офіційний сайт 1win http://visitkyiv.com.ua спортивні ставки, онлайн-казино, покер, live-ігри, швидкі висновки. Бонуси новим гравцям, мобільний додаток, цілодобова підтримка.
WilliamDix
| #
Мы готовы предложить дипломы психологов, юристов, экономистов и любых других профессий по доступным ценам. Дипломы производят на подлинных бланках Быстро и просто приобрести диплом о высшем образовании diplomd-magazinp.ru
Waynepooda
| #
This gaming hub is incredible with Elon Casino. elon casino
Michaelsoump
| #
ссылка на сайт казино р7 сегодня
Lazrjnr
| #
Где заказать диплом по необходимой специальности?
Купить диплом ВУЗа по невысокой цене возможно, обращаясь к проверенной специализированной компании.: diplommy.ru
JustinRen
| #
встраиваемые сейфы купить
JustinRen
| #
сейф встраиваемый в пол купить
diltiazem for chest pain
| #
diltiazem side effects took too many diltiazem 180 mg diltiazem price canada
can diltiazem cause foot swelling diltiazem 30 mg tablet when should you hold diltiazem
diltiazem foods to avoid
can diltiazem cause leg swelling does diltiazem lower heart rate diltiazem 30 mg tab lemmon
diltiazem cream 2% cost diltiazem side effects forum diltiazem vs amiodarone afib
1win_lukr
| #
1win.pro 1win6053.ru .
Bikuweloract
| #
На сайте https://apostolidi.ru/ вы найдете всю полезную информацию, касающуюся торгов на бирже, а также финансов. Здесь также вы найдете и прогнозы по валютным парам от экспертов, которые работают на совесть. Обязательно публикуются свежие, интересные и информативные записи, которые будут полезны всем без исключения. Также представлен и курс доллара, о котором нужно знать инвесторам. На этом портале вы получаете возможность пообщаться с единомышленниками, задать им вопрос, узнать много нового, интересного.
DavidNaing
| #
Tiger Fortune’s payouts are worth the hype. tigrinho demo
mua bán ma túy
| #
mua bán ma túy
A Brief History of Elemental Analysis » Artemis Analytical
Waynepooda
| #
Elon Casino’s futuristic theme is totally unique.
elon casino
Bahaxeldug
| #
На сайте https://pohudetlegko.ru/ представлена исчерпывающая, актуальная и полезная информация о том, как похудеть правильно, без вреда для организма и максимально эффективно. На сайте вы найдете вдохновляющие истории, советы звезд, которые выглядят младше своего возраста. Есть рецепты блюд, которые созданы из полезных, вкусных ингредиентов. Но при этом они помогут скинуть вес и привести себя в форму к летнему сезону. Рецепты простые и быстрые, а потому справиться с приготовлением пищи сможет каждый. Описаны и упражнения, обертывания для похудения.
Sazrhed
| #
Мы предлагаем дипломы любых профессий по разумным ценам. Стоимость может зависеть от выбранной специальности, года получения и образовательного учреждения. Всегда стараемся поддерживать для покупателей адекватную ценовую политику. Важно, чтобы дипломы были доступны для большого количества наших граждан. купить диплом мгоу
Trefmvq
| #
Приобрести документ о получении высшего образования вы сможете в нашей компании. diplomnie.com/kupit-prilozhenie-k-diplomu-2
https://массаж-в-анапе.рф/
| #
https://массаж-в-анапе.рф/
Dnrtzwz
| #
Где приобрести диплом специалиста?
Мы можем предложить дипломы любых профессий по приятным ценам. Мы можем предложить документы ВУЗов, которые находятся на территории всей Российской Федерации. Можно купить диплом от любого заведения, за любой год, включая сюда документы старого образца. Документы выпускаются на “правильной” бумаге высшего качества. Это позволяет делать государственные дипломы, которые невозможно отличить от оригинала. Документы заверяются необходимыми печатями и подписями. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Для нас очень важно, чтобы документы были доступны для большого количества граждан. institute-diplom.ru/kupit-diplom-v-kaluge-8
how to get generic brand levitra
| #
can you buy generic brand levitra
dragon money_bqmi
| #
казино dragon казино dragon .
Sazrlbz
| #
Мы готовы предложить дипломы любых профессий по приятным ценам. Основные преимущества заказа документов в нашей компании
Вы заказываете документ через надежную фирму. Это решение позволит сэкономить не только много средств, но и бесценное время.
На этом плюсы не заканчиваются, их гораздо больше:
• Дипломы печатаются на оригинальных бланках с печатями и подписями;
• Дипломы всех высших учебных заведений РФ;
• Стоимость значительно меньше чем пришлось бы заплатить на очном обучении в ВУЗе;
• Доставка как по столице, так и в другие регионы России.
Купить диплом ВУЗа– http://forumkoldovstva.listbb.ru/viewtopic.php?f=37&t=17208/ – forumkoldovstva.listbb.ru/viewtopic.php?f=37&t=17208
Waynepooda
| #
Elon Casino’s free spins are an absolute treat.
elon casino
bafapAmarM
| #
Проект Streetwear российским брендам уличной одежды посвящен. Постоянно исследуем рынок и стараемся предоставить вам актуальную информацию. Рассказываем, сколько стоит размещение в каталоге. Любой бренд модерацию на сайте перед размещением проходит. Ищете мужские русские бренды? Rustreetwear.ru – здесь можно узнать больше о проекте. Поддержать российских производителей и дизайнеров – это наша миссия. Если знаете бренд, которого нет в нашем каталоге, или желаете предложить сотрудничество, свяжитесь с нами через страницу контактов. Мы открыты для новых идей!
Diplomi_nhka
| #
аттестат 2000 года аттестат 2000 года .
find more
| #
find more
A Brief History of Elemental Analysis » Artemis Analytical
VanacrtMaymn
| #
На сайте https://grillhouse-spb.ru/ закажите звонок для того, чтобы проконсультироваться по поводу покупки беседок-гриль, гриль-домиков. Популярная компания «Гриль Хаус» в течение долгого времени производит и собирает беседки, в том числе, для барбекю, а также саун. Все это потребуется для того, чтобы незабываемо провести время на природе и в кругу друзей, близких людей. Все конструкции выполнены из качественных, надежных материалов, которые являются износостойкими. Прямо сейчас изучите каталог доступных для заказа вариантов.
internetCah
| #
провайдеры интернета в санкт-петербурге по адресу проверить
domashij-internet-spb002.ru
подключение интернета по адресу
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Медная мишень для распыления
DavidNaing
| #
This casino’s Tiger Fortune game is a standout. fortune tiger
DavidNaing
| #
This online casino’s Tiger Fortune is fantastic. fortune tiger demo gratis
Sazrsrl
| #
диплом мед образования купить
DavidNaing
| #
This online casino’s Tiger Fortune is top-tier. tiger fortune
buy motilium without rx
| #
how can i get cheap motilium no prescription where to buy generic motilium for sale where can i get cheap motilium without insurance
can i purchase motilium no prescription can i buy motilium pill cost motilium for sale
can i get motilium online
can i order cheap motilium how to buy cheap motilium for sale where buy motilium without prescription
can i get cheap motilium without dr prescription get cheap motilium online where can i get cheap motilium
Brianpew
| #
Доброго!
Как оформить развод через ЗАГС — это процесс, который можно осуществить без судебного разбирательства при наличии согласия обеих сторон. Мы поможем вам правильно подготовить все документы, включая заявление и свидетельства, необходимые для оформления развода. Юридическая помощь при разводе через ЗАГС гарантирует, что вы сможете пройти процесс быстро и без лишних шагов. Наши специалисты помогут вам с оформлением всех документов и объяснят, как избежать проблем. Обратитесь за помощью, чтобы оформить развод через ЗАГС без лишних трудностей.
Больше информации на сайте – https://capitallawdo.ru/
представительство в суде, все вопросы подряд пдд, госпошлина за подачу иска о лишении родительских прав
трудовой договор почасовая оплата труда образец, кем является потребитель по законодательству о защите прав потребителей, гражданское право консультация
Удачи!
Willieavamy
| #
The online flow is wild—stay sharp! craps game
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
Наша продукция:
Медный тройник РїРѕРґ пайку СЃ направленным отводом 76.1С…65С…1.6 РјРј 33.5С…36.5 РјРј мягкая пайка Рњ1 ГОСТ 32590-2013 Купить качественные медные тройники РїРѕРґ пайку для надежных Рё долговечных соединений РІ трубопроводных системах. Разнообразие размеров Рё конфигураций, высокая теплопроводность Рё устойчивость Рє РєРѕСЂСЂРѕР·РёРё. Простая установка Рё надежность соединения. Рдеальное решение для различных проектов. Редметсплав.СЂС„
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их происхождение. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы по мере того как находить ответы под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Полоса магниевая SISMg4640-10 – SS 141640 Порошок магниевый SISMg4640-07 – SS 144640 представляет СЃРѕР±РѕР№ высококачественный материал, используемый РІ различных отраслях, включая металлургическую Рё химическую. РћРЅ обеспечивает отличную проводимость Рё РЅРёР·РєСѓСЋ плотность, что делает его идеальным выбором для легких конструкций Рё сплавов.Ртот РїСЂРѕРґСѓРєС‚ отличается высокой чистотой Рё стабильностью, что гарантирует надежность РІ производственных процессах. Для тех, кто ищет эффективное решение для СЃРІРѕРёС… нужд, купить Порошок магниевый SISMg4640-07 – SS 144640 можно РїСЂСЏРјРѕ сейчас. РќРµ упустите возможность улучшить качество своей продукции!
Sazrnkh
| #
Мы изготавливаем дипломы любой профессии по разумным тарифам. Стоимость зависит от выбранной специальности, года выпуска и образовательного учреждения. Стараемся поддерживать для покупателей адекватную политику цен. Для нас важно, чтобы документы были доступными для большинства наших граждан. купить диплом с занесением в реестр в нижнем тагиле
Waynepooda
| #
The excitement never stops at Elon Casino – so thrilling!
elon casino
Trefswi
| #
Купить документ о получении высшего образования вы можете в нашем сервисе. diplom-zentr.com/kupit-diplom-tver
salexy
| #
Доска бесплатных объявлений https://salexy.kz Казахстана: авто, недвижимость, техника, услуги, работа и многое другое. Тысячи свежих объявлений каждый день — легко найти и разместить!
Waynepooda
| #
The graphics at Elon Casino are absolutely mind-blowing. elon casino
1win_yakr
| #
1win live http://www.1win6053.ru .
Robertarosy
| #
Привет всем!
Защита прав в суде — это важная юридическая услуга для тех, кто оказался в сложной ситуации и нуждается в защите своих интересов в суде. Мы представим ваши интересы в суде, подготовим все необходимые документы и будем сопровождать вас на протяжении всего судебного процесса. Юрист по защите прав в суде обеспечит, что ваше дело будет рассмотрено справедливо, а все факты учтены. Мы предлагаем профессиональную помощь по различным категориям дел — от гражданских до трудовых и административных. Обратитесь к нам для качественной юридической защиты.
Больше информации на сайте – https://businessfaq.ru
помощь при нарушении ПДД, какие документы нужны для оформления загранпаспорта детям, понятие и содержание брачного договора
банк зенит военная ипотека, закон о защите прав потребителей потребитель, автоюрист по страховке
Удачи!
Waynepooda
| #
Elon Casino’s gameplay is fierce and fast.
elon casino
how to get generic cymbalta tablets
| #
buy generic cymbalta without a prescription
DavidNaing
| #
The free spins in Tiger Fortune are awesome! tigrinho demo
Waynepooda
| #
Elon Casino is the star of online gaming!
elon casino
DonayrIngek
| #
На сайте https://vezuviy.shop/ вы сможете приобрести чугунные, стальные банные печи, нержавеющие, а также в облицовке, печи-камины, отопительные, каминные топки, дымоходы, порталы и многое другое. Вся продукция от производителя, а потому наценки очень маленькие. Такая техника считается идеальной не только с целью обогрева помещения, но и оформления пространства. А самое главное, что она отличается долгим сроком службы, надежностью из-за того, что в работе использовались высокотехнологичные, проверенные материалы.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние возможности.
Наши товары:
Мишень для распыления сплава AlCu
DavidNaing
| #
This online casino’s Tiger Fortune is top-notch. fortune tiger
1win_fnOn
| #
1win на телефон https://www.1win6054.ru .
Waynepooda
| #
Elon Casino’s free spins are an absolute treat.
elon casino
dragon money_xvmi
| #
drgn1n casino drgn1n casino .
1win_rcpi
| #
casino 1 win http://1win1001.top/ .
Waynepooda
| #
Elon Casino’s interface is smooth and super intuitive. elon casino
1win_ycpa
| #
1win login nigeria http://www.1win13.com.ng .
mostbet_llMa
| #
mostbet kg скачать на андроид http://www.mostbet6031.ru .
Willieavamy
| #
I won $200 on a slot online—still hyped! plinko
get cheap deltasone for sale
| #
can i buy cheap deltasone without rx order generic deltasone without rx how to get deltasone without prescription
can you buy generic deltasone buy cheap deltasone without rx where to buy deltasone tablets
buying deltasone pills
can i purchase generic deltasone online where buy generic deltasone can you get cheap deltasone without prescription
can i order deltasone no prescription cost of generic deltasone no prescription can i buy deltasone without rx
nvblfurn
| #
https://studio-servis.ru/the_articles/vmeste-veseley-igry-dlya-dvoih-na-sayte-igroutka-su.html игра примеры для 1 класса по математике
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наша продукция:
Титановая труба 68х3х4000 мм ОТ4-1 ГОСТ 22897-86 Откройте мир прочных и надежных титановых труб с компанией Редметсплав. Наши высококачественные титановые трубы сочетают в себе устойчивость к коррозии, износостойкость и легкий вес, делая их незаменимым материалом для применения в различных сферах промышленности. Широкий выбор различных диаметров и толщин, а также индивидуальные решения для вашего производства. Гарантированное качество и надежность поставок.
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их качество. Опытная поддержка – наш стандарт – мы на связи, чтобы ответить на ваши вопросы и адаптировать решения под специфику вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Рзделия РёР· РЅРёРѕР±РёСЏ БТЦ-Р’Р” – ГОСТ 10994-74 Рзделия РёР· РЅРёРѕР±РёСЏ БТЦ-Р’Р” – ГОСТ 10994-74 представляют СЃРѕР±РѕР№ высококачественные компоненты, произведенные РІ соответствии СЃ установленными стандартами. Рти изделия обладают отличными характеристиками, такими как высокая стойкость Рє РєРѕСЂСЂРѕР·РёРё Рё температурным воздействиям, что делает РёС… идеальными для применения РІ различных отраслях. Если вам необходимо купить Рзделия РёР· РЅРёРѕР±РёСЏ БТЦ-Р’Р” – ГОСТ 10994-74, РІС‹ можете быть уверены РІ РёС… надежности Рё долговечности. Наши изделия соответствуют всем техническим требованиям, что гарантирует РёС… высокий уровень безопасности Рё эффективности. Заказывайте лучшие решения для вашего бизнеса уже сегодня!
Waynepooda
| #
Elon Casino is the star of online gaming!
elon casino
internetCah
| #
интернет по адресу дома
domashij-internet-spb003.ru
провайдеры домашнего интернета санкт-петербург
sex
| #
sex
A Brief History of Elemental Analysis » Artemis Analytical
Michaelmub
| #
Доброго!
Составление исков необходимо для подачи жалоб в суд, и юрист поможет вам правильно оформить исковое заявление для получения нужного решения. Представительство в суде помогает вам отстоять свои права в суде, и юрист будет вашим представителем в процессе. Услуги юриста по банкротству помогут вам разобраться в процедуре банкротства, как для физических, так и для юридических лиц. Оформление брака с иностранцем требует соблюдения всех миграционных норм, и юрист поможет вам оформить все документы для регистрации брака. Сопровождение сделок по недвижимости включает помощь в вопросах покупки, продажи и аренды недвижимости, чтобы избежать юридических проблем.
Больше информации на сайте – https://consultantdo.ru/
юридическая помощь при увольнении, закон о защите прав потребителей ст 29 п 1, в противном случае я буду вынужден обратиться в суд за защитой своих прав
как уволить сотрудника по окончании срочного трудового договора правильно, лицензирование фармацевтической деятельности аптечных организаций осуществляет, оформление дарственной на авто
Удачи!
Waynepooda
| #
This gaming hub is a blast with Elon Casino. elon casino
Waynepooda
| #
The animations at Elon Casino are pure eye candy. elon casino
DavidNaing
| #
Tiger Fortune is the best game here, hands down! tigrinho demo
Robertfremi
| #
сейф для оружия купить в москве
Waynepooda
| #
Elon Casino stands out as a top-tier gaming hub. elon casino
Sazrdfk
| #
Всегда думал что купить диплом о высшем образовании это миф и нереально, но все оказалось не так, изначально искал информацию про: купить диплом в нижнекамске, купить диплом вуза в алма ате, купить диплом инженера специалиста, купить диплом колледжа образованиях, купить диплом медицинский университет, потом про дипломы вузов, подробнее здесь diplomybox.com/kupit-diplom-magistra-cherepovets
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Плоская мишень Fe+Mn
DavidNaing
| #
Tiger Fortune’s theme is a total vibe. fortune tiger 777
Diplomi_dzKt
| #
Приобрести диплом любого ВУЗа!
Мы можем предложить документы любых учебных заведений, которые находятся на территории всей Российской Федерации.
diplomk-v-krasnodare.ru/kupit-diplom-s-reestrom-bistro-i-bez-xlopot/
can i order cheap microzide tablets
| #
get microzide for sale
Waynepooda
| #
I love the futuristic feel at Elon Casino.
elon casino
Waynepooda
| #
This platform is top-notch with Elon Casino.
elon casino
1win_fupi
| #
casino en 1 win https://1win1001.top/ .
Sazrjbe
| #
Купить диплом университета по доступной цене можно, обратившись к надежной специализированной компании. Мы оказываем услуги по производству и продаже документов об окончании любых университетов Российской Федерации. Заказать диплом любого ВУЗа– freediplom.com/pokupka-diploma-s-zaneseniem-v-reestr-6/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Керамическая мишень для распыления оксида олова
mostbet_udPn
| #
мостбет скачать казино http://remsanteh.borda.ru/?1-6-0-00000047-000-0-0/ .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
поставляемая продукция:
Титановая труба 76х2.8х3000 мм ПТ1М ГОСТ 22897-86 Откройте мир прочных и надежных титановых труб с компанией Редметсплав. Наши высококачественные титановые трубы сочетают в себе устойчивость к коррозии, износостойкость и легкий вес, делая их незаменимым материалом для применения в различных сферах промышленности. Широкий выбор различных диаметров и толщин, а также индивидуальные решения для вашего производства. Гарантированное качество и надежность поставок.
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог высококачественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их качество. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы по мере того как предоставлять решения под специфику вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
Лист висмутовый CACIn903 – JIS H 2202-2016 Лист висмутовый CACIn903 – JIS H 2202-2016 представляет СЃРѕР±РѕР№ высококачественный материал, который используется РІ различных отраслях благодаря СЃРІРѕРёРј уникальным свойствам. Ртот РїСЂРѕРґСѓРєС‚ обладает отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью Рё хорошими механическими характеристиками, что делает его идеальным выбором для применения РІ электронике Рё медицине.Купить Лист висмутовый CACIn903 – JIS H 2202-2016 – значит обеспечить надежность Рё долговечность ваших проектов. Ртот лист соответствует современным стандартам, что гарантирует его высокую эффективность. РќРµ упустите возможность приобрести этот уникальный товар, который удовлетворит РІСЃРµ ваши потребности.
mostbet_xwPn
| #
служба поддержки мостбет номер телефона служба поддержки мостбет номер телефона .
Albertostomy
| #
Добрый день!
Правовая консультация по телефону позволяет получить помощь в любое время и из любого места. Юридическая помощь в бракоразводном процессе обеспечит вам поддержку и защиту интересов в сложных ситуациях. Консультация юриста по разводам поможет разобраться в юридических аспектах развода и делении имущества. Сопровождение сделок с недвижимостью обеспечит вам безопасность при заключении договоров. Юридическая помощь при оформлении недвижимости поможет вам правильно оформить все документы при покупке, продаже или аренде.
Больше информации на сайте – https://avtopravodo.ru/
оформление гражданства, срок регистрации в фнс ооо, срок давности по налогам для физических лиц на имущество
налог на имущество физических лиц налоговый кодекс, образец заявления на развод через суд с детьми и алиментами, оформление дарственной
Удачи!
mostbet_xvPn
| #
казино онлайн kg https://www.remsanteh.borda.ru/?1-6-0-00000047-000-0-0 .
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
поставляемая продукция:
Медный пресс тройник 15 мм М2РМ ГОСТ Р52948-2008 Приобретите медные тройники пресс из высококачественного медного сплава для надежной и простой установки в системах водоснабжения и отопления. Наслаждайтесь быстрой установкой, экономя время и средства, благодаря пресс-соединению. Гарантируйте долговечность и надежность в эксплуатации, используя тройники, которые подходят для различных типов систем и обеспечивают высокую теплопроводность и устойчивость к коррозии.
mostbet_yiMa
| #
мостбет зеркало mostbet6031.ru .
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Превосходное обслуживание – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы а также адаптировать решения под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
РўСЂСѓР±Р° кобальтовая СДП-3Рђ РўСЂСѓР±Р° кобальтовая СДП-3Рђ – это высококачественный РїСЂРѕРґСѓРєС‚, предназначенный для использования РІ различных областях, включая машиностроение Рё металлургическую промышленность. РћРЅР° отличается высокой прочностью Рё стойкостью Рє РєРѕСЂСЂРѕР·РёРё. Благодаря СЃРІРѕРёРј уникальным свойствам, труба может быть использована РІ агрессивных средах, что делает её идеальным выбором для профессионалов. Если РІС‹ ищете надежный Рё долговечный материал, то вам стоит купить РўСЂСѓР±Р° кобальтовая СДП-3Рђ. Ртот РїСЂРѕРґСѓРєС‚ отвечает всем современным требованиям Рё стандартам, обеспечивая надежность Рё эффективность РІ эксплуатации. РќРµ упустите возможность улучшить СЃРІРѕРё проекты!
Eanrgvd
| #
Для максимально быстрого продвижения по карьерной лестнице понадобится наличие диплома о высшем образовании. Быстро заказать диплом о высшем образовании у сильной компании: diplom-onlinex.com/kupit-attestat-11-klassov-bistro-i-bez-problem-3/
Dnrtkbd
| #
Где заказать диплом специалиста?
Мы предлагаем дипломы любой профессии по выгодным ценам. Мы можем предложить документы ВУЗов, которые находятся на территории всей России. Вы сможете заказать диплом от любого учебного заведения, за любой год, указав необходимую специальность и хорошие оценки за все дисциплины. Документы печатаются на бумаге самого высокого качества. Это позволяет делать государственные дипломы, которые невозможно отличить от оригиналов. Документы заверяются всеми требуемыми печатями и штампами. Стараемся поддерживать для покупателей адекватную политику цен. Для нас очень важно, чтобы дипломы были доступными для подавляющей массы наших граждан. kupit-diplomyz24.com/kupit-diplom-v-magadane-2-2
Alfredral
| #
Добрый день!
Юридическая помощь в трудовых спорах поможет вам защитить ваши права в случае нарушения условий трудового договора. Мы обеспечиваем поддержку при увольнении, а также при разногласиях по оплате труда и условиям работы. Наши специалисты окажут помощь в составлении исковых заявлений и защите ваших интересов в суде. Консультация по трудовому праву обеспечит вам понимание ваших прав и обязанностей в различных ситуациях. Обратитесь за помощью, и мы окажем вам юридическую поддержку по трудовым вопросам.
Больше информации на сайте – https://legallup.ru
наследство после смерти, подсудность о защите прав потребителей, билет 14 вопрос 20 пдд
может ли несовершеннолетний обратиться в суд за защитой своих прав, как отказаться от брака на сайте госуслуг регистрации, юрист по разделу имущества
Удачи!
where to buy cleocin online
| #
can you buy cleocin online cleocin t patient information sheet cleocin costco wholesale
cost of cleocin no prescription cleocin dosage for skin infections mechanism of action for cleocin
cleocin clindamycin phosphate cream
Cleocin 150 mg used for cleocin drug family cheat cleocin ovuli 100 mg
cleocin 100 mg suppository cleocin usage cleocin side effects in canada
BrockSmedo
| #
Здравствуйте!
Возврат страховки по кредиту — это важный вопрос, в котором мы предоставим юридическую помощь. Оформление гражданства — процесс, который требует точности и правильных действий. Оформление дарственной на квартиру должно быть выполнено в полном соответствии с законодательством. Автоюрист по лишению прав подскажет, как правильно действовать в случае изъятия прав. Помощь с военным билетом — важная услуга для военнослужащих.
Больше информации на сайте – https://agkonsult.ru
оформление долевой собственности, пенсионеры освобождаются от налога на имущество, документы для вступления в наследство после смерти матери
оформление скважины на воду для юридических лиц, нужно ли заверять брачный договор у нотариуса, подача иска
Удачи!
cost lisinopril without dr prescription
| #
Pills information. Cautions.
cost lisinopril without dr prescription
All trends of medication. Read information here.
1win_lmpi
| #
casino 1win 1win1001.top .
Waynepooda
| #
Elon Casino’s rewards are always a rush.
elon casino
1win_zgot
| #
1win,com 1win6042.ru .
mostbet_qdSa
| #
мостбет казино https://mostbet6032.ru .
Cazrttb
| #
Привет!
Без получения диплома очень трудно было продвинуться по карьерной лестнице. Сегодня же этот важный документ не дает никаких гарантий, что удастся найти престижную работу. Более важное значение имеют профессиональные навыки и знания специалиста, а также его опыт работы. Именно из-за этого решение о покупке диплома стоит считать выгодным и рациональным. Приобрести диплом университета babygirls016.copiny.com/question/details/id/1052761
1win_kbOa
| #
descărca 1win http://www.1win5010.ru .
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
поставляемая продукция:
Контакт РёР· драгоценных металлов серебряный 2С…8.5 РјРј РЎСЂ99.99 РўРЈ Рзысканные контакты РёР· золота, серебра Рё платины РѕС‚ Редметсплав. Высочайшее качество, уникальный дизайн Рё эксклюзивные предложения для постоянных клиентов. Добавьте роскошь Рё стиль вашему образу!
DavidNaing
| #
This casino’s Tiger Fortune game is legendary. fortune tiger
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена требуемыми документами, подтверждающими их происхождение. Опытная поддержка – наш стандарт – мы на связи, чтобы улаживать ваши вопросы и предоставлять решения под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
Проволока магниевая ZE41 – CSA HG.9 Полоса магниевая ZE41 – CSA HG.9 является высококачественным материалом, используемым РІ различных областях, таких как автомобилестроение Рё авиастроение. Отличается легким весом Рё высокой прочностью, что делает ее идеальным выбором для тех, кто ищет оптимальное сочетание этих характеристик. РћРЅР° обладает отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью Рё легко поддается механической обработке, что упрощает процесс изготовления изделий. Если РІС‹ хотите повысить эффективность СЃРІРѕРёС… проектов Рё приобрести надежные материалы, РЅРµ упустите возможность купить Полоса магниевая ZE41 – CSA HG.9. Рто решение, которое РІС‹ искали!
Waynepooda
| #
This platform is flawless with Elon Casino.
elon casino
internetCah
| #
недорогой интернет екатеринбург
domashij-internet-ekaterinburg001.ru
провайдеры интернета по адресу
1win_hoOn
| #
1 вин. 1win6054.ru .
DavidNaing
| #
Tiger Fortune keeps the excitement alive! fortune tiger demo
DavidNaing
| #
This online casino’s Tiger Fortune is a hit. fortune tiger demo
dragon money_ubsi
| #
dragonmoney зарабатывать dragonmoney зарабатывать .
dragon money_ekEt
| #
баланс драгон мани баланс драгон мани .
Sazrewu
| #
Мы можем предложить дипломы любой профессии по выгодным тарифам. Основные преимущества приобретения документов у нас
Вы покупаете диплом через надежную компанию. Это решение сэкономит не только деньги, но и время.
Преимуществ намного больше:
• Документы печатаются на настоящих бланках с печатями и подписями;
• Предлагаем дипломы любых учебных заведений России;
• Стоимость значительно ниже той, которую довелось бы платить за обучение в ВУЗе;
• Доставка как по столице, так и в любые другие регионы России.
Заказать диплом ВУЗа– http://freedost.com/read-blog/390_diplom-inzhenera-cena.html/ – freedost.com/read-blog/390_diplom-inzhenera-cena.html
1win_sbpa
| #
1 win nigeria 1win13.com.ng .
DanielHek
| #
https://kiteschoolhurghada.ru
nvblfurn
| #
https://elpix.ru/luchshie-onlajn-igry-dlja-malchikov-raznyh-vozrastov/ онлайн игры играть бесплатно без скачивания без регистрации
bitcevskoe
| #
Всё о кладбищах Видного bitcevskoe.ru/ Битцевское, Дрожжинское, Спасское, Жабкинское. Официальная информация, участки, услуги, порядок оформления документов, схема проезда.
Barrycromb
| #
https://1-board.ru/
DavidCatty
| #
Доброго!
Оформление гражданства — это важный процесс, требующий соблюдения всех требований миграционного законодательства, и юрист обеспечит вас поддержкой на каждом этапе. Оформление дарственной на квартиру — это процесс передачи права собственности на недвижимость, и юрист поможет вам в этом процессе. Автоюрист по лишению прав обеспечит защиту ваших интересов, если вы столкнулись с лишением водительских прав и хотите восстановить их. Помощь с военным билетом включает помощь в вопросах, связанных с военной службой и правами военнослужащих. Правовая консультация онлайн даст вам возможность получить консультацию, не выходя из дома, и решить все вопросы быстро.
Больше информации на сайте – https://juristywin.ru
защита прав в суде, правила ипотеки военной, открытие расчетного счета промсвязьбанк
налог на имущество подоходный, заявление о переходе на усн при регистрации ип, как вернуть права
Удачи!
mostbet_bzMa
| #
мосбет http://mostbet6031.ru .
Diplomi_yjka
| #
купить школьный диплом diplomys-vsem.ru .
1win_oopa
| #
win 1 https://www.1win6006.ru .
where can i get zofran no prescription
| #
Pills prescribing information. Long-Term Effects.
where can i get zofran no prescription
Best trends of medicament. Get information now.
1win_maot
| #
1вин войти 1вин войти .
HaroldGaR
| #
Sanctum.so is your all-in-one gateway to smart staking and instant liquidity on Solana. Whether you’re new to Solana or a long-time participant, Sanctum.so eliminates friction and complexity by offering an intuitive, powerful platform designed to optimize every move you make. With just a few clicks, users can stake their assets, maintain real-time access to liquidity, and enjoy seamless performance — all without giving up control. It’s the ultimate experience for those who value speed, simplicity, and full ownership of their digital journey.
Forget the old way of staking. Sanctum.so introduces a new standard where liquidity isn’t locked, where rewards flow without delays, and where one-click tools allow you to act quickly and confidently. Built to take full advantage of Solana’s speed and scalability, the platform makes staking as simple as connecting your wallet, choosing your path, and watching your assets grow. The elegant design, high-speed execution, and built-in transparency make Sanctum.so the most user-friendly Solana-native solution on the market.
Whether you’re looking to earn passive yield, actively manage your assets, or tap into Solana’s full performance, Sanctum.so adapts to your style. It offers real-time tracking, smart automation, and deep ecosystem integration that transforms staking from a chore into a strategic tool. Users benefit from deep liquidity, secure backend protocols, and an experience that requires zero technical knowledge — just results.
Sanctum.so empowers users at every level, from curious beginners to seasoned blockchain enthusiasts. Everything from asset management to reward harvesting is optimized behind the scenes, giving you more time and more return with less friction. You’re not just staking — you’re taking control of your future in a decentralized world.
In a sea of complex interfaces and fragmented tools, Sanctum.so stands out as a clean, focused, and highly effective platform for Solana users. It combines powerful staking mechanics with a beautiful, lightweight UI that just works. Fast. Simple. Reliable. This is staking reimagined for the modern user.
Welcome to a new era of staking. Welcome to a place where simplicity meets performance. Welcome to Sanctum.so — where your assets stay liquid, your rewards never sleep, and the future of Solana is at your fingertips.
Start staking now on Sanctum-sol.com
DavidNaing
| #
Tiger Fortune’s visuals are next-level cool. fortune tiger 777
Максим
| #
chinese-hitman-assassin.com https://chinese-hitman-assassin.com/
HaroldGaR
| #
Sanctum.so is your all-in-one gateway to smart staking and instant liquidity on Solana. Whether you’re new to Solana or a long-time participant, Sanctum.so eliminates friction and complexity by offering an intuitive, powerful platform designed to optimize every move you make. With just a few clicks, users can stake their assets, maintain real-time access to liquidity, and enjoy seamless performance — all without giving up control. It’s the ultimate experience for those who value speed, simplicity, and full ownership of their digital journey.
Forget the old way of staking. Sanctum.so introduces a new standard where liquidity isn’t locked, where rewards flow without delays, and where one-click tools allow you to act quickly and confidently. Built to take full advantage of Solana’s speed and scalability, the platform makes staking as simple as connecting your wallet, choosing your path, and watching your assets grow. The elegant design, high-speed execution, and built-in transparency make Sanctum.so the most user-friendly Solana-native solution on the market.
Whether you’re looking to earn passive yield, actively manage your assets, or tap into Solana’s full performance, Sanctum.so adapts to your style. It offers real-time tracking, smart automation, and deep ecosystem integration that transforms staking from a chore into a strategic tool. Users benefit from deep liquidity, secure backend protocols, and an experience that requires zero technical knowledge — just results.
Sanctum.so empowers users at every level, from curious beginners to seasoned blockchain enthusiasts. Everything from asset management to reward harvesting is optimized behind the scenes, giving you more time and more return with less friction. You’re not just staking — you’re taking control of your future in a decentralized world.
In a sea of complex interfaces and fragmented tools, Sanctum.so stands out as a clean, focused, and highly effective platform for Solana users. It combines powerful staking mechanics with a beautiful, lightweight UI that just works. Fast. Simple. Reliable. This is staking reimagined for the modern user.
Welcome to a new era of staking. Welcome to a place where simplicity meets performance. Welcome to Sanctum.so — where your assets stay liquid, your rewards never sleep, and the future of Solana is at your fingertips.
Start staking now on Sanctum-sol.com
dragon money_acEt
| #
dragon money рабочее зеркало dragon money рабочее зеркало .
mostbet_gySa
| #
aviator mostbet https://mostbet6032.ru/ .
dragon money_hcsi
| #
dragon мани dragon мани .
HaroldGaR
| #
Sanctum.so is your all-in-one gateway to smart staking and instant liquidity on Solana. Whether you’re new to Solana or a long-time participant, Sanctum.so eliminates friction and complexity by offering an intuitive, powerful platform designed to optimize every move you make. With just a few clicks, users can stake their assets, maintain real-time access to liquidity, and enjoy seamless performance — all without giving up control. It’s the ultimate experience for those who value speed, simplicity, and full ownership of their digital journey.
Forget the old way of staking. Sanctum.so introduces a new standard where liquidity isn’t locked, where rewards flow without delays, and where one-click tools allow you to act quickly and confidently. Built to take full advantage of Solana’s speed and scalability, the platform makes staking as simple as connecting your wallet, choosing your path, and watching your assets grow. The elegant design, high-speed execution, and built-in transparency make Sanctum.so the most user-friendly Solana-native solution on the market.
Whether you’re looking to earn passive yield, actively manage your assets, or tap into Solana’s full performance, Sanctum.so adapts to your style. It offers real-time tracking, smart automation, and deep ecosystem integration that transforms staking from a chore into a strategic tool. Users benefit from deep liquidity, secure backend protocols, and an experience that requires zero technical knowledge — just results.
Sanctum.so empowers users at every level, from curious beginners to seasoned blockchain enthusiasts. Everything from asset management to reward harvesting is optimized behind the scenes, giving you more time and more return with less friction. You’re not just staking — you’re taking control of your future in a decentralized world.
In a sea of complex interfaces and fragmented tools, Sanctum.so stands out as a clean, focused, and highly effective platform for Solana users. It combines powerful staking mechanics with a beautiful, lightweight UI that just works. Fast. Simple. Reliable. This is staking reimagined for the modern user.
Welcome to a new era of staking. Welcome to a place where simplicity meets performance. Welcome to Sanctum.so — where your assets stay liquid, your rewards never sleep, and the future of Solana is at your fingertips.
Start staking now on Sanctum-sol.com
dragon money_nton
| #
dragon money партнерка https://dragon-money10.com .
HaroldGaR
| #
Sanctum.so is your all-in-one gateway to smart staking and instant liquidity on Solana. Whether you’re new to Solana or a long-time participant, Sanctum.so eliminates friction and complexity by offering an intuitive, powerful platform designed to optimize every move you make. With just a few clicks, users can stake their assets, maintain real-time access to liquidity, and enjoy seamless performance — all without giving up control. It’s the ultimate experience for those who value speed, simplicity, and full ownership of their digital journey.
Forget the old way of staking. Sanctum.so introduces a new standard where liquidity isn’t locked, where rewards flow without delays, and where one-click tools allow you to act quickly and confidently. Built to take full advantage of Solana’s speed and scalability, the platform makes staking as simple as connecting your wallet, choosing your path, and watching your assets grow. The elegant design, high-speed execution, and built-in transparency make Sanctum.so the most user-friendly Solana-native solution on the market.
Whether you’re looking to earn passive yield, actively manage your assets, or tap into Solana’s full performance, Sanctum.so adapts to your style. It offers real-time tracking, smart automation, and deep ecosystem integration that transforms staking from a chore into a strategic tool. Users benefit from deep liquidity, secure backend protocols, and an experience that requires zero technical knowledge — just results.
Sanctum.so empowers users at every level, from curious beginners to seasoned blockchain enthusiasts. Everything from asset management to reward harvesting is optimized behind the scenes, giving you more time and more return with less friction. You’re not just staking — you’re taking control of your future in a decentralized world.
In a sea of complex interfaces and fragmented tools, Sanctum.so stands out as a clean, focused, and highly effective platform for Solana users. It combines powerful staking mechanics with a beautiful, lightweight UI that just works. Fast. Simple. Reliable. This is staking reimagined for the modern user.
Welcome to a new era of staking. Welcome to a place where simplicity meets performance. Welcome to Sanctum.so — where your assets stay liquid, your rewards never sleep, and the future of Solana is at your fingertips.
Start staking now on Sanctum-sol.com
DavidNaing
| #
Tiger Fortune has some seriously cool visuals. jogo do tigrinho demo
HaroldGaR
| #
Sanctum.so is your all-in-one gateway to smart staking and instant liquidity on Solana. Whether you’re new to Solana or a long-time participant, Sanctum.so eliminates friction and complexity by offering an intuitive, powerful platform designed to optimize every move you make. With just a few clicks, users can stake their assets, maintain real-time access to liquidity, and enjoy seamless performance — all without giving up control. It’s the ultimate experience for those who value speed, simplicity, and full ownership of their digital journey.
Forget the old way of staking. Sanctum.so introduces a new standard where liquidity isn’t locked, where rewards flow without delays, and where one-click tools allow you to act quickly and confidently. Built to take full advantage of Solana’s speed and scalability, the platform makes staking as simple as connecting your wallet, choosing your path, and watching your assets grow. The elegant design, high-speed execution, and built-in transparency make Sanctum.so the most user-friendly Solana-native solution on the market.
Whether you’re looking to earn passive yield, actively manage your assets, or tap into Solana’s full performance, Sanctum.so adapts to your style. It offers real-time tracking, smart automation, and deep ecosystem integration that transforms staking from a chore into a strategic tool. Users benefit from deep liquidity, secure backend protocols, and an experience that requires zero technical knowledge — just results.
Sanctum.so empowers users at every level, from curious beginners to seasoned blockchain enthusiasts. Everything from asset management to reward harvesting is optimized behind the scenes, giving you more time and more return with less friction. You’re not just staking — you’re taking control of your future in a decentralized world.
In a sea of complex interfaces and fragmented tools, Sanctum.so stands out as a clean, focused, and highly effective platform for Solana users. It combines powerful staking mechanics with a beautiful, lightweight UI that just works. Fast. Simple. Reliable. This is staking reimagined for the modern user.
Welcome to a new era of staking. Welcome to a place where simplicity meets performance. Welcome to Sanctum.so — where your assets stay liquid, your rewards never sleep, and the future of Solana is at your fingertips.
Start staking now on Sanctum-sol.com
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наши товары:
Гранулы ниобия 4N
Dnrtnmg
| #
Мы изготавливаем дипломы любой профессии по доступным ценам. Мы готовы предложить документы техникумов, которые находятся на территории всей России. Дипломы и аттестаты выпускаются на бумаге высшего качества. Это дает возможность делать настоящие дипломы, которые невозможно отличить от оригиналов. alternativaprofi.com/forum/viewthread.phpthread_id=35802
HaroldGaR
| #
Sanctum.so is your all-in-one gateway to smart staking and instant liquidity on Solana. Whether you’re new to Solana or a long-time participant, Sanctum.so eliminates friction and complexity by offering an intuitive, powerful platform designed to optimize every move you make. With just a few clicks, users can stake their assets, maintain real-time access to liquidity, and enjoy seamless performance — all without giving up control. It’s the ultimate experience for those who value speed, simplicity, and full ownership of their digital journey.
Forget the old way of staking. Sanctum.so introduces a new standard where liquidity isn’t locked, where rewards flow without delays, and where one-click tools allow you to act quickly and confidently. Built to take full advantage of Solana’s speed and scalability, the platform makes staking as simple as connecting your wallet, choosing your path, and watching your assets grow. The elegant design, high-speed execution, and built-in transparency make Sanctum.so the most user-friendly Solana-native solution on the market.
Whether you’re looking to earn passive yield, actively manage your assets, or tap into Solana’s full performance, Sanctum.so adapts to your style. It offers real-time tracking, smart automation, and deep ecosystem integration that transforms staking from a chore into a strategic tool. Users benefit from deep liquidity, secure backend protocols, and an experience that requires zero technical knowledge — just results.
Sanctum.so empowers users at every level, from curious beginners to seasoned blockchain enthusiasts. Everything from asset management to reward harvesting is optimized behind the scenes, giving you more time and more return with less friction. You’re not just staking — you’re taking control of your future in a decentralized world.
In a sea of complex interfaces and fragmented tools, Sanctum.so stands out as a clean, focused, and highly effective platform for Solana users. It combines powerful staking mechanics with a beautiful, lightweight UI that just works. Fast. Simple. Reliable. This is staking reimagined for the modern user.
Welcome to a new era of staking. Welcome to a place where simplicity meets performance. Welcome to Sanctum.so — where your assets stay liquid, your rewards never sleep, and the future of Solana is at your fingertips.
Start staking now on Sanctum-sol.com
Robertbut
| #
5 научных фактов о лжи, в которые трудно поверить
https://x.com/kiselev_igr/status/1909590459799716145
1win_itot
| #
1win регистрация https://1win6042.ru .
dragon money_jgsi
| #
драгон мани официальный сайт играть драгон мани официальный сайт играть .
Instant Withdrawal Casino
| #
Instant Withdrawal Casino
A Brief History of Elemental Analysis » Artemis Analytical
internetCah
| #
интернет провайдеры екатеринбург по адресу
domashij-internet-ekaterinburg002.ru
провайдеры интернета в екатеринбурге по адресу
Anonymous
| #
Your point of view caught my eye and was very interesting. Thanks. I have a question for you.
Williamfetap
| #
ремонт квартир в Балашихе Москва – динамичный мегаполис, где каждый стремится к созданию уютного и функционального пространства. Ремонт квартир в Москве – это не просто обновление интерьера, это инвестиция в комфорт и качество жизни. Мы предлагаем полный спектр услуг по ремонту квартир в Москве, от косметического обновления до капитальной перепланировки под ключ.
Brianslump
| #
Calibry Casino calibri casino app современное онлайн-казино с лицензией, щедрой бонусной системой и широким выбором игр. Участвуй в акциях, получай кэшбэк и выигрывай реальные деньги!
Xazremq
| #
Приобрести диплом о высшем образовании!
Мы можем предложить дипломы психологов, юристов, экономистов и любых других профессий по доступным тарифам. Вы заказываете документ через надежную фирму. : nadegda.listbb.ru/viewtopic.php?f=3&t=1419
where to get generic lopressor without prescription
| #
Pills information for patients. What side effects?
where to get generic lopressor without prescription
Some information about meds. Read now.
dragon money_miEt
| #
рабочее зеркало драгон мани рабочее зеркало драгон мани .
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
Наша продукция:
Титановый квадрат 14 мм ВТ5-1 ГОСТ 26492-85 Выберите идеальный титановый квадрат для высокоточных работ из нашего разнообразного ассортимента. Надежные, прочные и устойчивые к коррозии инструменты из высококачественного титана для металлообработки, строительства и ремонта. Разнообразие размеров и форм обеспечивает оптимальное решение для широкого спектра задач. Получите непревзойденные возможности для высокоточных работ с нашими титановыми квадратами.
GeorgeTom
| #
Source:
winmaxcasino.org
can i purchase cheap propecia
| #
order propecia without rx
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их качество. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы а также находить ответы под требования вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
Порошок висмутовый 302 – EN ISO 9453 Порошок висмутовый 302 – EN ISO 9453 является высококачественным материалом, предназначенным для применения РІ различных промышленных процессах. РћРЅ обладает отличной теплопроводностью Рё устойчивостью Рє окислению, что делает его идеальным выбором для пайки Рё РґСЂСѓРіРёС… технологий соединения. Рспользование данного порошка обеспечивает надежность Рё долговечность готовых изделий.Если РІС‹ ищете надежный Рё эффективный РїСЂРѕРґСѓРєС‚ для ваших нужд, покупая Порошок висмутовый 302 – EN ISO 9453, РІС‹ получаете высокую производительность Рё качество. РќРµ упустите возможность внести РІ СЃРІРѕР№ процесс улучшения СЃ помощью этого уникального порошка!
can i order generic florinef for sale
| #
how to buy florinef without rx
WomicGut
| #
На сайте https://mvpol.ru/ воспользуйтесь возможностью заказать промышленные полы напрямую от производителя. Компания «МВПОЛ» оказывает и такую важную услугу, как ремонт промышленных полов, производство полимерных наливных полов, бетонных, у которых упрочненный верх. Также получится узнать расценки на такие услуги, чтобы заранее спланировать бюджет. Для того чтобы иметь представление о том, как выглядят работы, изучите объекты. На все услуги предоставляются гарантии. Вся информация дается максимально быстро.
EugeneGam
| #
Sadece birkaç tıkla Pin-Up platformuna giriş yapabilir, canlı bahis ve casino keyfini mobil cihazında da yaşayabilirsin – kazanmaya başlamak için geç kalma! pin-up Türkiye çevrim
mostbet_trSa
| #
мостбет скачать http://mostbet6032.ru .
DavidNaing
| #
Tiger Fortune’s rewards make it worth playing. fortune tiger
1win_ufOa
| #
1win молдова 1win молдова .
BetaklDiz
| #
На сайте https://prometall.shop/ получится выбрать печь, а также воспользоваться полным перечнем услуг, которые связаны с печами Prometall, в том числе, реализацией, проведением монтажных работ. Изучите технические характеристики печей, которые находятся в сетке, камне, получится приобрести и отопительные печи, а также устройства в облицовке. Прямо сейчас вы сможете изучить хиты продаж – те товары, которые пользуются особой популярностью. Каталог товаров регулярно обновляется, чтобы вы приобрели лучшее.
1win_euSr
| #
1win.com.ci http://1win5011.ru .
Trefevf
| #
Приобрести диплом любого университета. Заказ диплома ВУЗа через надежную фирму дарит множество преимуществ. Данное решение помогает сберечь время и серьезные финансовые средства. jibharkepelo.copiny.com/question/details/id/1083754
JeffreyVed
| #
Привет всем!
Составление договора займа — это важная часть юридического сопровождения сделок, особенно если речь идет о крупных суммах. Помощь юриста в суде обеспечит вам надежную защиту на всех стадиях судебного разбирательства. Юридическая помощь при банкротстве поможет вам разобраться в процедуре банкротства и защитить свои интересы. Регистрация ООО требует выполнения ряда юридических процедур, и наш юрист поможет вам пройти их без ошибок. Открытие расчетного счета — это процесс, в котором юрист обеспечит правильное оформление всех документов.
Больше информации на сайте – https://capitalaw.ru/
страховые споры, адвокат по бракоразводным процессам воронеж, помощник адвоката москва вакансии по уголовным делам
налог на имущество рассчитать физических лиц, акт налоговой проверки пример, правовая консультация по телефону
Удачи!
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние достижения.
Наша продукция:
Гранулированный оксид титана
Zayebrot
| #
На сайте https://mvpol.ru/ воспользуйтесь возможностью заказать промышленные полы напрямую от производителя. Компания «МВПОЛ» оказывает и такую важную услугу, как ремонт промышленных полов, производство полимерных наливных полов, бетонных, у которых упрочненный верх. Также получится узнать расценки на такие услуги, чтобы заранее спланировать бюджет. Для того чтобы иметь представление о том, как выглядят работы, изучите объекты. На все услуги предоставляются гарантии. Вся информация дается максимально быстро.
nvblfurn
| #
https://studio-servis.ru/the_articles/vmeste-veseley-igry-dlya-dvoih-na-sayte-igroutka-su.html разновидность игры в бильярд 10 букв
ruleta americana online
| #
ruleta americana online
A Brief History of Elemental Analysis » Artemis Analytical
DavidNaing
| #
The animations in Tiger Fortune are incredible. fortune tiger 777
Cazrzic
| #
Здравствуйте!
Мы можем предложить дипломы любой профессии по приятным ценам. Цена зависит от определенной специальности, года выпуска и образовательного учреждения: diplomanrussians.com/
bokep viral
| #
bokep viral
A Brief History of Elemental Analysis » Artemis Analytical
BofadnyTum
| #
Компания «Ковка Винтаж» в течение длительного времени оказывает свои услуги и предлагает создать изысканные, завораживающие кованые изделия, которые украсят придомовую территорию, беседку, террасу, а также частный дом. На сайте https://kovka-vintazh.ru/ ознакомьтесь с полным ассортиментом изделий и работами компетентных и выдающихся мастеров.
Sazrlra
| #
Заказать диплом ВУЗа!
Купить диплом института по выгодной стоимости можно, обратившись к надежной специализированной фирме. Быстро заказать диплом: diplom-zentr.com/diplom-s-reestrom-kupit-tsena-garantiya-podlinnosti
bokep indo live
| #
bokep indo live
A Brief History of Elemental Analysis » Artemis Analytical
Uazrarm
| #
Здравствуйте!
Для некоторых людей, приобрести диплом университета – это острая необходимость, шанс получить хорошую работу. Но для кого-то – это понятное желание не терять время на учебу в институте. С какой бы целью вам это не потребовалось, наша компания готова помочь. Максимально быстро, профессионально и по разумной цене изготовим документ любого ВУЗа и любого года выпуска на подлинных бланках с реальными печатями.
Ключевая причина, почему многие люди прибегают к покупке документов, – получить хорошую должность. Предположим, способности и опыт позволяют человеку устроиться на желаемую работу, однако документального подтверждения квалификации нет. В случае если для работодателя важно присутствие “корочки”, риск потерять вакантное место очень высокий.
Приобрести документ института можно в нашей компании в столице. Мы предлагаем документы об окончании любых университетов Российской Федерации. Вы сможете получить диплом по любой специальности, любого года выпуска, включая документы Советского Союза. Гарантируем, что в случае проверки документов работодателем, подозрений не появится.
Ситуаций, которые вынуждают заказать диплом достаточно. Кому-то срочно потребовалась работа, и необходимо произвести хорошее впечатление на начальство при собеседовании. Другие хотят попасть в большую компанию, для того, чтобы повысить свой статус в социуме и в будущем начать собственное дело. Чтобы не тратить драгоценное время, а сразу начать эффективную карьеру, используя имеющиеся навыки, можно заказать диплом через интернет. Вы станете полезным в социуме, обретете финансовую стабильность в кратчайший срок- купить аттестат за 9 класс
Cazrtqy
| #
Купить диплом о высшем образовании!
Мы предлагаем документы учебных заведений, которые расположены в любом регионе РФ. Документы выпускаются на “правильной” бумаге самого высшего качества: finttech.ru/nastoyashhiy-diplom-lyubogo-uchebnogo-zavedeniya-kachestvennaya-pechat
Cazrnce
| #
Где заказать диплом по необходимой специальности?
Мы изготавливаем дипломы психологов, юристов, экономистов и других профессий по приятным ценам. Важно, чтобы документы были доступны для большого количества наших граждан. Приобрести диплом об образовании diplom-ryssia.com/kupit-diplom-texnikuma-s-zaneseniem-v-reestr-otzivi-24/
nvblfurn
| #
https://mdmotors.ru/populyarnost-i-uvlekatel-nost-onlayn-igr-na-dvoih/ сокровища пиратов играть онлайн бесплатно без регистрации сейчас
situs bokep
| #
situs bokep
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
Наша продукция:
Кольцо РёР· драгоценных металлов платиновое 45С…25С…5 РјРј ПлН95.5 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
Sazrabg
| #
Мы готовы предложить дипломы любых профессий по разумным ценам.
Вы заказываете документ через надежную и проверенную фирму. Приобрести диплом университета– http://mhealth-consulting.eu/employer/diplomy-grup-24/ – mhealth-consulting.eu/employer/diplomy-grup-24
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Превосходное обслуживание – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы по мере того как адаптировать решения под требования вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
Проволока вольфрамовая Р’Р”-25 Проволока вольфрамовая Р’Р”-25 – это качественный материал для различных производственных нужд. Рспользуется РІ электротехнике, машиностроении Рё металлургии. Обладает высокой прочностью Рё температурной стойкостью, что делает ее идеальным выбором для сварки Рё термической обработки. Наша проволока соответствует всем стандартам Рё требованиям, предоставляя надежность Рё долговечность. РќРµ упустите возможность сделать выгодное приобретение. Р’С‹ можете купить Проволока вольфрамовая Р’Р”-25 Рё обеспечить СЃРІРѕСЋ продукцию высококачественным материалом.
where can i buy generic ramipril price
| #
Drugs information leaflet. Short-Term Effects.
where can i buy generic ramipril price
Best about medicine. Get information here.
Hubertsek
| #
Looking for best prompts for stable diffusion? Findprompt.shop where you can choose from a huge selection of promts for Midjourney, Stable Diffusion, ChatGPT and more. Study the best recommendations for AI. You can sell your promts on our platform at a great price. Pay close attention to the choice of promts on the portal – it is the largest on the market. Our offers are unique, and the cost is affordable for everyone.
DavidNaing
| #
I can’t get enough of the Tiger Fortune soundtrack! tiger fortune
1win_japa
| #
1wi 1wi .
DavidNaing
| #
This online casino shines with Tiger Fortune. fortune tiger 777
link bokep
| #
link bokep
A Brief History of Elemental Analysis » Artemis Analytical
Diplomi_qgKt
| #
Приобрести диплом ВУЗа!
Мы можем предложить документы институтов, расположенных в любом регионе Российской Федерации.
diplomj-irkutsk.ru/kupit-diplom-ob-obrazovanii-s-reestrom-bez-truda/
dragon money_mbon
| #
поддержка dragon money поддержка dragon money .
mejores casinos online
| #
mejores casinos online
A Brief History of Elemental Analysis » Artemis Analytical
Dnrtxea
| #
Мы изготавливаем дипломы любых профессий по выгодным ценам. Мы можем предложить документы ВУЗов, которые находятся в любом регионе России. Дипломы и аттестаты выпускаются на бумаге высшего качества. Это дает возможность делать настоящие дипломы, которые невозможно отличить от оригиналов. agedcarepharmacist.com.au/employer/archive-diploma
1win_mtKa
| #
1win вход на сайт https://cah.forum24.ru/?1-13-0-00001695-000-0-0-1743258917 .
internetCah
| #
интернет провайдеры в екатеринбурге по адресу дома
domashij-internet-ekaterinburg003.ru
интернет по адресу дома
1win_ulKa
| #
1 win официальный сайт https://cah.forum24.ru/?1-13-0-00001695-000-0-0-1743258917 .
DavidNaing
| #
Tiger Fortune’s graphics are simply amazing. fortune tiger 777
DavidNaing
| #
Tiger Fortune’s features are super fun. fortune tiger bet
1win_nvKa
| #
1вин официальный сайт вход 1вин официальный сайт вход .
dragon money_qmsi
| #
dragon money зеркало dragon money зеркало .
1win_mqKr
| #
1win.kg 1win6043.ru .
HerbertGap
| #
Мир ремней
1win_imSr
| #
1 win md 1win5011.ru .
mostbet_jcmt
| #
мостбет скачать бесплатно http://mostbet6033.ru/ .
DavidNaing
| #
Tiger Fortune has some of the best odds I’ve seen. fortune tiger
ivonoSauro
| #
Ресурс hm.mnemosyne.ru выбрать ремень в подарок любимым людям предлагает. Мастера, создающие изделия, отличаются творческим потенциалом. Каждая модель с учетом модных трендов разрабатывается. Мы к покупателям всегда прислушиваемся. Идем при возможностях всегда на встречу. https://hm.mnemosyne.ru – тут сможете узнать, какой ремень в подарок руководителю, шефу или коллеге преподнести. Для изделий отбирается лучшая кожа. Оцените ручной работы неповторимость. Сделайте уже сейчас заказ на нашем портале. Пусть ваш стиль новыми красками засияет!
Trefrnz
| #
Заказать диплом об образовании. Покупка документа о высшем образовании через качественную и надежную компанию дарит множество достоинств. Данное решение позволяет сэкономить время и значительные финансовые средства. arzookanak0088.copiny.com/question/details/id/1083741
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Плоская мишень из иттрия, 3N, 4N
GeorgeTom
| #
Source:
– winmaxcasino.org
https://siconnet.com/knowledgebase/index.php/User:BriannaGadsdon4
| #
JBL’s soundbars are cinematic perfection!
https://yogaasanas.science/wiki/User:EmilioMoeller6
Toojddax
| #
On the site https://wallpapers4screen.com/ you can download wallpapers for desktop free. High Quality HD pictures wallpapers. High quality pictures and wallpapers! Choose one of many categories and download for free! Or check out the TOP downloadable images! You will definitely like them.
can i get cheap zithromax
| #
cost zithromax without a prescription
HerbertGap
| #
Мир ремней
YuwiyEnuts
| #
На сайте https://xn—-7sbjhqn0bhjc0lk.xn--p1ai/ получите юридическую консультацию по различным вопросам. Компания работает как с физическими, так и частными лицами. На все услуги установлены привлекательные расценки, чтобы воспользоваться ими смог каждый. Предприятие работает в этой сфере более 11 лет. В команде трудятся 11 специалистов, которые справятся с задачей независимо от сложности. К вашим услугам досудебный юрист, досудебное урегулирование, кредитный, семейный юрист, решение страховых споров.
can i purchase cheap artane no prescription
| #
order artane without dr prescription
Richardthema
| #
купить сейф оптом
Dnrtmpy
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и любых других профессий по приятным тарифам. Мы предлагаем документы техникумов, расположенных в любом регионе России. Документы делаются на “правильной” бумаге высшего качества. Это дает возможность делать государственные дипломы, которые не отличить от оригинала. artrp.5nx.ru/viewtopic.phpf=3&t=16477
pin up azerbaycan_veen
| #
pin up azerbaycan pin up azerbaycan .
mostbet_hnmt
| #
казино онлайн kg казино онлайн kg .
ciyovCoosy
| #
Вы находитесь на едином сайте аренды, где получится взять в прокат различную технику для выполнения самых разных работ. Вы сможете опубликовать объявление и предложить свою технику, чтобы ее заказал кто-то еще. https://arenda24.by/ – на сайте изучите категории, которые будут вам полезны: грузоперевозки, вспомогательное оборудование, режущий инструмент, строительные краны и многое другое. Вы сможете воспользоваться всеми опциями при условии, что зарегистрируетесь.
1win_rsKr
| #
1вин официальный мобильная https://1win6043.ru .
chusmeando
| #
chusmeando
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наша продукция:
Медная двухраструбная редукционная переходная муфта РїРѕРґ пайку 42С…40 РјРј мягкая пайка Рњ3Рњ ГОСТ 32590-2013 Выберите высококачественные медные двухраструбные редукционные муфты РїРѕРґ пайку РѕС‚ Редметсплав для надежного соединения медных труб различных диаметров. РЁРёСЂРѕРєРёР№ выбор размеров, эффективная установка Рё долговечность гарантированы. Рдеальное решение для систем отопления Рё водоснабжения.
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица товара подтверждена требуемыми документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – наш стандарт – мы на связи, чтобы ответить на ваши вопросы по мере того как адаптировать решения под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
Наши товары:
Труба титановая ВТ1-00 Пруток титановый ВТ1-00 — это высококачественный металлический элемент, обладающий замечательной прочностью и отличной коррозионной стойкостью. Он широко используется в аэрокосмической, медицинской и химической промышленности. Данный пруток отвечает всем современным требованиям, что делает его идеальным выбором для разнообразных конструкций. Свойства титана обеспечивают легкость и долговечность, что значительно расширяет области его применения. Если вам требуется надежный и высокотехнологичный материал, вы можете смело купить Пруток титановый ВТ1-00 для своих проектов. Наличие такого материала в своем арсенале гарантирует качественные результаты!
https://www.empresadeserviciosweb.com
| #
https://www.empresadeserviciosweb.com
A Brief History of Elemental Analysis » Artemis Analytical
DavidNaing
| #
Tiger Fortune’s payouts are always a surprise. fortune tiger 777
VernonSedia
| #
паническая атака симптомы Современный мир диктует свои условия, и порой нам необходима квалифицированная помощь, чтобы справиться с вызовами. Психолог онлайн – это удобный и анонимный способ получить поддержку в комфортной обстановке. Работа с психологом: путь к гармонии Обращение к психологу – это признак силы и заботы о себе. Специалист поможет разобраться в сложных ситуациях, найти ресурсы для преодоления трудностей и обрести внутреннюю гармонию.
PeterSlump
| #
https://telegra.ph/Best-Safe-Non-Gamstop-Casinos-in-the-UK-for-2023-03-27
Josephnat
| #
сейф офисный цены
Charleshag
| #
https://oxys-crema.net/
Jarioriji
| #
Где купить диплом специалиста?
Наша компания предлагаетбыстро и выгодно купить диплом, который выполняется на бланке ГОЗНАКа и заверен печатями, водяными знаками, подписями официальных лиц. Документ способен пройти любые проверки, даже при помощи профессиональных приборов. Решайте свои задачи быстро с нашим сервисом. Заказать диплом университета! fabnews.ru/forum/showthread.php?p=93248#post93248
Sazrzmf
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и других профессий по приятным тарифам. Стоимость будет зависеть от выбранной специальности, года выпуска и университета. Стараемся поддерживать для заказчиков адекватную ценовую политику. Для нас важно, чтобы документы были доступными для большого количества граждан. купить аттестат в пскове
Eanrlks
| #
Для быстрого продвижения по карьерной лестнице нужно наличие диплома о высшем образовании. Приобрести диплом университета у сильной организации: good-diplom.ru/kupite-diplom-v-ufe-bistro-i-bezopasno/
Jariorakg
| #
Где купить диплом по актуальной специальности?
Наши специалисты предлагают максимально быстро заказать диплом, который выполнен на оригинальной бумаге и заверен печатями, штампами, подписями должностных лиц. Документ способен пройти любые проверки, даже при помощи специального оборудования. Достигайте цели быстро с нашей компанией.
Купить диплом о высшем образовании kupit-diplom24.com/kupit-diplom-v-chelyabinske-18/
PeterSlump
| #
https://telegra.ph/Guide-to-Non-GamStop-Casinos-and-Their-Benefits3-03-27
Mazrrzk
| #
Мы можем предложить дипломы психологов, юристов, экономистов и любых других профессий по приятным тарифам. Стараемся поддерживать для клиентов адекватную политику цен. Важно, чтобы документы были доступными для подавляющей массы граждан.
Покупка документа, который подтверждает обучение в ВУЗе, – это грамотное решение. Заказать диплом о высшем образовании: peoplediplom.ru/kupit-diplom-meditsinskogo-kolledzha-4/
ww88
| #
หลังจากที่ผู้ใช้ทำการลงทะเบียนแล้ว สามารถเข้าสู่ระบบได้โดยการใช้ชื่อผู้ใช้และรหัสผ่านที่ได้ตั้งไว้ในขั้นตอนการสมัคร เมื่อเข้าสู่ระบบเรียบร้อยแล้ว ผู้เล่นสามารถเข้าถึงเกมคาสิโนออนไลน์ที่หลากหลาย ไม่ว่าจะเป็นสล็อต คาสิโนสด หรือกีฬา
และยังสามารถใช้โปรโมชั่นและโบนัสที่มีให้แก่ผู้ใช้ใหม่ได้ทันที
ZujizelPaw
| #
Популярная компания «Herbs & Flowers» предлагает огромный выбор искусственных цветов, которые идеально подходят как для украшения интерьера дома, так и офиса. В каталоге сайта https://herbsandflowers.ru/ вы найдете вертикальное озеленение, ампульные растения, а также деревья, ветви и многое другое.
internetCah
| #
подключить интернет казань
domashij-internet-kazan001.ru
домашний интернет тарифы казань
mostbet_inmt
| #
мосбет мосбет .
1win_aqKr
| #
1win ваучер http://1win6043.ru .
Lazrwde
| #
Заказать диплом любого ВУЗа!
Мы можем предложить дипломы любых профессий по приятным ценам— diplom-ryssia.com/kupite-attestati-za-11-klass-po-dostupnim-tsenam/
356bet
| #
356bet ให้ความสำคัญกับการถอนเงินของผู้ใช้เพื่อให้สมาชิกสามารถทำธุรกรรมได้อย่างสะดวกและรวดเร็ว โดยเว็บไซต์มีวิธีการถอนเงินที่ง่ายและปลอดภัย ผู้เล่นสามารถถอนเงินได้ผ่านช่องทางต่างๆ ที่รองรับ เช่น การโอนเงินผ่านธนาคาร หรือการใช้บริการ e-wallets ที่ได้รับความนิยมในปัจจุบัน ขั้นตอนการถอนเงินทำได้ง่ายๆ เพียงเข้าสู่ระบบ เลือกเมนูการถอนเงิน จากนั้นกรอกจำนวนเงินที่ต้องการถอนและเลือกวิธีการที่สะดวกที่สุด
การถอนเงินจะดำเนินการโดยอัตโนมัติภายในระยะเวลาที่กำหนด ซึ่งมักใช้เวลาไม่เกิน
24 ชั่วโมง ขึ้นอยู่กับวิธีการถอนที่เลือก
amado
| #
amado ให้ความสำคัญกับการถอนเงินของผู้ใช้เพื่อให้สมาชิกสามารถทำธุรกรรมได้อย่างสะดวกและรวดเร็ว โดยเว็บไซต์มีวิธีการถอนเงินที่ง่ายและปลอดภัย ผู้เล่นสามารถถอนเงินได้ผ่านช่องทางต่างๆ
ที่รองรับ เช่น การโอนเงินผ่านธนาคาร หรือการใช้บริการ
e-wallets ที่ได้รับความนิยมในปัจจุบัน ขั้นตอนการถอนเงินทำได้ง่ายๆ เพียงเข้าสู่ระบบ เลือกเมนูการถอนเงิน จากนั้นกรอกจำนวนเงินที่ต้องการถอนและเลือกวิธีการที่สะดวกที่สุด การถอนเงินจะดำเนินการโดยอัตโนมัติภายในระยะเวลาที่กำหนด ซึ่งมักใช้เวลาไม่เกิน 24 ชั่วโมง ขึ้นอยู่กับวิธีการถอนที่เลือก
mm88
| #
mm88 ให้ความสำคัญกับการถอนเงินของผู้ใช้เพื่อให้สมาชิกสามารถทำธุรกรรมได้อย่างสะดวกและรวดเร็ว โดยเว็บไซต์มีวิธีการถอนเงินที่ง่ายและปลอดภัย ผู้เล่นสามารถถอนเงินได้ผ่านช่องทางต่างๆ ที่รองรับ เช่น การโอนเงินผ่านธนาคาร หรือการใช้บริการ e-wallets
ที่ได้รับความนิยมในปัจจุบัน ขั้นตอนการถอนเงินทำได้ง่ายๆ เพียงเข้าสู่ระบบ เลือกเมนูการถอนเงิน จากนั้นกรอกจำนวนเงินที่ต้องการถอนและเลือกวิธีการที่สะดวกที่สุด การถอนเงินจะดำเนินการโดยอัตโนมัติภายในระยะเวลาที่กำหนด ซึ่งมักใช้เวลาไม่เกิน 24 ชั่วโมง ขึ้นอยู่กับวิธีการถอนที่เลือก
hranenie veshei_nnpa
| #
сервис хранения вещей сервис хранения вещей .
pin up azerbaycan_juen
| #
pinup azerbaycan pinup azerbaycan .
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-Free-Spins-Offers-and-Benefits2-03-27
golabgaf
| #
Ищете медтехника для дома интернет-магазин? Agsvv.ru/catalog/obluchateli_dlya_lecheniya у вас появится возможность приобрести для лечения кожных болезней облучатели по цене производителя с гарантией. ООО Хронос занимается производством облучателей для лечения кожных заболеваний с 2009-го года с доказанными методами лечения. Медицинские приборы от производителя славятся высочайшим качеством! Посмотрите наш ассортимент и назначение приборов. Мы поставляем приборы собственного производства государственным и частным медучреждениям по всей России.
Trefudi
| #
Выгодно заказать диплом ВУЗа. Приобретение документа о высшем образовании через надежную компанию дарит массу достоинств для покупателя. Это решение дает возможность сэкономить как дорогое время, так и серьезные деньги. feleempleo.es/employer/eonline-diploma
Thomasdrymn
| #
Upgrade your car into a masterpiece with our expert vehicle customization options. At Bergin Auto Works, we specialize in delivering high-quality customizations that exhibit your distinctive style. From chic body enhancements to robust performance boosts, our team provides excellent artistry. Check out our webpage https://berginautobody.com/ to explore our solutions and begin your car’s transformation now.
Leonardetemo
| #
аккаунты с балансом http://prodaja-akkauntov.ru
pk789
| #
pk789 ให้ความสำคัญกับการถอนเงินของผู้ใช้เพื่อให้สมาชิกสามารถทำธุรกรรมได้อย่างสะดวกและรวดเร็ว โดยเว็บไซต์มีวิธีการถอนเงินที่ง่ายและปลอดภัย ผู้เล่นสามารถถอนเงินได้ผ่านช่องทางต่างๆ ที่รองรับ เช่น การโอนเงินผ่านธนาคาร หรือการใช้บริการ e-wallets
ที่ได้รับความนิยมในปัจจุบัน ขั้นตอนการถอนเงินทำได้ง่ายๆ
เพียงเข้าสู่ระบบ เลือกเมนูการถอนเงิน จากนั้นกรอกจำนวนเงินที่ต้องการถอนและเลือกวิธีการที่สะดวกที่สุด การถอนเงินจะดำเนินการโดยอัตโนมัติภายในระยะเวลาที่กำหนด ซึ่งมักใช้เวลาไม่เกิน 24 ชั่วโมง ขึ้นอยู่กับวิธีการถอนที่เลือก
Iariorpjd
| #
Приобрести диплом университета по невысокой стоимости возможно, обращаясь к проверенной специализированной фирме. Купить документ университета можно в нашем сервисе. fastdiploms.com/gde-kupit-diplom-s-reestrom-bistro-i-nadezhno-4
Jamesneums
| #
услуги по продаже аккаунтов marketpleys-akkauntov-socsetei.ru
DanielPed
| #
маркетплейс для реселлеров marketpleysakkauntov.ru/
how can i get pamelor price
| #
buying generic pamelor online
me88
| #
หากผู้เล่นประสบปัญหาในการลงทะเบียนหรือการเข้าสู่ระบบ ทีมบริการลูกค้าของ me88 ก็พร้อมให้บริการตลอด 24 ชั่วโมงผ่านช่องทางต่างๆ ไม่ว่าจะเป็นแชทสด อีเมล หรือสายด่วน เพื่อให้ผู้เล่นได้รับความช่วยเหลืออย่างรวดเร็วและมีประสิทธิภาพ
PeterSlump
| #
https://telegra.ph/Best-Casino-No-Deposit-Bonus-Not-on-GamStop-03-27
168vip
| #
168vip ให้ความสำคัญกับการถอนเงินของผู้ใช้เพื่อให้สมาชิกสามารถทำธุรกรรมได้อย่างสะดวกและรวดเร็ว
โดยเว็บไซต์มีวิธีการถอนเงินที่ง่ายและปลอดภัย ผู้เล่นสามารถถอนเงินได้ผ่านช่องทางต่างๆ ที่รองรับ เช่น การโอนเงินผ่านธนาคาร หรือการใช้บริการ e-wallets ที่ได้รับความนิยมในปัจจุบัน ขั้นตอนการถอนเงินทำได้ง่ายๆ เพียงเข้าสู่ระบบ เลือกเมนูการถอนเงิน จากนั้นกรอกจำนวนเงินที่ต้องการถอนและเลือกวิธีการที่สะดวกที่สุด
การถอนเงินจะดำเนินการโดยอัตโนมัติภายในระยะเวลาที่กำหนด ซึ่งมักใช้เวลาไม่เกิน 24 ชั่วโมง ขึ้นอยู่กับวิธีการถอนที่เลือก
stake
| #
หลังจากที่ผู้ใช้ทำการลงทะเบียนแล้ว สามารถเข้าสู่ระบบได้โดยการใช้ชื่อผู้ใช้และรหัสผ่านที่ได้ตั้งไว้ในขั้นตอนการสมัคร เมื่อเข้าสู่ระบบเรียบร้อยแล้ว ผู้เล่นสามารถเข้าถึงเกมคาสิโนออนไลน์ที่หลากหลาย
ไม่ว่าจะเป็นสล็อต คาสิโนสด หรือกีฬา และยังสามารถใช้โปรโมชั่นและโบนัสที่มีให้แก่ผู้ใช้ใหม่ได้ทันที
Xazryis
| #
Купить диплом ВУЗа!
Мы предлагаем дипломы любой профессии по выгодным тарифам. Вы приобретаете документ через надежную компанию. : skvortsy.listbb.ru/viewtopic.php?f=31&t=3210
Lazrjoi
| #
Диплом любого ВУЗа Российской Федерации!
Без университета очень нелегко было продвигаться вверх по карьере. В связи с этим решение о покупке диплома можно считать целесообразным. Быстро и просто приобрести диплом об образовании rinadarmawangsa.copiny.com/question/details/id/1075233
buy generic hyzaar online
| #
cost of cheap hyzaar without dr prescription
DavidNaing
| #
Tiger Fortune’s free spins feature is a game-changer. fortune tiger demo
ygg
| #
ygg ให้ความสำคัญกับการถอนเงินของผู้ใช้เพื่อให้สมาชิกสามารถทำธุรกรรมได้อย่างสะดวกและรวดเร็ว โดยเว็บไซต์มีวิธีการถอนเงินที่ง่ายและปลอดภัย ผู้เล่นสามารถถอนเงินได้ผ่านช่องทางต่างๆ ที่รองรับ
เช่น การโอนเงินผ่านธนาคาร หรือการใช้บริการ e-wallets ที่ได้รับความนิยมในปัจจุบัน ขั้นตอนการถอนเงินทำได้ง่ายๆ เพียงเข้าสู่ระบบ เลือกเมนูการถอนเงิน จากนั้นกรอกจำนวนเงินที่ต้องการถอนและเลือกวิธีการที่สะดวกที่สุด การถอนเงินจะดำเนินการโดยอัตโนมัติภายในระยะเวลาที่กำหนด ซึ่งมักใช้เวลาไม่เกิน 24 ชั่วโมง ขึ้นอยู่กับวิธีการถอนที่เลือก
DavidNaing
| #
Tiger Fortune has some awesome surprise bonuses. fortune tiger
huay
| #
บริการลูกค้า huay – https://huay-778.com ออนไลน์ 24/7 ช่วยแก้ปัญหาผู้เล่นทุกคำถาม
Josephnat
| #
офисные сейфы
rauy
| #
rauy ให้ความสำคัญกับการถอนเงินของผู้ใช้เพื่อให้สมาชิกสามารถทำธุรกรรมได้อย่างสะดวกและรวดเร็ว โดยเว็บไซต์มีวิธีการถอนเงินที่ง่ายและปลอดภัย
ผู้เล่นสามารถถอนเงินได้ผ่านช่องทางต่างๆ
ที่รองรับ เช่น การโอนเงินผ่านธนาคาร หรือการใช้บริการ e-wallets ที่ได้รับความนิยมในปัจจุบัน
ขั้นตอนการถอนเงินทำได้ง่ายๆ เพียงเข้าสู่ระบบ เลือกเมนูการถอนเงิน จากนั้นกรอกจำนวนเงินที่ต้องการถอนและเลือกวิธีการที่สะดวกที่สุด การถอนเงินจะดำเนินการโดยอัตโนมัติภายในระยะเวลาที่กำหนด
ซึ่งมักใช้เวลาไม่เกิน 24 ชั่วโมง ขึ้นอยู่กับวิธีการถอนที่เลือก
w88
| #
w88 วิธีการสมัครและเข้าสู่ระบบสำหรับมือใหม่
Sazrxwh
| #
Купить диплом университета !
Приобретение диплома любого ВУЗа РФ у нас является надежным процессом, так как документ заносится в государственный реестр. Приобрести диплом о высшем образовании diplomg-kurerom.ru/kupit-diplom-s-zaneseniem-v-reestr-bistro-i-nadezhno-80
pin up azerbaycan_xyen
| #
pinup azerbaycan pinup azerbaycan .
Charleshag
| #
https://oxys-crema.net/
PeterSlump
| #
https://telegra.ph/Discussion-on-Non-Gamstop-Casinos-and-Player-Experiences1-03-27
marbo
| #
หลังจากที่ผู้ใช้ทำการลงทะเบียนแล้ว สามารถเข้าสู่ระบบได้โดยการใช้ชื่อผู้ใช้และรหัสผ่านที่ได้ตั้งไว้ในขั้นตอนการสมัคร
เมื่อเข้าสู่ระบบเรียบร้อยแล้ว ผู้เล่นสามารถเข้าถึงเกมคาสิโนออนไลน์ที่หลากหลาย ไม่ว่าจะเป็นสล็อต คาสิโนสด หรือกีฬา และยังสามารถใช้โปรโมชั่นและโบนัสที่มีให้แก่ผู้ใช้ใหม่ได้ทันที
123bet
| #
วิธีการลงทะเบียน 123bet พร้อมรับโบนัส 100$ สำหรับมือใหม่
hulk
| #
หลังจากที่ผู้ใช้ทำการลงทะเบียนแล้ว สามารถเข้าสู่ระบบได้โดยการใช้ชื่อผู้ใช้และรหัสผ่านที่ได้ตั้งไว้ในขั้นตอนการสมัคร เมื่อเข้าสู่ระบบเรียบร้อยแล้ว ผู้เล่นสามารถเข้าถึงเกมคาสิโนออนไลน์ที่หลากหลาย ไม่ว่าจะเป็นสล็อต คาสิโนสด หรือกีฬา และยังสามารถใช้โปรโมชั่นและโบนัสที่มีให้แก่ผู้ใช้ใหม่ได้ทันที
pg65
| #
หลังจากที่ผู้ใช้ทำการลงทะเบียนแล้ว สามารถเข้าสู่ระบบได้โดยการใช้ชื่อผู้ใช้และรหัสผ่านที่ได้ตั้งไว้ในขั้นตอนการสมัคร เมื่อเข้าสู่ระบบเรียบร้อยแล้ว ผู้เล่นสามารถเข้าถึงเกมคาสิโนออนไลน์ที่หลากหลาย ไม่ว่าจะเป็นสล็อต คาสิโนสด หรือกีฬา และยังสามารถใช้โปรโมชั่นและโบนัสที่มีให้แก่ผู้ใช้ใหม่ได้ทันที
1win_tzpi
| #
1win. casino. http://www.1win1001.top .
DonayrIngek
| #
На сайте https://vezuviy.shop/ вы сможете приобрести чугунные, стальные банные печи, нержавеющие, а также в облицовке, печи-камины, отопительные, каминные топки, дымоходы, порталы и многое другое. Вся продукция от производителя, а потому наценки очень маленькие. Такая техника считается идеальной не только с целью обогрева помещения, но и оформления пространства. А самое главное, что она отличается долгим сроком службы, надежностью из-за того, что в работе использовались высокотехнологичные, проверенные материалы.
yaxiceShoof
| #
Истринская мануфактура производит межкомнатные двери на заказ. При отменном качестве мебель компании по заманчивым ценам доступна. Мы творчески подходим к каждому заказу. Делаем красивую продукцию и ею гордимся. http://istradoors.ru – здесь представлены отзывы о нашей компании. Также на сайте перечислены часто задаваемые вопросы и ответы на них. С нами ведущие дизайнерские и архитектурные мастерские работают. Мы готовы любую задачу осуществить. Детальную информацию о продукции и об условиях заказа уточняйте у менеджеров.
m88
| #
หลังจากที่ผู้ใช้ทำการลงทะเบียนแล้ว สามารถเข้าสู่ระบบได้โดยการใช้ชื่อผู้ใช้และรหัสผ่านที่ได้ตั้งไว้ในขั้นตอนการสมัคร
เมื่อเข้าสู่ระบบเรียบร้อยแล้ว ผู้เล่นสามารถเข้าถึงเกมคาสิโนออนไลน์ที่หลากหลาย ไม่ว่าจะเป็นสล็อต คาสิโนสด
หรือกีฬา และยังสามารถใช้โปรโมชั่นและโบนัสที่มีให้แก่ผู้ใช้ใหม่ได้ทันที
Sazrubz
| #
Приобретение официального диплома через проверенную и надежную компанию дарит массу преимуществ для покупателя. Данное решение помогает сэкономить как дорогое время, так и значительные финансовые средства. Тем не менее, плюсов гораздо больше.Мы можем предложить дипломы психологов, юристов, экономистов и прочих профессий. Дипломы производят на подлинных бланках государственного образца. Доступная стоимость по сравнению с большими расходами на обучение и проживание. Приобретение диплома об образовании из российского университета станет мудрым шагом.
Заказать диплом: good-diplom.ru/visshee-obrazovanie-kupit-diplom-s-zaneseniem-6/
jbo
| #
jbo ให้ความสำคัญกับการถอนเงินของผู้ใช้เพื่อให้สมาชิกสามารถทำธุรกรรมได้อย่างสะดวกและรวดเร็ว
โดยเว็บไซต์มีวิธีการถอนเงินที่ง่ายและปลอดภัย ผู้เล่นสามารถถอนเงินได้ผ่านช่องทางต่างๆ ที่รองรับ เช่น การโอนเงินผ่านธนาคาร หรือการใช้บริการ e-wallets ที่ได้รับความนิยมในปัจจุบัน ขั้นตอนการถอนเงินทำได้ง่ายๆ เพียงเข้าสู่ระบบ เลือกเมนูการถอนเงิน จากนั้นกรอกจำนวนเงินที่ต้องการถอนและเลือกวิธีการที่สะดวกที่สุด การถอนเงินจะดำเนินการโดยอัตโนมัติภายในระยะเวลาที่กำหนด ซึ่งมักใช้เวลาไม่เกิน 24 ชั่วโมง ขึ้นอยู่กับวิธีการถอนที่เลือก
we88
| #
we88 สมัครสมาชิกใหม่ รับโบนัส 100$ ทันที ไม่มีเงื่อนไข
aspas
| #
หากผู้เล่นประสบปัญหาในการลงทะเบียนหรือการเข้าสู่ระบบ ทีมบริการลูกค้าของ aspas ก็พร้อมให้บริการตลอด 24 ชั่วโมงผ่านช่องทางต่างๆ ไม่ว่าจะเป็นแชทสด อีเมล หรือสายด่วน เพื่อให้ผู้เล่นได้รับความช่วยเหลืออย่างรวดเร็วและมีประสิทธิภาพ
Jarioruxg
| #
Где приобрести диплом по нужной специальности?
Наша компания предлагаетмаксимально быстро заказать диплом, который выполняется на оригинальном бланке и заверен мокрыми печатями, водяными знаками, подписями. Данный диплом пройдет любые проверки, даже с использованием специальных приборов. Решите свои задачи быстро и просто с нашим сервисом. Заказать диплом любого университета! mosreut.flybb.ru/viewtopic.php?f=2&t=541
bokep live
| #
bokep live
A Brief History of Elemental Analysis » Artemis Analytical
fun99
| #
fun99 ให้ความสำคัญกับการถอนเงินของผู้ใช้เพื่อให้สมาชิกสามารถทำธุรกรรมได้อย่างสะดวกและรวดเร็ว
โดยเว็บไซต์มีวิธีการถอนเงินที่ง่ายและปลอดภัย ผู้เล่นสามารถถอนเงินได้ผ่านช่องทางต่างๆ ที่รองรับ เช่น การโอนเงินผ่านธนาคาร หรือการใช้บริการ e-wallets ที่ได้รับความนิยมในปัจจุบัน ขั้นตอนการถอนเงินทำได้ง่ายๆ เพียงเข้าสู่ระบบ เลือกเมนูการถอนเงิน จากนั้นกรอกจำนวนเงินที่ต้องการถอนและเลือกวิธีการที่สะดวกที่สุด การถอนเงินจะดำเนินการโดยอัตโนมัติภายในระยะเวลาที่กำหนด ซึ่งมักใช้เวลาไม่เกิน 24 ชั่วโมง ขึ้นอยู่กับวิธีการถอนที่เลือก
Sazrubl
| #
купить диплом ставрополь
miami
| #
บริการลูกค้า miami พร้อมช่วยเหลือ 24
ชั่วโมง ทุกคำถามมีคำตอบ
WilliamIsofe
| #
Доброго!
Юридическая помощь при покупке жилья включает в себя проверку всех документов, сделок и договоров, а также защиту ваших интересов. Налоги при сдаче квартиры требуют правильного расчета и подачи декларации, и юрист поможет вам избежать налоговых ошибок. Регистрация дачного дома требует соблюдения всех норм, и юрист поможет вам оформить все документы на землю и строения. Автоюрист по каско обеспечит поддержку при возврате страховки или компенсации по условиям КАСКО. Юридическая консультация по ОСАГО поможет вам разобраться в условиях автогражданки и защитить свои права в случае аварии или спора с страховой компанией.
Больше информации на сайте – https://pravookey.ru
увольнение по статье, можно ли поменять неторжественную регистрацию брака на торжественную, ипотека военная от сбербанка
заявление на регистрацию ип онлайн, налоговая льгота по налогу на имущество организаций, консультация по правам потребителей
Удачи!
Jariorknk
| #
Где приобрести диплом по нужной специальности?
Мы предлагаем максимально быстро купить диплом, который выполняется на оригинальном бланке и заверен мокрыми печатями, штампами, подписями. Документ способен пройти любые проверки, даже с использованием специфических приборов. Решайте свои задачи быстро и просто с нашими дипломами.
Приобрести диплом о высшем образовании rusd-diplomj.ru/kupit-diplom-v-barnaule-9/
PeterSlump
| #
https://telegra.ph/Nonukcasinosuk-Review-of-Non-Gamstop-Casinos-03-27
789pro
| #
หากผู้เล่นประสบปัญหาในการลงทะเบียนหรือการเข้าสู่ระบบ ทีมบริการลูกค้าของ 789pro ก็พร้อมให้บริการตลอด 24 ชั่วโมงผ่านช่องทางต่างๆ
ไม่ว่าจะเป็นแชทสด อีเมล หรือสายด่วน เพื่อให้ผู้เล่นได้รับความช่วยเหลืออย่างรวดเร็วและมีประสิทธิภาพ
365bet
| #
365bet ให้ความสำคัญกับการถอนเงินของผู้ใช้เพื่อให้สมาชิกสามารถทำธุรกรรมได้อย่างสะดวกและรวดเร็ว โดยเว็บไซต์มีวิธีการถอนเงินที่ง่ายและปลอดภัย ผู้เล่นสามารถถอนเงินได้ผ่านช่องทางต่างๆ ที่รองรับ เช่น การโอนเงินผ่านธนาคาร
หรือการใช้บริการ e-wallets
ที่ได้รับความนิยมในปัจจุบัน ขั้นตอนการถอนเงินทำได้ง่ายๆ เพียงเข้าสู่ระบบ เลือกเมนูการถอนเงิน จากนั้นกรอกจำนวนเงินที่ต้องการถอนและเลือกวิธีการที่สะดวกที่สุด การถอนเงินจะดำเนินการโดยอัตโนมัติภายในระยะเวลาที่กำหนด ซึ่งมักใช้เวลาไม่เกิน 24 ชั่วโมง ขึ้นอยู่กับวิธีการถอนที่เลือก
Sazrmyo
| #
Купить документ о получении высшего образования можно у нас в столице. Мы оказываем услуги по продаже документов об окончании любых ВУЗов Российской Федерации. Вы сможете получить необходимый диплом по любым специальностям, любого года выпуска, включая документы старого образца. Гарантируем, что при проверке документов работодателем, подозрений не появится. asxdiplommy.com/kupit-diplom-s-zaneseniem-v-reestr-po-dostupnoj-tsene/
Mazrmpt
| #
Где купить диплом специалиста?
Мы оказываем услуги по изготовлению и продаже документов об окончании любых ВУЗов России. Документы изготавливаются на подлинных бланках. Рєveniaminv.flybb.ru/viewtopic.phpf=1&t=4288
DavidNaing
| #
The tiger roars in Tiger Fortune are epic. fortune tiger bet
pxj
| #
pxj ให้ความสำคัญกับการถอนเงินของผู้ใช้เพื่อให้สมาชิกสามารถทำธุรกรรมได้อย่างสะดวกและรวดเร็ว
โดยเว็บไซต์มีวิธีการถอนเงินที่ง่ายและปลอดภัย ผู้เล่นสามารถถอนเงินได้ผ่านช่องทางต่างๆ ที่รองรับ เช่น การโอนเงินผ่านธนาคาร หรือการใช้บริการ e-wallets ที่ได้รับความนิยมในปัจจุบัน ขั้นตอนการถอนเงินทำได้ง่ายๆ
เพียงเข้าสู่ระบบ เลือกเมนูการถอนเงิน จากนั้นกรอกจำนวนเงินที่ต้องการถอนและเลือกวิธีการที่สะดวกที่สุด การถอนเงินจะดำเนินการโดยอัตโนมัติภายในระยะเวลาที่กำหนด
ซึ่งมักใช้เวลาไม่เกิน 24 ชั่วโมง ขึ้นอยู่กับวิธีการถอนที่เลือก
22win
| #
22win สมัครสมาชิกใหม่วันนี้ รับโบนัสฟรี 100$
888bet
| #
หลังจากที่ผู้ใช้ทำการลงทะเบียนแล้ว สามารถเข้าสู่ระบบได้โดยการใช้ชื่อผู้ใช้และรหัสผ่านที่ได้ตั้งไว้ในขั้นตอนการสมัคร เมื่อเข้าสู่ระบบเรียบร้อยแล้ว ผู้เล่นสามารถเข้าถึงเกมคาสิโนออนไลน์ที่หลากหลาย ไม่ว่าจะเป็นสล็อต คาสิโนสด หรือกีฬา และยังสามารถใช้โปรโมชั่นและโบนัสที่มีให้แก่ผู้ใช้ใหม่ได้ทันที
Johnniemob
| #
Имморталы CS2 cs open case редкие скины, которые выделят тебя в матче. Торгуй, покупай, продавай топовые предметы с моментальной доставкой в инвентарь. Лучшие цены и безопасные сделки!
Robertmer
| #
Премиум скины для CS2 cs open case выделяйся в каждом раунде! Редкое оружие, эксклюзивные коллекции, эффектные раскраски и ножи. Только топовые предметы с мгновенной доставкой в Steam.
Davidcrafe
| #
Топ-дроп CS2 http://open-case-cs2.ru уже здесь! Самые редкие скины, ножи и эксклюзивы, которые действительно выпадают. Лучшие кейсы, обновления коллекций и советы для удачного открытия.
123vip
| #
123vip สมัครสมาชิกใหม่
รับโบนัส 100$ ทันที ไม่มีเงื่อนไข
internetCah
| #
интернет провайдеры в казани по адресу дома
domashij-internet-kazan002.ru
интернет по адресу
fun88
| #
วิธีสมัครสมาชิก fun88 และเข้าสู่ระบบอย่างง่ายดาย
euro
| #
euro – https://euro-778.com ตอบคำถามและช่วยเหลือผู้ใช้ตลอด 24 ชั่วโมง
บริการลูกค้าออนไลน์
qq188
| #
qq188 บริการลูกค้ามืออาชีพ 24/7 แก้ไขข้อสงสัยทันทีทุกเวลา
h25
| #
h25 บริการลูกค้ามืออาชีพ
24/7 แก้ไขข้อสงสัยทันทีทุกเวลา
1win_fvot
| #
1вин войти http://1win6042.ru/ .
best online casinos
| #
best online casinos
A Brief History of Elemental Analysis » Artemis Analytical
id888
| #
หลังจากที่ผู้ใช้ทำการลงทะเบียนแล้ว สามารถเข้าสู่ระบบได้โดยการใช้ชื่อผู้ใช้และรหัสผ่านที่ได้ตั้งไว้ในขั้นตอนการสมัคร เมื่อเข้าสู่ระบบเรียบร้อยแล้ว ผู้เล่นสามารถเข้าถึงเกมคาสิโนออนไลน์ที่หลากหลาย ไม่ว่าจะเป็นสล็อต คาสิโนสด หรือกีฬา และยังสามารถใช้โปรโมชั่นและโบนัสที่มีให้แก่ผู้ใช้ใหม่ได้ทันที
Sazrkxr
| #
Мы готовы предложить дипломы любых профессий по выгодным ценам.– diplomass.com/kupit-diplom-s-reestrom-otzivi-bistro-i-bezopasno/
PeterSlump
| #
https://telegra.ph/Best-Non-Gamstop-Casinos-in-the-UK-for-Players2-03-27
Mazrybl
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и любых других профессий по приятным ценам. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Важно, чтобы дипломы были доступными для большого количества наших граждан.
Заказ документа, который подтверждает окончание университета, – это грамотное решение. Купить диплом любого университета: zakaz-na-diplom.ru/diplom-texnikum-kupit-2/
ff88
| #
วิธีลงทะเบียน ff88 และเข้าสู่ระบบ พร้อมรับโบนัสทันที
Sazrvqu
| #
Мы изготавливаем дипломы любых профессий по приятным ценам. Стоимость зависит от выбранной специальности, года выпуска и образовательного учреждения. Стараемся поддерживать для заказчиков адекватную ценовую политику. Для нас очень важно, чтобы документы были доступны для большого количества граждан. отзывы где лучше купить диплом о высшем образовании
188bet
| #
188bet ให้ความสำคัญกับการถอนเงินของผู้ใช้เพื่อให้สมาชิกสามารถทำธุรกรรมได้อย่างสะดวกและรวดเร็ว โดยเว็บไซต์มีวิธีการถอนเงินที่ง่ายและปลอดภัย ผู้เล่นสามารถถอนเงินได้ผ่านช่องทางต่างๆ ที่รองรับ เช่น การโอนเงินผ่านธนาคาร หรือการใช้บริการ e-wallets ที่ได้รับความนิยมในปัจจุบัน ขั้นตอนการถอนเงินทำได้ง่ายๆ เพียงเข้าสู่ระบบ เลือกเมนูการถอนเงิน จากนั้นกรอกจำนวนเงินที่ต้องการถอนและเลือกวิธีการที่สะดวกที่สุด การถอนเงินจะดำเนินการโดยอัตโนมัติภายในระยะเวลาที่กำหนด ซึ่งมักใช้เวลาไม่เกิน 24 ชั่วโมง
ขึ้นอยู่กับวิธีการถอนที่เลือก
Lazrovc
| #
Купить диплом о высшем образовании!
Мы изготавливаем дипломы психологов, юристов, экономистов и любых других профессий по приятным тарифам— good-diplom.ru/gde-kupit-diplom-meditsinskij-4/
buy cheap lozol without prescription
| #
can i order generic lozol without prescription
RonaldSpurf
| #
Доброго!
Налогообложение доходов требует внимательности при оформлении всех налоговых деклараций и уплате налогов, и юрист поможет вам правильно рассчитать все обязательства. Юридическая проверка документов поможет вам убедиться в правильности и законности всех документов, перед их подачей в суд или другие инстанции. Помощь при продаже недвижимости обеспечит вам поддержку на всех этапах сделки — от подготовки документов до подписания договора. Услуги юриста по ДТП помогут вам защитить ваши права в случае ДТП и решить вопросы с возмещением ущерба. Автострахование требует правильного оформления страховых полисов и страховых случаев, и юрист поможет вам разобраться в условиях страхования.
Больше информации на сайте – https://tvokonsultant.ru/
налоговая проверка, регистрация договора купли продажи недвижимости в росреестре сроки, налоговый вычет за квартиру в 2023 когда подавать заявление на возврат подоходного налога за покупку
реестр лицензий росгвардии на охранную деятельность, лицензия на фармацевтическую деятельность реестр, возврат налогов
Удачи!
Towojddax
| #
On the site https://wallpapers4screen.com/ you can download wallpapers for desktop free. High Quality HD pictures wallpapers. High quality pictures and wallpapers! Choose one of many categories and download for free! Or check out the TOP downloadable images! You will definitely like them.
how can i get cheap persantine pills
| #
buy cheap persantine tablets
mc888
| #
หลังจากที่ผู้ใช้ทำการลงทะเบียนแล้ว สามารถเข้าสู่ระบบได้โดยการใช้ชื่อผู้ใช้และรหัสผ่านที่ได้ตั้งไว้ในขั้นตอนการสมัคร เมื่อเข้าสู่ระบบเรียบร้อยแล้ว ผู้เล่นสามารถเข้าถึงเกมคาสิโนออนไลน์ที่หลากหลาย ไม่ว่าจะเป็นสล็อต คาสิโนสด หรือกีฬา
และยังสามารถใช้โปรโมชั่นและโบนัสที่มีให้แก่ผู้ใช้ใหม่ได้ทันที
uea8
| #
uea8 ให้ความสำคัญกับการถอนเงินของผู้ใช้เพื่อให้สมาชิกสามารถทำธุรกรรมได้อย่างสะดวกและรวดเร็ว โดยเว็บไซต์มีวิธีการถอนเงินที่ง่ายและปลอดภัย ผู้เล่นสามารถถอนเงินได้ผ่านช่องทางต่างๆ ที่รองรับ เช่น การโอนเงินผ่านธนาคาร หรือการใช้บริการ e-wallets
ที่ได้รับความนิยมในปัจจุบัน ขั้นตอนการถอนเงินทำได้ง่ายๆ เพียงเข้าสู่ระบบ เลือกเมนูการถอนเงิน จากนั้นกรอกจำนวนเงินที่ต้องการถอนและเลือกวิธีการที่สะดวกที่สุด การถอนเงินจะดำเนินการโดยอัตโนมัติภายในระยะเวลาที่กำหนด ซึ่งมักใช้เวลาไม่เกิน 24 ชั่วโมง ขึ้นอยู่กับวิธีการถอนที่เลือก
pok9
| #
สมัคร pok9 วันนี้ รับโบนัส 100$ เข้าเล่นได้ทุกเกม
PeterSlump
| #
https://telegra.ph/Find-Reliable-Non-Gamstop-Casinos-in-2023-03-27
DavidNaing
| #
This casino’s Tiger Fortune slot is pure joy. fortune tiger bet
Jariorypq
| #
Где приобрести диплом специалиста?
Мы предлагаемвыгодно заказать диплом, который выполняется на оригинальной бумаге и заверен мокрыми печатями, штампами, подписями. Наш диплом способен пройти любые проверки, даже с применением специальных приборов. Решайте свои задачи максимально быстро с нашими дипломами. Приобрести диплом любого ВУЗа! ybeapp.com/read-blog/506_kupit-svidetelstvo-o-razvode.html
fan88
| #
หลังจากที่ผู้ใช้ทำการลงทะเบียนแล้ว สามารถเข้าสู่ระบบได้โดยการใช้ชื่อผู้ใช้และรหัสผ่านที่ได้ตั้งไว้ในขั้นตอนการสมัคร เมื่อเข้าสู่ระบบเรียบร้อยแล้ว ผู้เล่นสามารถเข้าถึงเกมคาสิโนออนไลน์ที่หลากหลาย
ไม่ว่าจะเป็นสล็อต คาสิโนสด หรือกีฬา
และยังสามารถใช้โปรโมชั่นและโบนัสที่มีให้แก่ผู้ใช้ใหม่ได้ทันที
DavidNaing
| #
Tiger Fortune is perfect for a quick gaming session. fortune tiger demo
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-2021-No-Deposit-Bonus-Guide1-03-27
1win_wipi
| #
casino 1 win casino 1 win .
Jariorbqg
| #
Где купить диплом специалиста?
Наша компания предлагает выгодно заказать диплом, который выполнен на оригинальной бумаге и заверен печатями, штампами, подписями. Наш диплом пройдет лубую проверку, даже при использовании специальных приборов. Достигайте свои цели быстро с нашими дипломами.
Заказать диплом любого университета poluchidiplom.com/kupit-diplom-v-yaroslavle-8/
Randyarcam
| #
Здравствуйте!
Оформление дарственной на землю требует юридической точности и соблюдения всех норм законодательства. Мы поможем вам подготовить все документы для передачи прав на земельный участок, будь то в рамках продажи или безвозмездной передачи. Юридическая помощь при оформлении дарственной обеспечит вам правильное оформление и защиту ваших интересов. Мы также предлагаем консультацию по вопросам налогообложения, связанных с дарением земли. Обратитесь к нам, и мы поможем вам правильно оформить дарственную.
Больше информации на сайте – https://legalyes.ru
как оформить лизинг, бесплатно юридическая консультация в самаре, бесплатная консультация юриста чита
согаз возврат страховки по кредиту при досрочном погашении согаз, сроки возврата денег за товар, юридическая помощь при увольнении
Удачи!
mostbet_ajSa
| #
скачать мостбет официальный сайт скачать мостбет официальный сайт .
PeterSlump
| #
https://telegra.ph/Top-Non-Gamstop-Casinos-in-the-UK-for-202311-03-27
pin up azerbaycan_foMa
| #
pin up azerbaycan pin up azerbaycan .
WonifdVef
| #
Компания «DELIVERY PLAY» предлагает выгодный прокат инвалидных колясок с быстрой доставкой и по привлекательным ценам. Каждый клиент получает возможность воспользоваться оперативной доставкой не только по Москве, но и области. На сайте https://deliveryplay.ru/arenda-invalidnyh-kolyasok изучите все модели, которые вы сможете заказать здесь.
1win_dvMr
| #
register with 1win website https://1win14.com.ng/ .
Sazrewj
| #
купить диплом техникума в магнитогорске
Zohagdjub
| #
На сайте https://ooomila.com изучите каталог такого оборудования, которое предназначено для организации животноводческих комплексов. Также вы сможете почитать и содержательную информацию о компании и ее достижениях. В каталоге вы найдете следующее оборудование: технологические линии, весовое оборудование, резиновые маты, крематоры и многое другое. На технику, которая находится в разделе, действуют гарантии. Несколько сотен единиц оборудования всегда находятся на складе, можно приобрести под заказ.
PeterSlump
| #
https://telegra.ph/Casinos-Not-on-Gamstop-No-Deposit-Bonus-Offers-03-27
Johnnytycle
| #
Привет всем!
Регистрация ООО поможет вам создать юридическое лицо для ведения бизнеса, соблюдая все законодательные требования. Открытие расчетного счета важно для организации, и юрист поможет вам выбрать банк и оформить все документы. Корпоративное право важно для обеспечения правильных и законных отношений внутри компании и с внешними партнерами. Консультация по НДС поможет вам разобраться с налогообложением и избежать штрафов и ошибок в расчетах. Составление отчетности важно для соблюдения налоговых и юридических норм, и юрист поможет вам в этом процессе.
Больше информации на сайте – https://legallurist.ru
вопросы по гражданству, юрист по дтп краснодар, моральный вред по трудовым спорам
закон 25 о защите прав потребителей возврат товара, нотариус москва на карте рядом со мной, юридическая помощь при лишении прав
Удачи!
Mazrrsv
| #
Мы изготавливаем дипломы любых профессий по приятным ценам. Стараемся поддерживать для клиентов адекватную ценовую политику. Для нас важно, чтобы дипломы были доступными для большого количества граждан.
Приобретение документа, который подтверждает обучение в университете, – это разумное решение. Приобрести диплом ВУЗа: kupitediplom0029.ru/diplom-kupit-texnikum-2/
DavidNaing
| #
Tiger Fortune’s theme is so adventurous. fortune tiger bet
Willieavamy
| #
I won $900 on a slot—still hyped! plinko online
DavidNaing
| #
Tiger Fortune’s rewards are always exciting. fortune tiger bet
biximoSmask
| #
Отопление и водоснабжение – компания, которая предоставляет профессиональные услуги. Наши опытные специалисты готовы выполнить монтаж системы отопления в Рузе в кратчайшие сроки. Создайте комфорт и тепло с нами в вашем доме. Ищете монтаж систем отопления? Ruzskiy-rayon.santex-uslugi.ru – тут можете уже сегодня с отзывами о нас ознакомиться. Гарантируем использование качественных материалов. Готовы превзойти ваши ожидания. Стараемся сделать услуги доступными для клиентов. Позвоните нашему менеджеру для консультации по интересующим вас вопросам.
Lazrrrt
| #
Быстро и просто заказать диплом института!
Мы изготавливаем дипломы любой профессии по приятным ценам— diplom-kaluga.ru/kupit-diplom-o-visshem-obrazovanii-zapolnennij-3/
Вадим
| #
chinessonsofanarchy-italia.com https://sonsofanarchy-italia.com
internetCah
| #
домашний интернет тарифы
domashij-internet-kazan003.ru
домашний интернет тарифы казань
PeterSlump
| #
https://telegra.ph/Legit-PayPal-Casinos-in-the-UK-Without-Gamstop-03-27
SorapnRah
| #
Компания «V8soft» предлагает облачный сервис для того, чтобы вы смогли вести бизнес удачно и максимально эффективно. На предприятии работают исключительно те профессионалы, у которых имеется весь необходимый опыт работы, полезные навыки. На сайте https://v8soft.ru/ изучите все возможности ПО, чтобы определить то, подходит ли вам решение.
where can i buy generic cipro pill
| #
where can i get cipro tablets
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние возможности.
Наши товары:
Гранулы магниевого фторида 4N
can i order inderal pills
| #
where to get generic inderal without insurance
DavidErows
| #
Вход на платформу — и ты уже на орбите. Всё движется мгновенно, интерфейс — по sci-fi.
Слоты — файлы, работают плавно.казино – игровые автоматы — это реальный хаб, где каждый спин — телепорт в глубину. А результат — как надо.
1win_oiMi
| #
1win букмекер 1win букмекер .
pin up azerbaycan_zeMa
| #
pin up azerbaycan pin up azerbaycan .
bankrotstvo v moskve_ttKi
| #
Если не можете платить по своим долгам, не нужно откладывать решение проблемы. Вы можете воспользоваться законной процедурой банкротства https://bankrotstvo-v-moskve95.ru .
1win_wyMi
| #
1 вин. https://cah.forum24.ru/?1-13-0-00001678-000-0-0 .
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-with-Paypal-Payment-Options-03-27
Uazreul
| #
Привет!
Для некоторых людей, приобрести диплом о высшем образовании – это необходимость, удачный шанс получить достойную работу. Но для кого-то – это желание не терять массу времени на учебу в ВУЗе. Что бы ни толкнуло вас на такой шаг, мы готовы помочь вам. Быстро, профессионально и по разумной стоимости сделаем документ любого ВУЗа и года выпуска на настоящих бланках с реальными подписями и печатями.
Ключевая причина, почему люди покупают документ, – получить определенную работу. К примеру, знания и опыт позволяют кандидату устроиться на желаемую работу, однако подтверждения квалификации не имеется. Когда работодателю важно наличие “корочки”, риск потерять место работы очень высокий.
Купить документ о получении высшего образования вы можете у нас в Москве. Мы предлагаем документы об окончании любых ВУЗов РФ. Вы получите диплом по любой специальности, любого года выпуска, в том числе документы СССР. Гарантируем, что в случае проверки документа работодателями, подозрений не появится.
Обстоятельств, которые вынуждают приобрести диплом о среднем образовании немало. Кому-то очень срочно нужна работа, и необходимо произвести впечатление на руководителя в процессе собеседования. Некоторые хотят попасть в серьезную компанию, чтобы повысить собственный статус и в дальнейшем начать свое дело. Чтобы не терять впустую годы жизни, а сразу начинать эффективную карьеру, используя врожденные таланты и приобретенные знания, можно купить диплом через интернет. Вы сможете быть полезным в обществе, получите денежную стабильность в максимально короткие сроки- диплом купить о высшем образовании
Richardthema
| #
купить сейф москва
Cazrdug
| #
Приветствую!
Мы изготавливаем дипломы психологов, юристов, экономистов и прочих профессий по доступным ценам. Стоимость зависит от той или иной специальности, года получения и ВУЗа: rdiplomm24.com/
1win_wqMr
| #
1win casino http://1win14.com.ng/ .
JamesJeape
| #
Добрый день!
Как вернуть товар по закону — это вопрос, с которым часто сталкиваются покупатели. Мы поможем вам разобраться в ваших правах как потребителя и вернуть товар, если он не соответствует условиям договора. Юрист по правам потребителей обеспечит, что вы получите возврат товара или компенсацию за его стоимость в случае нарушения ваших прав. Наши специалисты помогут вам составить заявление, подать жалобу и защитить ваши интересы в суде. Обратитесь за помощью, чтобы вернуть товар по закону.
Больше информации на сайте – https://uristvzakon.ru/
налоговый юрист, возникновение трудовых отношений, можно ли подать на алименты с рождения ребенка
как разделить квартиру в ипотеке с материнским капиталом при разводе есть дети, брачный договор фоксфорд, юридическая консультация онлайн
Удачи!
1win_fvMi
| #
1win футбол https://www.cah.forum24.ru/?1-13-0-00001678-000-0-0 .
DavidNaing
| #
This casino’s Tiger Fortune game is incredible. fortune tiger bet
1win_wlot
| #
1win кейсы http://1win6042.ru .
DavidNaing
| #
Tiger Fortune keeps me entertained all night. jogo do tigrinho demo
DavidNaing
| #
Tiger Fortune’s free spins are pure gold. tigrinho demo
PeterSlump
| #
https://telegra.ph/Best-Non-GamStop-Casinos-for-2023-03-27
Sazrlvj
| #
Мы можем предложить дипломы любой профессии по доступным тарифам.
Вы покупаете диплом в надежной и проверенной временем компании. Купить диплом о высшем образовании– http://trolleybus26.moibb.ru/viewtopic.phpf=3&t=1296/ – trolleybus26.moibb.ru/viewtopic.phpf=3&t=1296
Kennethzof
| #
ДПДГ ПТСР Панические атаки Современный мир диктует свои условия, и порой нам необходима квалифицированная помощь, чтобы справиться с вызовами. Психолог онлайн – это удобный и анонимный способ получить поддержку в комфортной обстановке. Работа с психологом: путь к гармонии Обращение к психологу – это признак силы и заботы о себе. Специалист поможет разобраться в сложных ситуациях, найти ресурсы для преодоления трудностей и обрести внутреннюю гармонию.
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наша продукция:
Медное двухраструбное колено под пайку 180 градусов 8х6х0.6 мм 6.8х8.8 мм твердая пайка М1т ГОСТ Р52922-2008 Купите надежные медные двухраструбные колена под пайку от ведущего производителя. Обеспечьте эффективное отопление и водоснабжение с прочными и устойчивыми к воздействию внешних факторов коленами из меди.
GeorgeTom
| #
Source:
winmaxcasino.org
pin up azerbaycan_wwMa
| #
pinup azerbaycan pinup azerbaycan .
Sazrzwp
| #
купить аттестат в минске
1win_ueKr
| #
1 вин 1 вин .
mostbet_xdmt
| #
mostbet скачать на телефон бесплатно андроид https://mostbet6033.ru .
Rodneytug
| #
Топовые скины CS2 csgo open case ru от легендарных ножей до эксклюзивных обложек на AWP. Красота, стиль, престиж. Оцени крутой дроп и подбери скин, который подходит именно тебе.
Michaelweing
| #
Лучшие скины CS2 open csgo case яркие, редкие, премиальные. Собери коллекцию, прокачай инвентарь и покажи всем свой вкус. Обзор самых крутых скинов с актуальными ценами и рейтингами.
ThomasAutof
| #
Топовые скины CS2 open case cs ru от легендарных ножей до эксклюзивных обложек на AWP. Красота, стиль, престиж. Оцени крутой дроп и подбери скин, который подходит именно тебе.
texedseics
| #
Интернет-бутик Feringer.shop занимается продажей печей для бань и саун. Часто устраиваем акции для привлечения новых покупателей. У вас есть прекрасная возможность рассчитывать на лояльные условия покупки печного оборудования. Расширяем выбор и улучшаем свой сервис. Ищете установка печи ферингер? Feringer.shop – здесь есть отзывы и каталог, в котором имеются доступные разновидности печного оборудования. Оцените параметры каждой модели. Мы все пожелания и потребности наших заказчиков учитываем. Готовы грамотно вас проконсультировать.
Cazrmry
| #
Где купить диплом специалиста?
Мы изготавливаем дипломы любых профессий по приятным тарифам. Важно, чтобы документы были доступными для большинства наших граждан. Купить диплом о высшем образовании diplomus-spb.ru/kupit-ofitsialnij-diplom-s-zaneseniem-v-reestr-14/
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их происхождение. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы по мере того как находить ответы под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в гибкости нашего предложения
поставляемая продукция:
Порошок кобальтовый Stellite 4LC Порошок кобальтовый Stellite 4LC представляет СЃРѕР±РѕР№ высококачественный материал, предназначенный для термической обработки Рё защиты РѕС‚ РёР·РЅРѕСЃР°. РћРЅ обладает отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ устойчивостью, что делает его идеальным выбором для применения РІ harsh условиях. Благодаря своей высокой прочности Рё износостойкости, порошок РїРѕРґС…РѕРґРёС‚ для изготовления режущего инструмента Рё деталей, работающих РїСЂРё высоких температурах. Если РІС‹ хотите повысить эффективность СЃРІРѕРёС… производственных процессов, рекомендуем купить Порошок кобальтовый Stellite 4LC уже сегодня. Ртот материал успешно применяется РІ авиастроении, машиностроении Рё РґСЂСѓРіРёС… отраслях, что подтверждает его надежность Рё качество.
mostbet_ylMa
| #
скачать мостбет https://www.mostbet6031.ru .
PeterSlump
| #
https://telegra.ph/Exploring-Casinos-Not-on-GamStop-for-Players-03-27
JustinDug
| #
Добрый день!
Легализация документов поможет вам пройти процесс официального признания документов в других странах. Помощь в приватизации недвижимости требует знания всех юридических аспектов этого процесса, и юрист поможет вам с оформлением. Составление брачного договора обеспечит защиту ваших интересов при вступлении в брак, особенно в вопросах раздела имущества. Юридическая помощь при лишении прав поможет вам восстановить водительские права, если они были лишены незаконно. Оформление опеки требует соблюдения всех юридических норм, чтобы обеспечить интересы ребенка и сторон, оформляющих опеку.
Больше информации на сайте – https://obrascidoc.ru/
как оформить банкротство, декларация по налогу на имущество организаций, как оформить договор по доверенности образец
иск образец по защите прав потребителей, ускоренное оформление загранпаспорта, сопровождение бизнеса
Удачи!
DavidNaing
| #
Tiger Fortune’s features are a total win. fortune tiger demo
Willieavamy
| #
Online casinos should push safe play more. plinko
1win_jxMr
| #
register with 1win website https://1win14.com.ng/ .
1win_tbOa
| #
1win.com 1win.com .
DavidNaing
| #
Tiger Fortune is the best slot in this casino’s lineup. fortune tiger demo gratis
Eanrgzz
| #
Для эффективного продвижения вверх по карьерной лестнице понадобится наличие официального диплома о высшем образовании. Быстро и просто приобрести диплом университета у проверенной организации: rusd-diplomj.ru/diplom-kupit-6/
PeterSlump
| #
https://telegra.ph/No-Gamstop-Casino-Insights-and-Alternatives-03-27
DavidNaing
| #
Tiger Fortune’s wild symbols are my favorite! fortune tiger demo
ScottMop
| #
Привет всем!
Консультация по трудовому праву поможет вам разобраться в вопросах, связанных с трудовой деятельностью. Автоюрист по штрафам подскажет, как правильно действовать при наличии штрафов. Юридическая помощь при оформлении наследства — это процесс, который требует внимательности и грамотных действий. Оформление долевой собственности — сложный процесс, и мы поможем вам справиться с ним. Гражданский брак и имущество — вопрос, который требует юридического подхода.
Больше информации на сайте – https://llawdo.ru/
автоюрист по каско, коды вычетов по подоходному налогу рб расшифровка, пошлина на регистрацию права собственности на машиноместо
о защите прав потребителей закон рф 2300 1, есть ли в мфц нотариус в москве, налоги при продаже квартиры
Удачи!
1win_sjSr
| #
cazino md https://www.1win5011.ru .
order cheap prasugrel online
| #
can you get prasugrel online
PeterSlump
| #
https://telegra.ph/Top-Casinos-Not-on-Gamstop-UK-With-No-Deposit-Bonus1-03-27
DavidNaing
| #
The tiger roars in Tiger Fortune make it so fun! fortune tiger 777
can i order cheap plendil tablets
| #
buying generic plendil without dr prescription
https://hifrequency.live/community/profile/audryheller0208/
| #
https://hifrequency.live/community/profile/audryheller0208/ квартира на сутки Гродно
Diplomi_plKt
| #
Купить диплом ВУЗа!
Мы готовы предложить документы институтов, которые расположены в любом регионе РФ.
diplom-kaluga.ru/kupite-diplom-bakalavra-s-vneseniem-v-reestr-bistro-2/
mostbet_atSa
| #
mostbet kg скачать https://www.mostbet6032.ru .
Vufamciz
| #
На сайте https://rustreetwear.ru вы найдете огромное количество интересных, разнообразных и эксклюзивных брендов, которые создают роскошную уличную одежду на любые случаи жизни. Есть как совсем не известные марки, которые только начинают свой путь, так и гиганты мировой индустрии. Перед вами самый впечатляющий каталог, где вы найдете не только одежду, но и аксессуары. Каталог обновляется ежедневно, чтобы вы обязательно нашли для себя лучший вариант. Здесь установлены доступные расценки.
cenigalob
| #
Если вы находитесь в поисках качественного, надежного и практичного стального профиля, то обратитесь на предприятие ООО “Валцпроф”, где вы найдете продукцию в широком ассортименте. В этой компании вы также сможете заказать и выпадающие пороги, которые идеально подходят для дверей различного типа. https://waltzprof.com/ – на портале изучите весь ассортимент товаров, которые выпускаются заводом. Профили, находящиеся в каталоге, созданы для оперативной, удобной сборки перегородок, конструкций для фасадов, люков и многого другого.
internetCah
| #
провайдеры интернета по адресу
domashij-internet-msk001.ru
какие провайдеры интернета есть по адресу москва
Iariorqvo
| #
Заказать диплом ВУЗа по доступной стоимости можно, обратившись к проверенной специализированной фирме. Приобрести документ университета вы можете у нас в Москве. diplomt-nsk.ru/kupit-diplom-s-vneseniem-v-reestr-bistro-i-nadezhno-34
nvblfurn
| #
https://timelady.ru/1368-onlayn-igry.html какие игры можно поиграть на двоих
PeterSlump
| #
https://telegra.ph/Casino-Free-Spins-No-Deposit-Options-Off-Gamstop-03-27
dragon money_fjsi
| #
онлайн-казино драгон мани онлайн-казино драгон мани .
dragon money_cnEt
| #
драгон мани официальный сайт драгон мани официальный сайт .
Basketballpuns#puns
| #
Hi joke lovers!
Pass these jokes around like a no-look assist—funny and unexpected. basketball puns
Steal some smiles with basketball play on words that pass the vibe check. Fast breaks, faster laughs.
Toda la información en el enlace – п»їhttps://ontheborder.com.au/the-history-of-tequila/#comment-2550
Wishing you lots of funny moments!
CliftonCab
| #
Здравствуйте!
Налоговая консультация поможет вам правильно оформить налоги при продаже квартиры и других объектов недвижимости. Мы поможем вам разобраться в вопросах налогообложения доходов, связанных с продажей имущества. Юридическая помощь при налогообложении недвижимости обеспечит правильное заполнение всех документов. Мы предоставляем консультации по налогу на имущество, а также по налоговым вычетам. Обратитесь к нам за помощью, и мы поможем вам избежать налоговых ошибок.
Больше информации на сайте – https://moepravodo.ru
защита интересов в суде, какие нужны документы для возврата налога за лечение ребенка, автострахование курск
анапа юрист бесплатная консультация, билет 7 вопрос 13 пдд, консультация по НДС
Удачи!
PeterSlump
| #
https://telegra.ph/Guide-to-Non-Gamstop-Casino-Sister-Sites1-03-27
1win_peOa
| #
descărca 1win https://1win5010.ru/ .
DavidNaing
| #
Tiger Fortune has some seriously cool visuals. fortune tiger bet
HegitnMut
| #
ООО «Хронос» считается прогрессивным разработчиком медицинской техники, которая используется в клиниках, различных учреждениях, где важна точность, высокое качество обслуживания. Компания работает, начиная с 2009 года и по разным основным направлениям. На сайте https://agsvv.ru вы сможете с ними ознакомиться. Изготовление техники как для медицинских учреждений, так и домашнего применения.
XaxoztHiets
| #
«REDICO» представляет собой международный центр сертификации, а также лицензирования. Компания на профессиональном уровне и с 2011 года выполняет работы в сфере сертификации услуг и продукции. На сайте https://redico.ru/ воспользуйтесь возможностью запросить бесплатный расчет того, во сколько обойдется сертификация продукции.
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-No-Deposit-Bonus-Guide-20231-03-27
mostbet_ivKn
| #
мостбет казино мостбет казино .
online casinos new zealand
| #
online casinos new zealand
A Brief History of Elemental Analysis » Artemis Analytical
mostbet_boKn
| #
mostber http://eisberg.forum24.ru/?1-5-0-00002579-000-0-0-1743260427/ .
DavidNaing
| #
This online casino’s Tiger Fortune is top-notch. tiger fortune
YuwiyEnuts
| #
На сайте https://xn—-7sbjhqn0bhjc0lk.xn--p1ai/ получите юридическую консультацию по различным вопросам. Компания работает как с физическими, так и частными лицами. На все услуги установлены привлекательные расценки, чтобы воспользоваться ими смог каждый. Предприятие работает в этой сфере более 11 лет. В команде трудятся 11 специалистов, которые справятся с задачей независимо от сложности. К вашим услугам досудебный юрист, досудебное урегулирование, кредитный, семейный юрист, решение страховых споров.
PeterSlump
| #
https://telegra.ph/Best-Casinos-Not-on-GamStop-for-Unlimited-Play-03-27
mostbet_klKn
| #
мост бет eisberg.forum24.ru/?1-5-0-00002579-000-0-0-1743260427 .
cumosemorn
| #
Отопление и водоснабжение – компания, которая на предоставлении качественных услуг специализируется. В области монтажа отопительных систем у нас опыт и глубокие знания. Следим за современными технологиями, чтобы вам передовые решения предлагать. Подходим к каждому проекту индивидуально. Ищете монтаж систем отопления? Krasnogorsk.santex-uslugi.ru – портал, где опубликованы отзывы о нас и указаны цены. Применяем оборудование от лучших производителей и надежные материалы. Успешно выполнили множество работ. Готовы комфортный дом для вашей семьи создать.
1win_srMa
| #
1win 1win .
Kennethzof
| #
Экспрессы Мир ставок – это захватывающая сфера, где азарт и аналитика переплетаются в стремлении предсказать исход спортивных событий. Это игра, где интуиция встречается с расчетом, а знание предмета становится ключом к успеху. От футбола до тенниса, от баскетбола до хоккея – ставки на спорт охватывают огромный спектр дисциплин. Каждая игра, каждый матч – это новая возможность проверить свою проницательность и, возможно, выиграть.
mostbet_yfMa
| #
mostbet kg скачать http://mostbet6031.ru/ .
DavidNaing
| #
Tiger Fortune is my lucky game this month! tigrinho demo
generic finasteride 1mg buying
| #
where can i get finasteride without a prescription
JamesSes
| #
Dubayda “Portu Pati” n?dir v? niy? ham? bundan dan?s?r?
https://x.com/IrinaPavlovna84/status/1909960841606070387
Diplomi_wyKt
| #
Заказать диплом любого университета!
Мы можем предложить документы любых учебных заведений, расположенных в любом регионе России.
diplom4you.com/kupit-diplom-vuza-s-vneseniem-v-reestr-bistro-i-nadezhno/
PeterSlump
| #
https://telegra.ph/Explore-New-Non-Gamstop-Casinos-for-Thrilling-Gaming-03-27
can i order prandin for sale
| #
can i get cheap prandin
DavidNaing
| #
Tiger Fortune is my lucky charm at this casino. tiger fortune
dragon money_kwEt
| #
как вывести с драгон мани https://dragon-money27.com/ .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Мишень для распыления ванадия
DavidNaing
| #
Tiger Fortune’s payouts keep me smiling. tiger fortune
1win_pwOa
| #
descărca 1win descărca 1win .
DavidNaing
| #
The free spins in Tiger Fortune are awesome! fortune tiger 777
1win_wgKr
| #
1вин про 1win6043.ru .
Zayebrot
| #
На сайте https://mvpol.ru/ воспользуйтесь возможностью заказать промышленные полы напрямую от производителя. Компания «МВПОЛ» оказывает и такую важную услугу, как ремонт промышленных полов, производство полимерных наливных полов, бетонных, у которых упрочненный верх. Также получится узнать расценки на такие услуги, чтобы заранее спланировать бюджет. Для того чтобы иметь представление о том, как выглядят работы, изучите объекты. На все услуги предоставляются гарантии. Вся информация дается максимально быстро.
PeterSlump
| #
https://telegra.ph/PayPal-Accepted-at-Non-GamStop-Casinos-Available-Online-03-27
dragon money_yysi
| #
баланс драгон мани баланс драгон мани .
mostbet_vymt
| #
mostbet kg скачать https://www.mostbet6033.ru .
DavidNaing
| #
The Tiger Fortune game is so easy to play. tiger fortune
DavidNaing
| #
The animations in Tiger Fortune are incredible. tiger fortune
Sazrzhc
| #
Приобретение документа о высшем образовании через надежную компанию дарит множество достоинств. Данное решение помогает сэкономить время и существенные денежные средства. Впрочем, только на этом выгода не ограничивается, преимуществ значительно больше.Мы изготавливаем дипломы психологов, юристов, экономистов и прочих профессий. Дипломы производятся на подлинных бланках государственного образца. Доступная стоимость сравнительно с большими тратами на обучение и проживание. Заказ диплома об образовании из российского университета станет выгодным шагом.
Заказать диплом о высшем образовании: diplomt-v-samare.ru/kupit-diplom-o-visshem-obrazovanii-zanosim-v-reestr/
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наши товары:
Рнструментальная круглая РїРѕРєРѕРІРєР° 48 РјРј 9Р“2Р¤ ГОСТ 1133-71 РЁРёСЂРѕРєРёР№ выбор круглых РїРѕРєРѕРІРѕРє для профессиональной обработки металла. Рнструментальная круглая РїРѕРєРѕРІРєР° высочайшего качества СЃ разнообразием размеров Рё форм. Повышенная прочность, износостойкость Рё превосходное качество изготовления. Обеспечивает отличное сцепление СЃ материалом Рё эффективную работу оборудования. Покупайте РїСЂСЏРјРѕ сейчас!
Daviddougs
| #
Смотри топ скинов CS2 cs2-open-case.ru/ самые красивые, редкие и желанные облики для оружия. Стиль, эффект и внимание на сервере гарантированы. Обновляем коллекцию каждый день!
GeorgeObjer
| #
Смотри топ скинов CS2 https://case-cs2-open.ru самые красивые, редкие и желанные облики для оружия. Стиль, эффект и внимание на сервере гарантированы. Обновляем коллекцию каждый день!
Williampeaft
| #
Иммортал-дроп CS2 open-case-csgo.ru/ только для избранных. Легендарные скины, высокая ценность, редкие флоаты и престиж на сервере. Пополни свою коллекцию настоящими шедеврами.
PeterSlump
| #
https://telegra.ph/Best-Non-Gamstop-Casinos-UK-No-Deposit-Bonus-Offers-03-27
DavidNaing
| #
I’ve had some great wins on Tiger Fortune recently. jogo do tigrinho demo
1win_wzMa
| #
1win молдова 1win молдова .
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена требуемыми документами, подтверждающими их качество. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы и предоставлять решения под требования вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Лист магниевый AZ-81A – РўРЈ 1714-001-00545484-96 Лента магниевая AZ-81A – РўРЈ 1714-001-00545484-96 является высококачественным материалом, предназначенным для использования РІ различных отраслях. РћРЅР° отличается отличной прочностью Рё высокой РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью, что делает ее идеальным выбором для элементов конструкции Рё РґСЂСѓРіРёС… применений. Р—Р° счет своей легкости Рё гибкости, лента обеспечивает удобство РІ работе. Если РІС‹ ищете надежный материал, купить Лента магниевая AZ-81A – РўРЈ 1714-001-00545484-96 станет правильным решением для вашего проекта. Обеспечьте долговечность Рё эффективность СЃРІРѕРёС… изделий СЃ этой лентой!
Diplomi_rsKt
| #
Приобрести диплом о высшем образовании!
Мы готовы предложить документы любых учебных заведений, которые находятся в любом регионе России.
diplomk-vo-vladivostoke.ru/kupit-diplom-o-visshem-obrazovanii-bistro-i-prosto-2/
BetaklDiz
| #
На сайте https://prometall.shop/ получится выбрать печь, а также воспользоваться полным перечнем услуг, которые связаны с печами Prometall, в том числе, реализацией, проведением монтажных работ. Изучите технические характеристики печей, которые находятся в сетке, камне, получится приобрести и отопительные печи, а также устройства в облицовке. Прямо сейчас вы сможете изучить хиты продаж – те товары, которые пользуются особой популярностью. Каталог товаров регулярно обновляется, чтобы вы приобрели лучшее.
Mazront
| #
Где приобрести диплом по актуальной специальности?
Получаемый диплом со всеми печатями и подписями отвечает запросам и стандартам Министерства образования и науки, неотличим от оригинала – даже со специально предназначенным оборудованием. Не стоит откладывать личные цели на продолжительные годы, реализуйте их с нашей компанией – отправляйте заявку на изготовление документа уже сегодня! Получить диплом о среднем специальном образовании – не проблема! myweektour.ru/gotovyiy-diplom-s-nuzhnyimi-rekvizitami-polnyiy-komplekt-dokumentov
PeterSlump
| #
https://telegra.ph/Free-Spins-No-Deposit-Casinos-Not-on-GamStop-03-27
Mazrbji
| #
Где приобрести диплом по необходимой специальности?
Мы предлагаем документы об окончании любых университетов России. Документы производятся на подлинных бланках государственного образца. Рєrealestate.kctech.com.np/profile/sonjacalwell7
BetaklDiz
| #
На сайте https://prometall.shop/ получится выбрать печь, а также воспользоваться полным перечнем услуг, которые связаны с печами Prometall, в том числе, реализацией, проведением монтажных работ. Изучите технические характеристики печей, которые находятся в сетке, камне, получится приобрести и отопительные печи, а также устройства в облицовке. Прямо сейчас вы сможете изучить хиты продаж – те товары, которые пользуются особой популярностью. Каталог товаров регулярно обновляется, чтобы вы приобрели лучшее.
Лев каз
| #
Лев Казино – это ваше место для захватывающих азартных игр и шанс стать победителем. Здесь каждый найдет что-то по своему вкусу: от классических слотов до захватывающих игр с живыми дилерами. Присоединяйтесь к нам и начните выигрывать прямо сейчас, наслаждаясь качественным игровым процессом.
Почему именно https://levclub-play.site/? Все ваши данные защищены, а выплаты всегда своевременные и без скрытых комиссий. Регулярные акции, турниры и бонусы для новых и постоянных игроков делают игру еще более увлекательной. Мы предлагаем высокие шансы на победу и бонусы, которые помогут вам увеличить свои выигрыши.
Более 500 игр от известных провайдеров.
Еженедельные бонусы и специальные предложения для наших игроков.
Мгновенные депозиты и быстрые выплаты.
Увлекательные турниры с крупными призовыми фондами.
Начните играть в Лев Казино и наслаждайтесь выгодными предложениями и большими шансами на успех.
Sazrchl
| #
Заказать документ университета можно в нашем сервисе. Мы оказываем услуги по продаже документов об окончании любых ВУЗов Российской Федерации. Вы сможете получить диплом по любым специальностям, любого года выпуска, в том числе документы образца СССР. Гарантируем, что при проверке документа работодателями, никаких подозрений не возникнет. diplomskiy.com/kupit-diplom-s-zaneseniem-v-reestr-bez-problem-3/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Плоская мишень Sio для распыления
PeterSlump
| #
https://telegra.ph/Non-GamStop-Casinos-Bonus-Offers-and-Benefits-Explained1-03-27
Uazrgbw
| #
Доброго времени суток!
Для многих людей, приобрести диплом университета – это необходимость, шанс получить выгодную работу. Впрочем для кого-то – это желание не терять массу времени на учебу в ВУЗе. С какой бы целью вам это не потребовалось, наша фирма готова помочь. Максимально быстро, профессионально и по разумной цене сделаем документ любого года выпуска на государственных бланках с реальными подписями и печатями.
Ключевая причина, почему многие люди покупают документы, – желание занять хорошую должность. К примеру, способности и опыт позволяют человеку устроиться на работу, однако подтверждения квалификации нет. В том случае если работодателю важно присутствие “корочки”, риск потерять место работы довольно высокий.
Купить документ о получении высшего образования можно у нас в Москве. Мы оказываем услуги по производству и продаже документов об окончании любых университетов РФ. Вы сможете получить диплом по любой специальности, включая документы СССР. Гарантируем, что при проверке документа работодателями, подозрений не появится.
Ситуаций, которые вынуждают заказать диплом немало. Кому-то прямо сейчас требуется работа, а значит, нужно произвести особое впечатление на начальство при собеседовании. Некоторые задумали попасть в престижную компанию, для того, чтобы повысить свой статус в социуме и в будущем начать свой бизнес. Чтобы не терять массу времени, а сразу начать эффективную карьеру, применяя имеющиеся навыки, можно заказать диплом прямо в онлайне. Вы станете полезным в социуме, обретете финансовую стабильность в максимально короткие сроки- купить диплом о высшем образовании
Davidedges
| #
Вся информация о теплицах, начиная от строительства и фундамента и заканчивая правильным освещением и отоплением. А также выращивание в теплицах культур, подбор сортов и удобрений https://portalteplic.ru/
PeterSlump
| #
https://telegra.ph/Top-Non-GamStop-Curacao-Casino-Sites-for-Players-03-27
pin up azerbaycan_kuOa
| #
pin up az?rbaycan pin up az?rbaycan .
Cazrsgo
| #
Добрый день!
Мы изготавливаем дипломы любой профессии по приятным ценам. Стоимость будет зависеть от той или иной специальности, года получения и образовательного учреждения: rdiplomans.com/
1win_ecMa
| #
portofele electronice casino 1win5012.ru .
DavidNaing
| #
This online casino’s Tiger Fortune is a gem. tiger fortune
Peterjef
| #
СМС активации оптом
internetCah
| #
провайдеры по адресу дома
domashij-internet-msk002.ru
проверить провайдера по адресу
1win_njPi
| #
1wi http://sebezh.borda.ru/?1-10-0-00000117-000-0-0-1743052058/ .
pin up azerbaycan_ogen
| #
pinup azerbaycan pinup azerbaycan .
1win_gnMi
| #
1win https://www.1win6007.ru .
Sazrasl
| #
Мы изготавливаем дипломы любой профессии по приятным ценам.
Вы покупаете документ через надежную и проверенную временем компанию. Приобрести диплом о высшем образовании– http://cart-gurus.com/employer/diplomy-grup-24/ – cart-gurus.com/employer/diplomy-grup-24
PeterSlump
| #
https://telegra.ph/Discover-Non-Gamstop-Casinos-for-Unrestricted-Gaming-03-27
1win_euPi
| #
1vin казино http://sebezh.borda.ru/?1-10-0-00000117-000-0-0-1743052058/ .
Sazrdji
| #
Мы готовы предложить дипломы любой профессии по выгодным ценам.– diplomh-40.ru/schnell-kupit-diplom-magistra-s-provodkoj-bez-lishnix-zatrat/
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наша продукция:
Пружина из драгоценных металлов палладиевая 1000х10х0.2 мм ПДСР-20 ТУ Выберите идеальные пружины из золота, серебра или платины, чтобы дополнить ваше украшение. Найдите пружины с изысканным дизайном и прочностью. Они предлагают широкий выбор для того, чтобы подчеркнуть уникальность вашего украшения.
mostbet_lbSa
| #
mostbet apk скачать https://mostbet6011.ru .
Gunuktpen
| #
Популярный сервис «Zoomy» предлагает воспользоваться такой популярной услугой, как аренда мотоциклов в Пхукете, Краби, Банг-Ламунг и других районах Таиланда. На сайте https://zoomy.travel уточните то, в каких районах Таиланда вы сможете взять мотоцикл в аренду.
1win_fzPi
| #
1вин http://www.sebezh.borda.ru/?1-10-0-00000117-000-0-0-1743052058 .
PeterSlump
| #
https://telegra.ph/New-Casinos-Without-GamStop-for-Unrestricted-Play-03-27
Williamcoice
| #
Быстрый и удобный фундаментные работы калькулятор рассчитайте стоимость и объем работ за пару минут. Онлайн-калькулятор поможет спланировать бюджет, сравнить варианты и избежать лишних затрат.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
“ерамическая мишень CeO2, плоская форма
mamonovskoe
| #
Мамоновское кладбище mamonovskoe.ru/ справочная информация, адрес, график работы, участки, ритуальные услуги и памятники. Всё, что нужно знать, собрано на одном сайте.
Cazrgxp
| #
Где приобрести диплом специалиста?
Мы готовы предложить дипломы любой профессии по приятным ценам. Важно, чтобы документы были доступны для подавляющей массы граждан. Приобрести диплом о высшем образовании diplomk-v-krasnodare.ru/kupit-diplom-s-zaneseniem-18/
Williamjulty
| #
новости фк краснодар на сегодня krasnodar-news35.ru/
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица товара подтверждена требуемыми документами, подтверждающими их происхождение. Дружелюбная помощь – наш стандарт – мы на связи, чтобы разрешать ваши вопросы а также адаптировать решения под специфику вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
Наши товары:
Молибден ЦСДМ Молибден ЦСДМ – это высококачественный РїСЂРѕРґСѓРєС‚, который используется РІ различных отраслях, включая металлургию Рё производство сплавов. Благодаря СЃРІРѕРёРј уникальным свойствам, таким как высокая прочность Рё термостойкость, молибден идеально РїРѕРґС…РѕРґРёС‚ для создания изделий, работающих РІ экстремальных условиях. Ртот металл устойчив Рє РєРѕСЂСЂРѕР·РёРё Рё способен выдерживать высокие температуры, что делает его незаменимым РІ современных технологиях.Если РІС‹ ищете надежный Рё высококачественный материал, то купить Молибден ЦСДМ – ваше лучшее решение. Продукция отвечает всем международным стандартам Рё прошла необходимые проверки. РќРµ упустите возможность обеспечить СЃРІРѕР№ проект качественными материалами!
Williamexect
| #
Лизинг — долгосрочная аренда с правом выкупа. Собственником остаётся лизингодатель, что снижает риски для клиента. Платежи часто ниже кредитных, а НДС включается в сумму, что выгодно для бизнеса. Лизинговые платежи учитываются как расходы, уменьшая налог на прибыль: Бин-Лизинг
Peterjef
| #
Register WhatsApp number for free
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Online-Casino-Lightning-Roulette-Guide5-03-27
Toojddax
| #
On the site https://wallpapers4screen.com/ you can download wallpapers for desktop free. High Quality HD pictures wallpapers. High quality pictures and wallpapers! Choose one of many categories and download for free! Or check out the TOP downloadable images! You will definitely like them.
PirojokLog
| #
dark web drug marketplace darkmarket list
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Керамическая мишень для распыления оксида магния
DavidNaing
| #
Tiger Fortune’s rewards are worth every spin. jogo do tigrinho demo
DavidNaing
| #
I can’t stop playing Tiger Fortune – it’s too good! fortune tiger bet
pin up azerbaycan_dnOa
| #
pinup azerbaycan pinup azerbaycan .
rawegbup
| #
МосСтройАльянс специалисты компании готовы решить задачу любой сложности. Мы на осуществлении фасадных, уборочных и кровельных работ специализируемся. Предлагаем самые выгодные цены. Вы результатом останетесь довольны. Ищете монтаж крыши цены в москве? Mos-stroi-alians.ru – здесь можете ознакомиться с отзывами наших клиентов. Для работы применяем исключительно материалы высшего качества. Ценим ваше время. За соблюдение сроков мы ответственность несем. Позвоните нам по телефону на портале. Проконсультируем и ответим на интересующие вас вопросы.
PeterSlump
| #
https://telegra.ph/Best-Non-Gamstop-Casino-Sites-for-UK-Players-03-27
how can i get generic mestinon price
| #
how to get cheap mestinon pill
1win_xgMi
| #
1вин официальный сайт http://1win6007.ru .
how can i get generic amoxil without insurance
| #
how to take 875 mg of amoxil
1win_tjSr
| #
1win moldova https://1win5011.ru .
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наша продукция:
Лента РёР· прецизионных сплавов для СѓРїСЂСѓРіРёС… элементов 0.28×47 РјРј РЎРџ22 ГОСТ 14117-85 Купите ленту РёР· прецизионных сплавов для СѓРїСЂСѓРіРёС… элементов РїРѕ выгодной цене РЅР° Редметсплав.СЂС„. Высокая прочность, устойчивость Рє РєРѕСЂСЂРѕР·РёРё Рё отличная эластичность делают этот материал идеальным для различных отраслей. Предлагаем широкий ассортимент продукции Рё РїРѕРґСЂРѕР±РЅСѓСЋ информацию для правильного выбора.
mostbet_isSa
| #
mostbet kg скачать mostbet6011.ru .
PeterSlump
| #
https://telegra.ph/Exploring-Non-Gamstop-Casino-Fruity-Slots-Adventures-03-27
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Вращающаяся мишень для распыления вольфрама
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
поставляемая продукция:
Кольцо РёР· драгоценных металлов палладиевое 9С…8С…0.1 РјРј РџРґР82-18 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их качество. Дружелюбная помощь – наш стандарт – мы на связи, чтобы ответить на ваши вопросы и предоставлять решения под специфику вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наша продукция:
Пруток вольфрамовый РР’Рў-15 Пруток вольфрамовый РР’Рў-15 – это высококачественный материал, используемый РІ различных отраслях, включая авиацию Рё производство электроники. Его уникальные характеристики, такие как высокая температура плавления (3422В°C) Рё отличная коррозионная стойкость, делают его идеальным выбором для demanding приложений. Пруток обладает хорошей механической прочностью Рё легко поддается обработке. Если РІС‹ ищете надежный Рё долговечный материал, стоит рассмотреть возможность купить Пруток вольфрамовый РР’Рў-15. РћРЅ станет отличным дополнением Рє вашим проектам Рё обеспечит стабильную работу РІ экстремальных условиях.
PeterSlump
| #
https://telegra.ph/No-Gamstop-Online-Casinos-for-Unlimited-Gaming-03-27
pin up azerbaycan_lfOa
| #
pinup azerbaycan pinup azerbaycan .
czmicro
| #
микронаушник аренда микронаушник купить
1win_sxMi
| #
скачать 1win с официального сайта http://1win6007.ru .
ivonoSauro
| #
Ресурс hm.mnemosyne.ru выбрать ремень в подарок любимым людям предлагает. Мастера, которые изделия создают, творческим потенциалом отличаются. Каждая модель разрабатывается с учетом модных трендов. Мы к покупателям всегда прислушиваемся. При возможностях всегда идем на встречу. https://hm.mnemosyne.ru – тут сможете узнать, какой ремень в подарок руководителю, шефу или коллеге преподнести. Для изделий отбирается лучшая кожа. Оцените неповторимость ручной работы. Сделайте уже сейчас заказ на нашем портале. Пусть ваш стиль новыми красками засияет!
PeterSlump
| #
https://telegra.ph/Top-Non-Gamstop-Casinos-for-UK-Players-2023-03-27
jasdam
| #
микронаушник Чехия http://jasdam.cz
DavidNaing
| #
I love the jungle energy of Tiger Fortune. tiger fortune
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их качество. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы а также находить ответы под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Порошок кобальтовый РђРўР”-14 Порошок кобальтовый РђРўР”-14 – это высококачественный материал, применяемый РІ различных отраслях, включая металлургию Рё производство суперсплавов. Его отличает высокая устойчивость Рє РєРѕСЂСЂРѕР·РёРё Рё износостойкость, что делает его незаменимым для создания деталей, работающих РІ экстремальных условиях. Если РІС‹ стремитесь Рє долговечности Рё надежности СЃРІРѕРёС… продуктов, РїРѕРєСѓРїРєР° Порошка кобальтового РђРўР”-14 станет отличным решением. Ртот порошок идеально РїРѕРґС…РѕРґРёС‚ для создания металлических композитов Рё может использоваться РІ разных технологиях нанесения. РќРµ упустите возможность купить Порошок кобальтовый РђРўР”-14 РїСЂСЏРјРѕ сейчас Рё повысить качество своей работы!
mostbet_otSa
| #
mosbet https://mostbet6011.ru/ .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
поставляемая продукция:
Латуное наружное стопорное пружинное кольцо M33 30.5С…1.5 РјРј ЛС DIN 471 Латунные наружные стопорные кольца РїРѕ DIN – широкий выбор высококачественных крепежных изделий РёР· латуни для промышленных Рё строительных применений. Большой ассортимент размеров. Высокая надежность Рё долговечность. Заказывайте РїСЂСЏРјРѕ сейчас РЅР° сайте Редметсплав.
DavidNaing
| #
Tiger Fortune makes every win feel huge. fortune tiger bet
PeterSlump
| #
https://telegra.ph/Casino-Options-Without-Gamstop-Restrictions-03-27
internetCah
| #
подключение интернета москва
domashij-internet-msk003.ru
лучший интернет провайдер москва
Willieavamy
| #
Online casinos should speed—wait off! doradobet
pin up azerbaycan_soen
| #
pinup azerbaycan pinup azerbaycan .
1win_bfpt
| #
1win вход 1win6008.ru .
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их качество. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы а также находить ответы под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
Наши товары:
Лист титановый Р’Рў32 Лента титановая Р’Рў32 сочетает РІ себе высокую прочность Рё легкость, что делает ее идеальным выбором для различных промышленных Рё строительных приложений. Ртот материал устойчив Рє РєРѕСЂСЂРѕР·РёРё Рё выдерживает экстремальные температуры, что обеспечивает длительный СЃСЂРѕРє службы. Благодаря СЃРІРѕРёРј уникальным свойствам, лента широко используется РІ авиации, машиностроении Рё медицине.Если РІС‹ ищете надежное решение для вашего проекта, РІС‹ можете купить Лента титановая Р’Рў32 РїСЂСЏРјРѕ сейчас. Рто оптимальный вариант для тех, кто ценит качество Рё эффективность РІ работе.
1win_jmSr
| #
portofele electronice casino 1win5011.ru .
PeterSlump
| #
https://telegra.ph/Guide-to-UK-Non-GamStop-Casinos-for-Players3-03-27
cost gabapentin without prescription
| #
where to buy cheap gabapentin prices
DavidNaing
| #
I won 50 free spins on Tiger Fortune yesterday! tiger fortune
sozavAceno
| #
Пороги автоматического типа являются идеальным вариантом при условии, что осуществить монтажные работы стационарного не удастся либо нежелательно. В данный момент можно изучить каталог всех вариантов, которые предлагает предприятие «SMARTПОРОГ», что позволит подобрать решение, которое определенно подойдет. https://smartporog.ru/ – на портале ознакомьтесь с достоинствами, на которые вы сможете рассчитывать при установке порогов внутри помещения. Такие конструкции потребуются с той целью, чтобы в помещение не попадали солнечные лучи, посторонние запахи. Считается отличным вариантом для организации ванной комнаты, например.
1win_rmMr
| #
1win sportsbook https://1win14.com.ng/ .
buy cheap macrobid for sale
| #
buy cheap macrobid without prescription
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-No-Deposit-Bonus-Offers-Explained7-03-27
DavidNaing
| #
I love how Tiger Fortune mixes luck and fun. jogo do tigrinho demo
DonayrIngek
| #
На сайте https://vezuviy.shop/ вы сможете приобрести чугунные, стальные банные печи, нержавеющие, а также в облицовке, печи-камины, отопительные, каминные топки, дымоходы, порталы и многое другое. Вся продукция от производителя, а потому наценки очень маленькие. Такая техника считается идеальной не только с целью обогрева помещения, но и оформления пространства. А самое главное, что она отличается долгим сроком службы, надежностью из-за того, что в работе использовались высокотехнологичные, проверенные материалы.
PeterSlump
| #
https://telegra.ph/Best-Casinos-Not-on-Gamstop-with-No-Deposit-Required-03-27
Sazrqaj
| #
Покупка документа о высшем образовании через надежную фирму дарит ряд достоинств для покупателя. Это решение позволяет сэкономить как личное время, так и серьезные денежные средства. Однако, только на этом выгода не ограничивается, плюсов гораздо больше.Мы предлагаем дипломы любой профессии. Дипломы изготавливаются на оригинальных бланках. Доступная цена по сравнению с серьезными тратами на обучение и проживание в чужом городе. Покупка диплома о высшем образовании из российского института будет выгодным шагом.
Приобрести диплом о высшем образовании: diplom4you.com/kupit-diplom-o-visshem-obrazovanii-reestr-bistro-i-nadezhno-2/
BofadnyTum
| #
Компания «Ковка Винтаж» в течение длительного времени оказывает свои услуги и предлагает создать изысканные, завораживающие кованые изделия, которые украсят придомовую территорию, беседку, террасу, а также частный дом. На сайте https://kovka-vintazh.ru/ ознакомьтесь с полным ассортиментом изделий и работами компетентных и выдающихся мастеров.
Juliusplomy
| #
Здравствуйте!
Легализация документов поможет вам пройти процесс официального признания документов в других странах. Помощь в приватизации недвижимости требует знания всех юридических аспектов этого процесса, и юрист поможет вам с оформлением. Составление брачного договора обеспечит защиту ваших интересов при вступлении в брак, особенно в вопросах раздела имущества. Юридическая помощь при лишении прав поможет вам восстановить водительские права, если они были лишены незаконно. Оформление опеки требует соблюдения всех юридических норм, чтобы обеспечить интересы ребенка и сторон, оформляющих опеку.
Больше информации на сайте – https://zashitapravi.ru
оформление наследства, какие документы нужны для нотариуса для оформления доверенности на продажу квартиры, оформление визы в китай в санкт петербурге
увольнение по статье за прогул, возврат за кружки детей налога, юридическая помощь пенсионерам
Удачи!
PeterSlump
| #
https://telegra.ph/Explore-Online-Casinos-Not-on-Gamstop-in-2023-03-27
Cazruaz
| #
Мы можем предложить дипломы психологов, юристов, экономистов и любых других профессий по приятным тарифам. Купить свидетельство о браке — kyc-diplom.com/svidetelstvo-o-brake.html
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние возможности.
Наши товары:
Мишень Диспрозия 3N, плоская форма”
Cazrnvh
| #
Купить диплом любого университета можем помочь. Купить диплом специалиста в Ярославле – diplomybox.com/kupit-diplom-spetsialista-v-yaroslavle
https://padlet.com/
| #
https://padlet.com/
A Brief History of Elemental Analysis » Artemis Analytical
Mazruoz
| #
Где приобрести диплом специалиста?
Полученный диплом с приложением отвечает стандартам Министерства образования и науки, никто не отличит его от оригинала – даже со специально предназначенным оборудованием. Не откладывайте свои мечты и задачи на пять лет, реализуйте их с нами – отправьте заявку на диплом сегодня! Диплом о среднем образовании – не проблема! domovou.3nx.ru/viewtopic.phpp=14248#14248
PeterSlump
| #
https://telegra.ph/No-Deposit-Non-Gamstop-Casinos-for-Players-03-27
DavidNaing
| #
I love the jungle vibe of Tiger Fortune – so immersive! fortune tiger bet
Mazrytv
| #
Где приобрести диплом по актуальной специальности?
Мы предлагаем документы об окончании любых университетов России. Документы изготавливаются на фирменных бланках государственного образца. Рєforum.artinvestment.ru/member.phpu=1807451
RobertRor
| #
Здравствуйте!
Оформление дарственной на авто требует внимательности при оформлении документов для передачи прав на транспортное средство. Регистрация договора аренды поможет вам правильно оформить договор аренды недвижимости. Проверка контрагента поможет вам избежать рисков при заключении договоров с новыми партнерами. Правовая помощь по трудовому спору обеспечит защиту ваших интересов при возникновении трудовых конфликтов с работодателем. Налоговая проверка — это процесс, в котором вам может понадобиться помощь юриста для защиты ваших прав.
Больше информации на сайте – https://yslugiyurista.ru
юрист по ДТП, налог при продаже квартиры когда не надо платить, бесплатный юрист консультация онлайн бесплатно без регистрации
договорное регулирование трудовых отношений это, юридическая помощь инвалидам 3 группы, возврат налогов
Удачи!
Willieavamy
| #
I lost time online—swift pass! minas
1win_nkKr
| #
1win партнерская программа вход [url=1win6043.ru]1win6043.ru[/url] .
PeterSlump
| #
https://telegra.ph/PayPal-Non-Gamstop-Casino-Sites-for-Safe-Betting-03-27
Sazrozw
| #
Приобрести документ института можно в нашем сервисе. Мы оказываем услуги по продаже документов об окончании любых ВУЗов России. Вы получите необходимый диплом по любым специальностям, включая документы СССР. Даем 100% гарантию, что в случае проверки документа работодателем, подозрений не появится. asxdiplommy.com/kupit-diplom-s-zaneseniem-v-reestr-po-nizkoj-tsene-4/
WilliamDix
| #
Мы изготавливаем дипломы любой профессии по приятным ценам. Дипломы изготавливаются на подлинных бланках государственного образца Приобрести диплом об образовании dip-lom-rus.ru
DavidNaing
| #
The casino’s Tiger Fortune slot has great winning potential. tiger fortune
Michaelfar
| #
Привет всем!
Юрист по семейным делам поможет вам решить вопросы, связанные с разводами, разделом имущества, алиментами и другими семейными спорами. Мы предоставляем консультации по всем аспектам семейного права и обеспечиваем защиту ваших интересов в суде. Если вам необходима помощь при оформлении развода или соглашений по алиментам, наши юристы окажут необходимую поддержку. Юридическая помощь при семейных спорах обеспечит вам уверенность в правомерности всех действий. Обратитесь за консультацией, чтобы решить семейные вопросы с минимальными последствиями.
Больше информации на сайте – https://besturisty.ru/
регистрация права собственности на квартиру, пошлина за регистрацию договора аренды в росреестре, сбербанк каско автострахование
номер и серия свидетельства о регистрации ип, где можно открыть наследственное дело, как вернуть права
Удачи!
Lazrwtf
| #
Где заказать диплом по необходимой специальности?
Приобрести диплом института по невысокой стоимости вы можете, обращаясь к проверенной специализированной компании.: 10000diplomov.ru
Davidtix
| #
Привет всем!
Правовая помощь по трудовым вопросам необходима для того, чтобы защитить ваши интересы на рабочем месте. Мы поможем вам составить трудовой договор, оформить увольнение по статье и защитить ваши права в трудовых спорах. Юридическая помощь при трудовых спорах поможет вам избежать нарушений ваших прав работодателем. Мы также обеспечиваем помощь в составлении исковых заявлений и претензий. Обратитесь за консультацией по трудовому праву, и мы поможем вам решить вашу проблему.
Больше информации на сайте – https://voproslaw.ru/
юридическое сопровождение бизнеса, налог на имущество статья налогового кодекса, юрист по медицинским вопросам новосибирск
как киргизу получить гражданство рф, юридическая помощь при разводе и разделе имущества, оформление наследства
Удачи!
get prothiaden pills
| #
cost of cheap prothiaden for sale
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наши товары:
Титановый шестигранник 85 РјРј Р’Рў14 ГОСТ 8560-78 Познакомьтесь СЃ широким ассортиментом титановых шестигранников РѕС‚ Редметсплав.СЂС„, предлагающих надежность, высокую прочность Рё устойчивость Рє экстремальным условиям. Рспользуйте титановые шестигранники для авиационных систем, медицинского оборудования, химических процессов Рё машиностроения. Подберите оптимальный размер Рё конфигурацию для вашего проекта!
PeterSlump
| #
https://telegra.ph/Best-Non-UK-Casinos-Not-on-GamStop-in-2023-03-27
can i get generic zebeta without rx
| #
where can i buy generic zebeta no prescription
mostbet_kkmt
| #
mostbet chrono mostbet6033.ru .
bet77
| #
Aproveite a oferta exclusiva do bet77 para novos usuários e receba 100$ de
bônus ao se registrar! Este bônus de boas-vindas permite que você experimente uma vasta
gama de jogos de cassino online sem precisar gastar imediatamente.
Com o bônus de 100$, você poderá explorar jogos como roleta, blackjack, caça-níqueis e muito mais, aumentando suas chances de vitória desde o primeiro minuto.
Não perca essa chance única de começar com um valor significativo
– cadastre-se agora!
internetCah
| #
интернет по адресу нижний новгород
domashij-internet-nizhnij-novgorod001.ru
недорогой интернет нижний новгород
DavidNaing
| #
Tiger Fortune’s theme is a total vibe. fortune tiger 777
DavidNaing
| #
Tiger Fortune’s payouts are always a surprise. fortune tiger 777
mazusTor
| #
Alkomarket-dubai.ru предлагает коллекцию алкогольных напитков на разный вкус и кошелек. Наш основной приоритет – качество продукции, мы о здоровье своих клиентов заботимся. Закажите на дом уже сейчас алкоголь с доставкой. Наслаждайтесь удобством! Ищете alcohol delivery dubai? Alkomarket-dubai.ru – тут большой выбор вина, скотча, водки и виски, а также иных премиальных напитков от популярных мировых брендов. Бережно относимся к покупателям, заботясь об их потребностях. Поможем для вас и на подарок близким выбрать достойные варианты.
PeterSlump
| #
https://telegra.ph/Guide-to-Non-Gamstop-Casinos-and-Their-Benefits1-03-27
yaxiceShoof
| #
Истринская мануфактура межкомнатные двери на заказ производит. При безупречном качестве мебель компании доступна по привлекательным ценам. Мы творчески подходим к каждому заказу. Делаем красивую продукцию и ею гордимся. http://istradoors.ru – тут отзывы о нашей компании предоставлены. Кроме этого на портале часто задаваемые вопросы и ответы на них перечислены. С нами работают ведущие архитектурные и дизайнерские мастерские. Мы готовы любую задачу осуществить. Подробную информацию о продукте, а также об условиях заказа вы можете у менеджеров уточнить.
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена требуемыми документами, подтверждающими их происхождение. Превосходное обслуживание – наш стандарт – мы на связи, чтобы разрешать ваши вопросы и адаптировать решения под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
РљСЂСѓРі магниевый AZ91 – CSA HG.10 Фольга магниевая AZ91 – CSA HG.10 представляет СЃРѕР±РѕР№ высококачественный материал, подходящий для множества применений РІ различных отраслях. РћРЅР° отлична своей легкостью Рё прочностью, что делает ее идеальным выбором для авиации, автомобилестроения Рё РґСЂСѓРіРёС… технологических процессов. Уникальные свойства магния обеспечивают хорошую РєРѕСЂСЂРѕР·РёРѕРЅРЅСѓСЋ стойкость Рё отличную обрабатываемость. Если вам необходимо сочетание легкости Рё прочности, фольга магниевая AZ91 – CSA HG.10 станет оптимальным решением. РќРµ упустите возможность купить Фольга магниевая AZ91 – CSA HG.10 Рё улучшить эффективность ваших проектов.
Davidedges
| #
Сайт строительной тематики с полезными и актуальными статьями о ремонте, строительстве, дизайне. Также полезные советы и лайфхаки по стройке https://geekometr.ru/
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-Bonus-Offers-and-Benefits-03-27
DavidNaing
| #
Tiger Fortune’s bonuses are wild and exciting. fortune tiger demo gratis
DavidNaing
| #
Tiger Fortune brings the jungle to life! fortune tiger demo gratis
1win_gupt
| #
1win букмекер http://1win6008.ru .
Sazrtjy
| #
Заказать диплом о высшем образовании!
Заказать диплом института по выгодной цене возможно, обращаясь к надежной специализированной компании. Быстро заказать диплом о высшем образовании: diplomh-40.ru/ofitsialnij-diplom-s-reestrom-vozmozhno-li-kupit
Sazrtae
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и других профессий по выгодным тарифам.– diplomt-v-samare.ru/schnell-kupit-diplom-magistra-s-provodkoj-bez-lishnix-zatrat/
1win_kxKr
| #
1win сайт https://1win6043.ru .
PeterSlump
| #
https://telegra.ph/Understanding-Non-GamStop-Casinos-for-Players-03-27
Willieavamy
| #
I won $1400 on a slot—hype high! plinko
Willieavamy
| #
I love the poker range online—new always! plinko
1win_rgPt
| #
1 цшт http://1win6009.ru/ .
1win_gqMr
| #
1win website 1win website .
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наши товары:
Труба из драгоценных металлов платиновая 150х7х1 мм ПлПд95-5 ТУ Гарантированное качество и изысканный дизайн – купите драгоценные трубы для ювелирных украшений, машиностроения и архитектуры. Широкий выбор и высокое качество! Доставка по России.
Jarioragy
| #
Где приобрести диплом по нужной специальности?
Наша компания предлагаетбыстро и выгодно купить диплом, который выполняется на бланке ГОЗНАКа и заверен мокрыми печатями, водяными знаками, подписями должностных лиц. Документ пройдет любые проверки, даже с использованием специфических приборов. Достигайте своих целей быстро с нашим сервисом. Купить диплом о высшем образовании! 4division.ru/viewtopic.php?f=13&t=10035
DavidNaing
| #
The graphics in Tiger Fortune are absolutely stunning. tigrinho demo
Zohagdjub
| #
На сайте https://ooomila.com изучите каталог такого оборудования, которое предназначено для организации животноводческих комплексов. Также вы сможете почитать и содержательную информацию о компании и ее достижениях. В каталоге вы найдете следующее оборудование: технологические линии, весовое оборудование, резиновые маты, крематоры и многое другое. На технику, которая находится в разделе, действуют гарантии. Несколько сотен единиц оборудования всегда находятся на складе, можно приобрести под заказ.
PeterSlump
| #
https://telegra.ph/Discover-Non-Gamstop-Online-Casinos-for-Safe-Play-03-27
WomicGut
| #
На сайте https://mvpol.ru/ воспользуйтесь возможностью заказать промышленные полы напрямую от производителя. Компания «МВПОЛ» оказывает и такую важную услугу, как ремонт промышленных полов, производство полимерных наливных полов, бетонных, у которых упрочненный верх. Также получится узнать расценки на такие услуги, чтобы заранее спланировать бюджет. Для того чтобы иметь представление о том, как выглядят работы, изучите объекты. На все услуги предоставляются гарантии. Вся информация дается максимально быстро.
WilliamVoink
| #
Ищете безопасный VPN? Попробуйте https://amneziavpn.org — open-source решение для анонимности и свободы в интернете. Полный контроль над соединением и личными данными.
Константин
| #
Falschgeld kaufen Verkauf Euro https://www.google.com.tn/url?q=https://falschgeldkaufen.org
Josephlor
| #
Обеспечь анонимность с amnezia vpn. Защита трафика, собственный сервер, простая установка и отсутствие слежки. Отличный выбор для тех, кто ценит свободу в интернете.
1win_msEa
| #
1win войти 1win войти .
MartinKig
| #
nanosluchatka mikrosluchatko-cena.cz/
mostbet_ijmt
| #
mostbet https://mostbet6033.ru/ .
how can i get generic deltasone
| #
where to buy generic deltasone without dr prescription
PeterSlump
| #
https://telegra.ph/No-Deposit-Bonuses-at-Non-Gamstop-Casinos-Explained8-03-27
DavidNaing
| #
The animations in Tiger Fortune are incredible. fortune tiger demo
how to get cheap prograf tablets
| #
prograf 0.5 mg capsules
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их происхождение. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы и адаптировать решения под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
Фольга магниевая ISO-MgAl2Mn – ISO 16220 РўСЂСѓР±Р° магниевая ISO-MgAl2Mn – ISO 16220 представляет СЃРѕР±РѕР№ современный материал, обеспечивающий высокую прочность Рё легкость конструкций. РћРЅР° идеально РїРѕРґС…РѕРґРёС‚ для применения РІ авиации, автомобилестроении Рё РґСЂСѓРіРёС… отраслях, требующих надежности Рё долговечности. Каждая труба РїСЂРѕС…РѕРґРёС‚ строгий контроль качества, что гарантирует ей долговечность РІ эксплуатации. Приобретая РўСЂСѓР±Р° магниевая ISO-MgAl2Mn – ISO 16220, РІС‹ получаете РЅРµ только высококачественный РїСЂРѕРґСѓРєС‚, РЅРѕ Рё отличное соотношение цены Рё качества. РќРµ упустите возможность купить РўСЂСѓР±Р° магниевая ISO-MgAl2Mn – ISO 16220 Рё улучшить СЃРІРѕРё проекты уже сегодня!
Jariorqts
| #
Где заказать диплом специалиста?
Мы предлагаем выгодно и быстро заказать диплом, который выполнен на бланке ГОЗНАКа и заверен печатями, водяными знаками, подписями. Наш документ пройдет любые проверки, даже при использовании специального оборудования. Достигайте свои цели быстро и просто с нашей компанией.
Приобрести диплом любого университета diplomus-spb.ru/kupit-diplom-v-novosibirske-19/
1win_teKr
| #
1win ваучер 1win6043.ru .
PeterSlump
| #
https://telegra.ph/Non-Gamstop-UK-Casino-Sites-for-Players-in-2023-03-27
ZujizelPaw
| #
Популярная компания «Herbs & Flowers» предлагает огромный выбор искусственных цветов, которые идеально подходят как для украшения интерьера дома, так и офиса. В каталоге сайта https://herbsandflowers.ru/ вы найдете вертикальное озеленение, ампульные растения, а также деревья, ветви и многое другое.
DavidNaing
| #
Tiger Fortune’s jungle theme is so well done. tigrinho demo
pin up azerbaycan_ioMl
| #
pin up az?rbaycan pin up az?rbaycan .
internetCah
| #
подключить домашний интернет нижний новгород
domashij-internet-nizhnij-novgorod002.ru
домашний интернет подключить нижний новгород
PeterSlump
| #
https://telegra.ph/Best-Non-Gamstop-Casinos-in-the-UK-for-20236-03-27
DavidNaing
| #
Tiger Fortune’s visuals are a feast for the eyes. fortune tiger demo
Willieavamy
| #
Online casinos should curb—care play! plinko
Willieavamy
| #
Online casinos are great for practice—nice! Vai de Bet
Willieavamy
| #
I wish online cut bets—low fun! minas
DavidNaing
| #
The tiger-themed bonuses in Tiger Fortune rock! fortune tiger demo gratis
sklad_eosn
| #
склад для индивидуального хранения вещей склад для индивидуального хранения вещей .
PeterSlump
| #
https://telegra.ph/Exploring-UK-Casinos-Without-Gamstop-Restrictions-03-27
mostbet_plmt
| #
mostbet kg скачать на андроид http://mostbet6033.ru .
1win_kkPt
| #
1 вин 1 вин .
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casino-Review-Features-and-Insights1-03-27
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Плоская мишень Cofeb
onebra
| #
Aposte com 100$ de Bônus no onebra – Cadastre-se e Aproveite!
SorapnRah
| #
Компания «V8soft» предлагает облачный сервис для того, чтобы вы смогли вести бизнес удачно и максимально эффективно. На предприятии работают исключительно те профессионалы, у которых имеется весь необходимый опыт работы, полезные навыки. На сайте https://v8soft.ru/ изучите все возможности ПО, чтобы определить то, подходит ли вам решение.
esportesdasorte
| #
No esportesdasorte, Cadastre-se e Ganhe 100$ para
Começar a Jogar!
mikrocz.cz
| #
mikro sluchatko http://mikrocz.cz
Lazrmhc
| #
Заказать диплом ВУЗа!
Мы изготавливаем дипломы любой профессии по приятным ценам— diplomist.com/kupit-meditsinskij-diplom-v-moskve-bez-lishnix-trudnostej/
RonaldPit
| #
Сантехник Юго-Восточный https://santekhnik-moskva.blogspot.com/p/south-eastern-administrative-okrug-of.html административный округ Москвы (ЮВАО). В состав Юго-Восточного административного округа входят районы: Выхино-Жулебино, Капотня, Кузьминки, Лефортово, Люблино, Марьино, Некрасовка, Нижегородский район, Печатники, Рязанский район, Текстильщики, Южнопортовый район.
pgwin
| #
Novo no pgwin – https://www.pgwin-br.com? Faça o Registro e Ganhe 100$ Imediatos!
infinity bet
| #
Registre-se no infinity bet – https://infinitybet-br.com e Comece com 100$ de Bônus para Apostar
blaze
| #
No blaze, novos usuários
podem aproveitar um bônus de 100$ ao se registrar no site!
Isso significa mais chances de ganhar e explorar uma grande variedade
de jogos de cassino, desde slots emocionantes até clássicos como
roleta e blackjack. Com o bônus de boas-vindas, você começa com um saldo extra, o que
aumenta suas chances de sucesso. Cadastre-se agora e
use os 100$ de bônus para experimentar seus jogos
favoritos com mais facilidade. Aproveite a oferta
e comece sua aventura no cassino agora mesmo!
PatrickBor
| #
Доброго!
Строительство домов каркасных — это современный и экономичный способ возведения жилья. Мы специализируемся на строительстве каркасных домов, которые становятся все более популярными благодаря своей энергоэффективности и быстроте возведения. Каркасные дома идеально подходят для людей, ценящих комфорт и экологичность. Наши специалисты помогут вам создать дом, который будет служить вам долгие годы. мастер вызов на дом ремонт холодильников, дом отделка внутри деревянного, квартиры в москве от застройщика рядом с метро с отделкой Строительство домов каркасных — это экономия и надежность.
Больше информации на сайте – https://postroikado.ru/
программу для строительства дома, бетонный дом отделка, ремонт дома в липецке
ремонт в квартире под ключ в москве, пеноблок строительство дома цена, отделка стены в доме
Удачи!
DavidNaing
| #
This casino’s Tiger Fortune game is flawless. tigrinho demo
1win_ogEa
| #
1 вин официальный сайт вход https://www.1win6044.ru .
PeterSlump
| #
https://telegra.ph/Top-Non-Gamstop-Casinos-for-UK-Players-20231-03-27
prodentim official
| #
prodentim a groundbreaking probiotic supplement uniquely designed to support oral health and promote robust gums and teeth.
pin up azerbaycan_qnMl
| #
pin up azerbaycan pin up azerbaycan .
prostavive
| #
prostavive maintaining prostate health is crucial for men’s overall wellness, especially as they grow older.
betleao
| #
No betleao, novos usuários podem aproveitar um
bônus de 100$ ao se registrar no site! Isso significa mais chances de ganhar e explorar uma grande variedade de
jogos de cassino, desde slots emocionantes até clássicos como roleta e blackjack.
Com o bônus de boas-vindas, você começa com um saldo extra, o que aumenta suas chances de sucesso.
Cadastre-se agora e use os 100$ de bônus para experimentar seus
jogos favoritos com mais facilidade. Aproveite a
oferta e comece sua aventura no cassino agora mesmo!
buy cheap lamictal price
| #
buying lamictal without insurance
how to get cheap allopurinol no prescription
| #
can you buy allopurinol without dr prescription
bwin
| #
100$ de Bônus no bwin – https://bwin-br.com: Registre-se
Agora e Aproveite a Oferta!
dobrowin
| #
Ao se cadastrar no dobrowin, você ganha um bônus
de 100$ para começar sua jornada no cassino com
o pé direito! Não importa se você é um novato ou um apostador
experiente, o bônus de boas-vindas é a oportunidade
perfeita para explorar todas as opções que o
site tem a oferecer. Jogue seus jogos favoritos, descubra novas opções de
apostas e aproveite para testar estratégias sem risco, já que o bônus ajuda
a aumentar suas chances de ganhar. Cadastre-se hoje e
comece com 100$!
PeterSlump
| #
https://telegra.ph/Discover-New-Non-Gamstop-Casinos-in-the-UK-03-27
WilliamPioxy
| #
Доброго!
Нотариус в Москве — это специалист, который поможет вам с заверением важных документов. Юрист по уголовным делам окажет необходимую помощь, если вы столкнулись с обвинениями или расследованием. Юридическая помощь при аресте обеспечит вашу защиту и соблюдение всех прав. Составление апелляции — это важный этап в судебном процессе, и наш юрист поможет вам оформить все необходимые документы. Юридическая консультация пенсионерам предоставит необходимые разъяснения по вопросам пенсионных выплат и социальных льгот.
Больше информации на сайте – https://zakonlw.ru/
оформление юридического лица, подоходный налог на детей вычет, образец трудовой договор сдельная оплата труда образец
налог от сдачи квартиры сколько, сколько с неработающего алименты на 1 ребенка, семейное право консультация
Удачи!
TimothySpamp
| #
Чеки для отчётности https://t.me/kupitchekiru в Москве — быстро, конфиденциально и с гарантией. Подтверждающие документы для отчёта, бухгалтерии, авансовых отчётов. Оперативная подготовка и доставка.
dobrowin-brasil.com
| #
No dobrowin [dobrowin-brasil.com], Cadastre-se e Ganhe 100$ para Começar a Jogar!
dobrowin
| #
dobrowin: 100$ de Bônus para Novos Jogadores
– Cadastre-se Já!
888
| #
O 888 oferece uma excelente oportunidade para quem deseja começar sua experiência
no cassino online com um bônus de 100$ para novos
jogadores! Ao se registrar no site, você garante esse bônus exclusivo que pode ser utilizado em diversos jogos
de cassino, como slots, roleta e poker. Esse é o momento perfeito para explorar o
mundo das apostas com um saldo extra, aproveitando ao máximo suas apostas sem precisar investir um grande valor
logo de início. Não perca essa oportunidade e cadastre-se já!
гостевые дома в Вардане
| #
гостевые дома в Вардане
Sazruup
| #
купить диплом училища в самаре
DavidNaing
| #
This casino’s Tiger Fortune game keeps me coming back for more. fortune tiger 777
PeterSlump
| #
https://telegra.ph/Guide-to-UK-Licensed-Non-Gamstop-Casinos-in-2023-03-27
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наша продукция:
Медный однораструбный отвод 45 градусов под пайку 27.4х23х0.9 мм 18.4х20.4 мм мягкая пайка М3РТ ГОСТ Р52922-2008 Приобретите высококачественные Медные отводы 45 градусов под пайку от компании Редметсплав.рф и улучшите вашу трубопроводную систему. Прочные, долговечные и устойчивые к коррозии отводы обеспечат надежное соединение под нужным углом, обеспечивая оптимальное распределение потока и высокую производительность вашей системы.
DavidNaing
| #
Tiger Fortune’s energy is off the charts. tigrinho demo
Trefkly
| #
Заказать диплом любого университета. Приобретение диплома ВУЗа через надежную фирму дарит ряд преимуществ. Это решение помогает сберечь время и серьезные финансовые средства. vetreriameliante.it/index.php/kforum/jm-slideshow-module/11444-img-https-i-posti#11445
MarioDup
| #
Drinks delivery предоставляет большой выбор алкогольных напитков и доступные цены. Наш основной приоритет – это качество. Заботимся о своей репутации. Продаем только то, в чем уверены. С нами вы легко выберете нужный товар. Ищете drinks delivery dubai? Deliverydubaidrinks.com – тут имеется водка, пиво, джин, шампанское, бризер, ром, текила, абсент, коньяк, вино, ликер, виски. Выполняем оперативную доставку алкоголя. Мы любим свое дело и всегда прислушиваемся к желаниям покупателей. На любой вопрос мы ответим и выбрать для вас подходящие напитки поможем.
pin up azerbaycan_hrMl
| #
pinup azerbaycan pinup azerbaycan .
mostbet_omKn
| #
mosbet mosbet .
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-with-Free-Spins-Bonuses-03-27
ClintonCag
| #
Онлайн-казино 7k — это продвинутая платформа для игр с ставками с большим выбором активностей. Здесь можно найти лучшие игровые автоматы, классические игры и игры с живыми дилерами с квалифицированными дилерами. Интерфейс плавный и простой в навигации, что делает игру удобной и приятной. На сайт казино предусмотрены бонусные бонусы для участников. В целом, ресурс предоставляет уникальный опыт для каждого игрока.
KennyHibia
| #
Доброго!
Как вернуть права? Автоюрист поможет вам решить вопросы, связанные с восстановлением водительских прав после лишения. Помощь в сборе документов — это важный процесс при подаче различных заявлений или жалоб. Составление жалобы поможет вам правильно оформить документ и подать его в соответствующие инстанции. Помощь при нарушении ПДД — это услуга, которая поможет вам защитить свои права в случае штрафов или других нарушений. Автоюрист по возврату прав обеспечит вас необходимой юридической поддержкой в случае лишения водительских прав.
Больше информации на сайте – https://konsultantok.ru
справка 2-НДФЛ, порядок рассмотрения судом трудовых споров, если ребенок остается с отцом после развода мать платит алименты
закон на возврат товара в течении 14 дней без объяснения причин право, стоимость регистрации брака в загсе, оформление земли
Удачи!
Jariorynb
| #
Где приобрести диплом по нужной специальности?
Наши специалисты предлагаютвыгодно заказать диплом, который выполняется на бланке ГОЗНАКа и заверен мокрыми печатями, водяными знаками, подписями. Документ пройдет любые проверки, даже при помощи профессиональных приборов. Решите свои задачи максимально быстро с нашим сервисом. Заказать диплом любого университета! babygirls020.copiny.com/question/details/id/1068426
nvblfurn
| #
https://blogcamp.com.ua/ru/2025/04/igry-dlja-devochek-volshebnyj-mir-fantazii-i-veselja/ популярные игры для девушек на пк
internetCah
| #
интернет провайдеры нижний новгород
domashij-internet-nizhnij-novgorod003.ru
интернет по адресу нижний новгород
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их происхождение. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы и адаптировать решения под особенности вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
Рзделия РёР· кобальта Рљ1Рђ Рзделия РёР· кобальта Рљ1Рђ – это высококачественные компоненты, используемые РІ различных отраслях, включая медицину Рё машиностроение. Кобальт обладает отличной прочностью Рё устойчивостью Рє РєРѕСЂСЂРѕР·РёРё, что делает его идеальным для создания долговечных решений. Р’ нашем ассортименте РІС‹ найдете разнообразные изделия, отвечающие самым строгим стандартам. Если вас заинтересовали преимущества Рё качество этих изделий, покупая РёС… Сѓ нас, РІС‹ делаете правильный выбор. РќРµ упустите возможность купить Рзделия РёР· кобальта Рљ1Рђ Рё обеспечить СЃРІРѕР№ проект надежными материалами.
roleta online
| #
Aproveite a oferta exclusiva do roleta online para novos usuários e receba 100$ de bônus ao se registrar!
Este bônus de boas-vindas permite que você experimente uma vasta gama
de jogos de cassino online sem precisar gastar imediatamente.
Com o bônus de 100$, você poderá explorar jogos como roleta, blackjack,
caça-níqueis e muito mais, aumentando suas
chances de vitória desde o primeiro minuto. Não perca essa chance única de começar
com um valor significativo – cadastre-se agora!
PeterSlump
| #
https://telegra.ph/Best-Non-Gamstop-Casinos-Accepting-PayPal-Payments-03-27
1win_etEa
| #
1win ваучер http://1win6044.ru/ .
DavidNaing
| #
I love the tiger-themed graphics in Tiger Fortune. tigrinho demo
buy cheap viagra_kcSr
| #
viagra https://www.cheapviagrausaonline.com .
Virgildig
| #
система управління фінансами
pin up azerbaycan_nrMa
| #
pin up azerbaycan pin up azerbaycan .
1win_reMi
| #
1 win https://www.1win6007.ru .
PeterSlump
| #
https://telegra.ph/Explore-the-Latest-Non-Gamstop-Casinos-Today-03-27
Kennethzof
| #
песня море
Roberthails
| #
Самые интересные новости Киева – честно, вовремя и без фейков. Будьте в курсе свежих новостей Киева и узнавайте заранее об интересных мероприятиях: новости киева сейчас
DavidNaing
| #
The excitement of Tiger Fortune never fades. tiger fortune
Willieavamy
| #
Online casinos feel chancy—watch out! blue wizard
mostbet_haSa
| #
mostbet.kg https://www.mostbet6011.ru .
where can i get microzide pill
| #
cost of microzide for sale
can i purchase cheap atarax without dr prescription
| #
where can i buy generic atarax price
DavidNaing
| #
The Tiger Fortune game runs so smoothly on my phone. tigrinho demo
DavidNaing
| #
The sound effects in Tiger Fortune are spot on. fortune tiger
PeterSlump
| #
https://telegra.ph/No-Gamstop-Casino-Options-in-the-UK-03-27
Sazrmmh
| #
Всегда думал что купить диплом о высшем образовании это миф и нереально, но все оказалось не так, изначально искал информацию про: куплю диплом делом, купить диплом в иваново, купить диплом в оренбурге, купить диплом слесаря, диплом купить сад, потом про дипломы вузов, подробнее здесь diplomybox.com/kupit-attestata-v-kurske
Willieavamy
| #
Online casino ads roar—hush down! aviator
DavidNaing
| #
The animations in Tiger Fortune are incredible. fortune tiger demo gratis
Jariorsua
| #
Где заказать диплом специалиста?
Мы предлагаем быстро и выгодно заказать диплом, который выполняется на оригинальном бланке и заверен печатями, водяными знаками, подписями официальных лиц. Данный документ пройдет любые проверки, даже при использовании специально предназначенного оборудования. Достигайте своих целей быстро и просто с нашей компанией.
Заказать диплом о высшем образовании diplomt-v-chelyabinske.ru/kupit-diplom-v-spb-16/
JesusNes
| #
Официальный веб-сайт казино Рояль предлагает располагающую обстановку для поклонников азартных игр. С его изящным дизайном и премиальным выбором игр, Рояль является великолепным местом для тех, кто высоко ценит качественное азартное развлечение. Исследуйте широкий спектр игровых автоматов и настольных игр на основном веб-сайте https://royalcasino-slots.com/.
Казино Рояль гарантирует первоклассное обслуживание клиентов и поддержку. Отзывчивая команда поддержки в наличии круглосуточно, оборудована откликнуться на любые возникающие вопросы и исправить проблемы, предоставляя непрерывную игру и удовлетворение.
PeterSlump
| #
https://telegra.ph/Reliable-Non-GamStop-Casinos-for-Safe-Online-Gaming1-03-27
Iariorado
| #
Заказать диплом института по невысокой цене вы сможете, обращаясь к проверенной специализированной фирме. Купить документ о получении высшего образования можно в нашем сервисе. diplomaj-v-tule.ru/kupit-diplom-magistra-s-provodkoj-3
1win_aemt
| #
1win online http://1win15.com.ng .
buy cheap viagra_rxSr
| #
viagra cheap http://buyviagrat.com .
Hijamenetly
| #
На сайте https://pilmi.ru вы найдете огромное количество фильмов самых разных жанров, включая комедии, мелодрамы, драмы, романтические. Для того чтобы подыскать наиболее подходящий вариант, нужно воспользоваться специальным каталогом. Вашему вниманию представлены и ожидаемые любопытные новинки этого года, которые произведут эффект. Все это можно просматривать в любое время и на различных устройствах. Прямо сейчас воспользуйтесь возможностью посмотреть азиатское кино. Используйте поиск для облегчения выбора.
JacobAdhep
| #
Доброго!
Если болит ухо, что делать, если боль появляется при изменении температуры воздуха? Сильные перепады температуры могут вызвать боль в ухе, особенно при простудах и воспалениях. В таком случае важно обратиться к врачу, чтобы исключить инфекцию или повреждения барабанной перепонки. Врач может порекомендовать средства для защиты ушей от холода или тепла. Лечение должно быть направлено на снятие воспаления и предотвращение осложнений. Не стоит игнорировать такие симптомы, так как они могут привести к серьезным заболеваниям.
Больше информации на сайте – https://medikasv.ru/
вздутие живота у ребенка, записаться к лору ульяновск, лейла адамян гинеколог биография
консультация онколога онлайн, ростов гинеколог эндокринолог, диета при диабете
Удачи!
PeterSlump
| #
https://telegra.ph/Best-UK-Casinos-with-No-Deposit-Free-Spins-Outside-Gamstop-03-27
brlwin
| #
Bônus de 100$ ao Registrar-se no brlwin – Comece a Apostar Já!
Dnrthqd
| #
Мы изготавливаем дипломы любой профессии по невысоким тарифам. Мы можем предложить документы ВУЗов, которые расположены на территории всей России. Дипломы и аттестаты делаются на бумаге самого высокого качества. Это дает возможности делать государственные дипломы, не отличимые от оригинала. lovn1world.com/read-blog/14295_kupit-diplom-s-zaneseniem-v-reestr-stoimost.html
Sazrtbs
| #
Заказать диплом на заказ можно через сайт компании. lavorare.eu/companies/frees-diplom
Garrettshoob
| #
Премиум скины CS2 https://cs2-case-simulator.ru топовые облики для AWP, AK-47, M4, ножей и перчаток. Яркий стиль, редкие коллекции, эксклюзивные дизайны. Укрась свою игру и выделяйся в каждой катке!
PeterSlump
| #
https://telegra.ph/Guide-to-Non-Gamstop-Curacao-Casino-Sites-03-27
Stanleyred
| #
Хочешь Dragon Lore http://cs-case-simulator.ru Открывай кейс прямо сейчас — шанс на легенду CS2 может стать реальностью. Лучшие скины ждут тебя, рискни и получи топовый дроп!
Jamesmip
| #
Glock-18 Fade http://case-simulator-csgo.ru один из самых ярких и редких скинов для стартового пистолета в CS2. Плавный градиент, премиум-качество и высокий спрос среди коллекционеров.
how can i get cheap glucophage
| #
Meds information sheet. Effects of Drug Abuse.
how can i get cheap glucophage
Some about medication. Get now.
Sazrsmd
| #
Выгодно заказать диплом университета!
Приобрести диплом ВУЗа по выгодной стоимости возможно, обращаясь к проверенной специализированной компании. Купить диплом: diplomt-v-samare.ru/kupit-diplom-s-provodkoj-tsena-i-garantiya-podlinnosti
profsox
| #
Требуется прогон сайта недорого? Cервис ProfLinks.ru ориентирован на размещение вечных ссылок с ИКС от 10; «вечных», в нашем случае, на постоянной основе и с разовой оплатой; «ИКС от 10», означает, что индекс качества сайтов, присваиваемый ресурсам поисковой системой Яндекс, будет минимум от 10 пунктов, в зависимости от выбранного Вами тарифного плана или доп. услуги.
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-in-the-UK-at-Betz-Casino1-03-27
sex lồn không lông
| #
sex lồn không lông
A Brief History of Elemental Analysis » Artemis Analytical
buy cheap viagra_hhSr
| #
buy viagra http://www.buyviagrat.com .
internetCah
| #
домашний интернет тарифы
domashij-internet-novosibirsk001.ru
какие провайдеры по адресу
DavidNaing
| #
Tiger Fortune keeps me entertained all night. tigrinho demo
Cazrjgf
| #
Приобрести диплом любого ВУЗа!
Мы готовы предложить документы ВУЗов, которые расположены в любом регионе Российской Федерации. Документы делаются на “правильной” бумаге высшего качества: jobs.theelitejob.com/employer/aurus-diploms
mostbet_vqKn
| #
mostbet casino http://mostbet6012.ru .
Sazrkjc
| #
Приобрести диплом ВУЗа !
Приобретение диплома ВУЗа России в нашей компании – надежный процесс, потому что документ заносится в реестр. Приобрести диплом любого ВУЗа diplom-top.ru/kupit-diplom-menedzhera-s-zaneseniem-v-reestr-18
1win_knMa
| #
1win.com.ci https://1win5012.ru/ .
GeorgeTom
| #
Source:
cyberlocman.ru
PeterSlump
| #
https://telegra.ph/Advantages-of-Non-Gamstop-Casinos-for-Players-03-27
Diplomi_inka
| #
купить диплом политеха купить диплом политеха .
1win_utmt
| #
1win bet deposit http://1win15.com.ng .
Sogiphvor
| #
Академия «Арчибальд» занимается обучением грумингу. Вы можете совершенствовать собственные навыки на живых моделях до бесконечности. Преподаватели отвечают на все вопросы. Дарим методические материалы, учебники, а также видеолекции. Окунитесь в атмосферу, с которой расставаться не хочется. Ищете курсы груминга в москва? Grooming-dream.ru – здесь представлены реальные отзывы клиентов. Мы лучших груминг-специалистов выпускаем. Предлагаем инструменты в период обучения. Если вам интересна новая профессия – Арчибальд, вот то место, которое вам подойдет.
can i buy ditropan without dr prescription
| #
buying ditropan without dr prescription
where to buy generic dapsone without dr prescription
| #
buying generic dapsone without rx
pague bet
| #
Aproveite a oferta exclusiva do pague bet para novos usuários e receba 100$ de bônus ao se
registrar! Este bônus de boas-vindas permite que você
experimente uma vasta gama de jogos de cassino online sem precisar gastar imediatamente.
Com o bônus de 100$, você poderá explorar jogos como
roleta, blackjack, caça-níqueis e muito mais, aumentando suas chances de vitória desde
o primeiro minuto. Não perca essa chance única de começar com um
valor significativo – cadastre-se agora!
Sazrizo
| #
Приобрести диплом университета по доступной цене вы можете, обращаясь к проверенной специализированной фирме. Мы оказываем услуги по производству и продаже документов об окончании любых ВУЗов России. Заказать диплом любого университета– diplomj-irkutsk.ru/kachestvennij-diplom-visshego-obrazovaniya-po-dostupnoj-tsene-2/
PeterSlump
| #
https://telegra.ph/Explore-New-Non-Gamstop-Casinos-for-Exciting-Wins1-03-27
pin up azerbaycan_aaMa
| #
pin up azerbaijan pin up azerbaijan .
Iariordkh
| #
Приобрести диплом университета по невысокой стоимости можно, обращаясь к проверенной специализированной компании. Купить документ ВУЗа можно у нас. kupitediplom.ru/kupit-diplom-s-zaneseniem-v-reestr-bistro-i-bezopasno-18
mostbet_qqSa
| #
мостбет мобильная версия скачать мостбет мобильная версия скачать .
Corey_sah
| #
Корпоративные сувениры – это важный элемент делового этикета и эффективный инструмент продвижения бренда. Они помогают укрепить лояльность клиентов и подчеркнуть корпоративную культуру внутри компании. Важно, чтобы сувениры были не только качественными, но и соответствовали имиджу вашей компании. Если вы ищете оригинальные и практичные решения с возможностью брендирования, рекомендуем ознакомиться с широким ассортиментом на сайте: https://fermerzag.ru/izgotovlenie-metallicheskih-znachkov-metodom-himicheskogo-travleniya/
mostbet_qrKn
| #
мотбет мотбет .
PeterSlump
| #
https://telegra.ph/No-Deposit-Bonuses-at-Non-Gamstop-Casinos-Explained9-03-27
Trefcuz
| #
Купить диплом об образовании. Приобретение диплома через надежную компанию дарит ряд плюсов для покупателя. Данное решение дает возможность сэкономить время и серьезные деньги. babygirls015.copiny.com/question/details/id/1083418
BlaineKeesk
| #
StatTrak AK-47 Vulcan get-skin-cs2.ru/ мощный стиль и счётчик убийств в одном скине. Агрессивный дизайн, сине-чёрная цветовая гамма и высокая ценность на рынке CS2. Идеальный выбор для бойца с характером!
1win_qdMi
| #
1 вин скачать http://www.1win6007.ru .
PeterSlump
| #
https://telegra.ph/PayPal-Accepted-at-Non-Gamstop-Casinos-03-27
1win_mpmt
| #
1win football http://1win15.com.ng .
Kevinspoda
| #
пнд пластик листовой цена
Williamsag
| #
M4A4 Emperor https://get-skin-cs.ru/ эффектный скин в стиле королевской власти. Яркий синий фон, золотые детали и образ императора делают этот скин настоящим украшением инвентаря в CS2.
muzetGuimi
| #
Delivery-dubai.com предлагает широкий ассортимент разных видов алкоголя. На сайте можно ознакомиться с имеющейся продукцией в каталоге с ценами. Это даст возможность под ваш повод напиток выбрать подходящий. Выполняем быструю доставку алкоголя. Будем рады видеть вас в числе клиентов! Ищете алкоголь в дубае с доставкой? Delivery-dubai.com – тут широкий выбор алкоголя представлен. Вы отыщите для себя все то, что принести удовольствие может. Предлагаем товары исключительного качества. Для заказа напишите нам в мессенджеры: WhatsApp, Telegram.
Nikerson#nosserck[WascuwyripyfdiBI,2,5]
| #
?Hola fanaticos del casino
Muchos de los mejores casinos online sin licencia estГЎn optimizados para mГіviles y tablets. licencia internacional La experiencia de usuario es fluida y moderna.
Los juegos de casino sin licencia incluyen opciones innovadoras y proveedores poco conocidos. Esto aГ±ade variedad a tu experiencia de juego. Es perfecto para quienes buscan salir de lo tradicional.
Mas detalles en el enlace – п»їhttps://casinossinlicenciaenespana.guru/
?Que tengas excelentes exitos!
DavidNaing
| #
This online casino hit the mark with Tiger Fortune. fortune tiger demo gratis
Lazrjoi
| #
Купить диплом о высшем образовании!
Мы изготавливаем дипломы любой профессии по приятным ценам— diplomp-irkutsk.ru/diplom-o-srednem-med-obrazovanii-kupit-2/
PeterSlump
| #
https://telegra.ph/Best-Non-Gamstop-Casinos-to-Play-in-2021-03-27
DavidNaing
| #
Tiger Fortune has the best energy of any game. fortune tiger demo gratis
Vincenthic
| #
AK-47 Case Hardened get-skins-cs2 классика CS2 с уникальным закалённым узором. Каждый скин отличается: от редких фулл-блю до золотых комбинаций. Настоящая находка для коллекционера и трейдера.
buy generic coreg pills
| #
Drugs prescribing information. What side effects can this medication cause?
buy generic coreg pills
Best about meds. Read here.
Sazrxfv
| #
Приобрести диплом под заказ в столице вы имеете возможность используя официальный портал компании. blog.nataraj.ru/~/Interest/Купитьдипломофициальногообразца
SydneyShurb
| #
софиты кровли В мире современного строительства выбор материалов играет ключевую роль в долговечности и эстетическом облике зданий. Компания “СтройКомплекс” предлагает широкий ассортимент кровельных и фасадных материалов, отвечающих самым высоким стандартам качества. Для обустройства кровли мы предлагаем: металлочерепицу, известную своей прочностью и долговечностью; профлист и профнастил, оптимальные для промышленных и коммерческих объектов; мягкую и гибкую черепицу, идеальные для частного домостроения, а также элегантную фальцевую кровлю.
Iariorqpk
| #
Заказать диплом ВУЗа по невысокой стоимости возможно, обратившись к надежной специализированной компании. Купить документ университета можно в нашей компании в Москве. diplom-zentr.com/kupit-diplom-s-zaneseniem-v-reestr-stoimost-8
PeterSlump
| #
https://telegra.ph/Find-Reliable-Non-Gamstop-Casino-Sister-Sites-Today-03-27
sex hiep dam gai cap 3
| #
sex hiep dam gai cap 3
A Brief History of Elemental Analysis » Artemis Analytical
Sazrdoi
| #
купить диплом о высшем образовании в екатеринбург
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние достижения.
Наша продукция:
Высокочистые гранулы Y2O3
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casino-Reviews-and-Player-Insights-03-27
Kevinspoda
| #
https://plastictek.ru/
Richardedice
| #
Забудьте о скучных evenings и рутинной жизни! На нашем сайте вы можете познакомиться с красивыми, страстными девушками из Омска, готовыми к новым знакомствам и приключениям. Они ждут вас с нетерпением, чтобы подарить множество ярких впечатлений и романтических встреч https://night24-omsk.com/
PeterSlump
| #
https://telegra.ph/Top-Non-Gamstop-Casinos-in-the-UK-for-202314-03-27
pin up azerbaycan_sjOa
| #
pinup azerbaycan pinup azerbaycan .
DavidNaing
| #
Tiger Fortune’s bonus rounds are the best part. fortune tiger
internetCah
| #
подключить домашний интернет в новосибирске
domashij-internet-novosibirsk002.ru
подключить интернет в квартиру новосибирск
Joshuawardy
| #
Деньги онлайн займ занять деньги онлайн
Brianfup
| #
Займ на карту без отказа без проверки мгновенно моментальный займ на карту без проверок круглосуточно
DavidNaing
| #
This casino’s Tiger Fortune slot is a joy. fortune tiger demo gratis
Sikapent#nonick[KywcuwypipyfdiBI,2,5]
| #
¡Hola seguidores del azar
Casinos sin licencia de EspaГ±a destacan por ofrecer retiros instantГЎneos. casinossinlicenciaenespana No necesitas esperar dГas para cobrar tus ganancias. Verifica si hay lГmites semanales o mensuales.
Los juegos de casino sin licencia ofrecen jackpots progresivos, juegos con crupier y torneos exclusivos. Es una experiencia rica y completa. No olvides revisar RTP y volatilidad.
Consulta el enlace para más información – п»їhttps://casinossinlicenciaenespana.guru/
¡Por muchos momentos de risa!
PeterSlump
| #
https://telegra.ph/Discover-Non-Gamstop-Casinos-at-Betz-Casino-UK-03-27
Dnrtggv
| #
Мы изготавливаем дипломы любой профессии по приятным ценам. Мы готовы предложить документы ВУЗов, расположенных в любом регионе РФ. Дипломы и аттестаты выпускаются на “правильной” бумаге самого высокого качества. Это позволяет делать государственные дипломы, которые невозможно отличить от оригинала. swellenjobs.co.za/employer/archive-diploma
fasciopraktika
| #
Запишитесь на курсы для массажистов и прокачайте свою профессию. Современные методики, опытные преподаватели, доступная цена и максимальная польза для практики.
minocycline buy
| #
Meds information sheet. Drug Class.
minocycline buy
Everything news about drug. Read now.
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наши товары:
Рнструментальная круглая РїРѕРєРѕРІРєР° 63 РјРј РҐ12Р’РњР¤ ГОСТ 1133-71 РЁРёСЂРѕРєРёР№ выбор круглых РїРѕРєРѕРІРѕРє для профессиональной обработки металла. Рнструментальная круглая РїРѕРєРѕРІРєР° высочайшего качества СЃ разнообразием размеров Рё форм. Повышенная прочность, износостойкость Рё превосходное качество изготовления. Обеспечивает отличное сцепление СЃ материалом Рё эффективную работу оборудования. Покупайте РїСЂСЏРјРѕ сейчас!
Сasinossinlicenciaen#espana[WcuwyripyfdiBI,2,5]
| #
?Hola fanaticos del casino
Un casino sin licencia te permite jugar sin restricciones de depГіsitos o retiros. licencia internacional Esto lo hace ideal para jugadores con estrategias avanzadas. Revisa los lГmites de la plataforma antes de empezar.
Casinos sin licencia de EspaГ±a permiten depГіsitos mГnimos bajos. Es una buena forma de empezar sin arriesgar mucho. Ideal para nuevos jugadores que quieren probar.
Mas detalles en el enlace – п»їhttp://casinossinlicenciaenespana.guru
?Que tengas excelentes ventajas!
where to get generic valtrex without prescription
| #
get valtrex pill
Willieavamy
| #
Online gambling can spiral—stay in control! aviator
can i buy avodart without rx
| #
avodart bible
PeterSlump
| #
https://telegra.ph/Live-Casino-Options-Beyond-GamStop-Restrictions-03-27
Sazriwe
| #
Приобрести диплом возможно используя сайт компании. skymix2018.forumex.ru/viewtopic.phpf=14&t=946
1win_lkPt
| #
1вин кг https://1win6009.ru/ .
Richardedice
| #
На нашем сайте собраны наиболее горячие девушки Омска, которые рады новым встречам и интимному общению. Более 2500 анкет реальных девушек помогут вам выбрать ту, с которой захочется провести время и найти возбуждение и интригу в каждом моменте https://night24-omsk.com/
1win_ynKr
| #
1вин официальный мобильная http://1win6043.ru/ .
JimmieMer
| #
Здравствуйте!
Если болит ухо, что делать, если боль появилась после длительного воздействия шума? Боль в ухе после длительного воздействия громкого шума может быть признаком шума травматического повреждения слуха, например, шума от музыки, работающей техники или других источников громкого звука. Важно избегать дальнейших повреждений слуха, используя защитные наушники или беруши в будущем. Если боль не проходит или сопровождается ухудшением слуха, необходимо обратиться к врачу-отоларингологу для диагностики и назначения лечения.
Больше информации на сайте – https://ovrachahl.ru
родинка стала больше, для сухости глаз капли, гинеколог эндокринолог новороссийск
диета сахарный диабет 2 типа, лор детский в серпухове, гастрит у подростков
Удачи!
Richardevabs
| #
read https://datadex.ink/
realsbet
| #
100$ de Bônus para Apostar no realsbet!
Cadastre-se Agora!
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-What-You-Need-to-Know-03-27
where buy cheap plan b without rx
| #
Medicines prescribing information. Effects of Drug Abuse.
where buy cheap plan b without rx
All news about pills. Get information here.
GeorgeTom
| #
Source:
cyberlocman.ru
DavidNaing
| #
I hit a bonus round in Tiger Fortune today – wow! fortune tiger
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их качество. Превосходное обслуживание – наш стандарт – мы на связи, чтобы разрешать ваши вопросы по мере того как находить ответы под требования вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
Магний AZ80A – ASTM B107 Магний AZ80A – ASTM B107 – это высококачественный легкий сплав, который обладает отличной прочностью Рё РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью. РћРЅ широко используется РІ авиастроении Рё автомобилестроении благодаря СЃРІРѕРёРј превосходным механическим свойствам. Ртот материал прекрасно обрабатывается Рё сваривается, что делает его идеальным для создания сложных конструкций. Если РІС‹ хотите купить Магний AZ80A – ASTM B107, получите надежный Рё долговечный РїСЂРѕРґСѓРєС‚ для СЃРІРѕРёС… проектов. Заказывая этот сплав, РІС‹ выбираете оптимальное сочетание качества Рё доступности. РќРµ упустите возможность обеспечить СЃРІРѕСЋ работу надежным материалом уже сегодня.
DavidNaing
| #
Tiger Fortune’s free spins are a game-changer. fortune tiger 777
Sazrbdm
| #
Быстро и просто заказать диплом любого института!
Заказать диплом института по выгодной стоимости возможно, обращаясь к надежной специализированной компании. Заказать диплом: diplomh-40.ru/ofitsialnij-diplom-o-visshem-obrazovanii-garantiya-uspexa
PeterSlump
| #
https://telegra.ph/Best-Non-Gamstop-Casinos-Accepting-Paypal-for-Players-03-27
Diplomi_moKt
| #
Купить диплом ВУЗа!
Мы предлагаем документы университетов, расположенных на территории всей РФ.
fastdiploms.com/kupit-diplom-v-moskve-s-zaneseniem-v-reestr-4/
Peterwrimb
| #
Привет всем!
Если болит ухо после плавания, что делать? Это может быть связано с попаданием воды в ухо или развитием воспаления наружного слухового прохода. Для снятия дискомфорта можно попробовать осторожно высушить ухо ватным тампоном или воспользоваться каплями для ушей. Однако, если боль не проходит, лучше обратиться к врачу, чтобы исключить инфекцию или травму. Не стоит откладывать визит к специалисту, особенно если появляются другие симптомы, такие как зуд или гнойные выделения.
Больше информации на сайте – https://prostudailor.ru/
шишка под мышкой, курканина светлана николаевна гинеколог ярославль, головокружение при вставании с кровати
блинов илья викторович гастроэнтеролог, педиатр чита детский, отзывы о врачах
Удачи!
MerrillMuT
| #
dig this https://universalx.us/
PeterSlump
| #
https://telegra.ph/Exploring-Casino-Options-Beyond-Gamstop-Restrictions-03-27
pin up azerbaycan_bhOa
| #
pin up azerbaijan pin up azerbaijan .
1win_vvpt
| #
игра ракета на деньги 1win http://www.1win6008.ru .
PeterSlump
| #
https://telegra.ph/No-Gamstop-Casino-Options-for-Players-03-27
DavidNaing
| #
The bonuses in Tiger Fortune are so exciting! fortune tiger demo gratis
hedinic
| #
Блог о маркетплейсах «OOWA» предлагает ознакомиться с актуальной, интересной и познавательной информацией, которая касается удачных продаж на маркетплейсах, обзоров товаров и услуг. На портале регулярно публикуются ценные и содержательные рекомендации от экспертов, которые помогут начать лучше разбираться в тонкостях такого бизнеса. Ищете продажа б/у вещей на ozon? На oowa.ru регулярно публикуются ценные и содержательные рекомендации от экспертов, которые помогут начать лучше разбираться в тонкостях такого бизнеса. Почитайте о самых востребованных товарах с маркетплейсов.
cost cheap mestinon for sale
| #
can i purchase cheap mestinon price
how can i get detrol without prescription
| #
where buy detrol tablets
PeterSlump
| #
https://telegra.ph/Safe-Online-Casinos-in-the-UK-Without-GamStop-03-27
can i order cheap celebrex without dr prescription
| #
Medication information. Generic Name.
can i order cheap celebrex without dr prescription
Some information about medicine. Read here.
internetCah
| #
интернет тарифы новосибирск
domashij-internet-novosibirsk003.ru
провайдер по адресу
daliyeTig
| #
Market-delivery-dubai.ru доставку алкоголя предлагает. У вас есть возможность купить виски, ром, вино, текилу, бризер, абсцент, коньяк, водку, пиво, джин, ликер. Мы гарантируем приемлемые цены. Доверие покупателей заслужили. Ищете dubai alcohol delivery? Market-delivery-dubai.ru – здесь есть то, что вам нужно. Регулярно работаем над ассортиментом. Прекрасно знаем потребности своих клиентов. Заказы принимаем через Whatsapp. Осуществляем доставку любого вида алкогольной продукции высокого качества в короткие сроки. Нужна помощь? Свяжитесь с нами.
Robertcor
| #
Привет всем!
Если болит ухо, что делать, если боль не прекращается после лечения? Если боль в ухе не исчезает после применения препаратов, это может указывать на необходимость корректировки лечения или более тщательной диагностики. Важно обратиться к врачу для повторного осмотра и назначения дополнительных исследований, например, анализов или МРТ. Врач может изменить подход к лечению, назначив другие препараты или физиотерапевтические процедуры. Непрекращающаяся боль может быть признаком хронического воспаления, которое требует длительного и комплексного лечения. Своевременное обращение к врачу поможет исключить осложнения и достичь полноценного выздоровления.
Больше информации на сайте – https://konsmediic.ru/
онкомаркеры по анализу крови, онколог вакансии москва, педиатры в геленджике
казань гинеколог узи, зенон отзывы врачей, шум в голове
Удачи!
Willieavamy
| #
I wish online eased bets—low go! vincispin
DavidNaing
| #
This online casino’s Tiger Fortune is flawless. fortune tiger
XaiUNecope
| #
На сайте https://officepro54.ru представлено огромное количество мебели, которая идеально подходит для организации офисного пространства. Вся она выполнена из инновационных, уникальных материалов, а потому прослужит очень долго, не синтезирует в воздух опасных веществ. В разделе вы найдете кабинет руководителя, функциональные и вместительные столы для переговоров, шкафы-купе, сейфы, акустические системы, ученическую мебель и многое другое. Постоянно действуют акции. На продукцию установлены привлекательные цены.
Sazrpsr
| #
Приобрести диплом о высшем образовании !
Приобретение диплома ВУЗа России у нас является надежным делом, поскольку документ заносится в государственный реестр. Купить диплом о высшем образовании diplom-ryssia.com/kupit-diplom-visshego-obrazovaniya-s-zaneseniem-v-reestr-25
1win_vvKr
| #
зайти в 1вин зайти в 1вин .
PeterSlump
| #
https://telegra.ph/Guide-to-Non-Gamstop-Casinos-for-Players-Worldwide-03-27-2
DavidNaing
| #
Tiger Fortune’s payouts keep me coming back for more. fortune tiger demo
DavidNaing
| #
This casino’s Tiger Fortune game is a gem. tiger fortune
talon777
| #
Aproveite 100$ de Bônus no talon777 ao
Criar Sua Conta!
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-No-Deposit-Bonuses-Explained-03-27
fazobetai
| #
100$ de Bônus para Apostar no fazobetai!
Cadastre-se Agora!
Mazrdhf
| #
Мы можем предложить дипломы любых профессий по приятным тарифам. Всегда стараемся поддерживать для клиентов адекватную ценовую политику. Важно, чтобы документы были доступны для большого количества граждан.
Приобретение документа, подтверждающего окончание ВУЗа, – это выгодное решение. Заказать диплом университета: diplommy.ru/kupit-diplom-novogo-obraztsa-10/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние возможности.
Наша продукция:
Плоская мишень NiV
goinbet
| #
Ganhe 100$ para Apostar no goinbet ao Se Registrar Hoje!
seroquel for morning anxiety
| #
Drug information for patients. Cautions.
seroquel for morning anxiety
Some about pills. Get information here.
1win_dipt
| #
1вин официальный сайт https://1win6008.ru/ .
Mazrcob
| #
Быстро приобрести диплом ВУЗа!
Мы оказываем услуги по продаже документов об окончании любых ВУЗов Российской Федерации. Документы производят на фирменных бланках. godscoop.com/read-blog/631_kupit-diplom-tehnikuma-s-zaneseniem-v-reestr-otzyvy.html
PeterSlump
| #
https://telegra.ph/Best-Casinos-Not-on-Gamstop-with-No-Deposit-Bonuses4-03-27
how can i get generic benicar without a prescription
| #
Medication information leaflet. Generic Name.
how can i get generic benicar without a prescription
Some news about pills. Get now.
DavidNaing
| #
I love the thrill of Tiger Fortune’s jackpots. fortune tiger
DavidNaing
| #
Tiger Fortune’s features are pure genius. fortune tiger
Cazrdrn
| #
Купить диплом университета!
Мы предлагаем документы ВУЗов, расположенных в любом регионе Российской Федерации. Документы выпускаются на бумаге самого высокого качества: feleempleo.es/employer/aurus-diploms
kepirdrind
| #
В случае если желаете узнать самую ценную, полезную и важную информацию о трейдинге, вложениях, то нужно зайти на портал, где вы отыщете все необходимое на такую тему. Здесь доступным языком рассказывается о том, как правильно вести торговлю на внебиржевом рынке. http://fxidea.com/ рекомендует воспользоваться акциями каждому новичку. Скорее ознакомьтесь с индикаторами, отзывами трейдеров, самыми эффективными стратегиями. Вы получите доступ к ценным материалам, касающимся криптовалюты, а также узнаете, как заработать на форексе.
PeterSlump
| #
https://telegra.ph/Explore-Non-Gamstop-Casinos-in-Curacao-Today-03-27
JustinVar
| #
Привет всем!
Когда болит ухо, что делать, если оно стало чувствительным к шуму? Повышенная чувствительность к звукам, сопровождающаяся болью в ухе, может быть симптомом воспаления слухового нерва или других заболеваний. В таком случае важно как можно скорее обратиться к врачу для диагностики. Врач может провести аудиометрическое исследование для оценки слуха и определить точную причину боли. Лечение может включать противовоспалительные препараты и средства для защиты слуха. Не стоит откладывать визит к специалисту, чтобы избежать ухудшения состояния.
Больше информации на сайте – https://vrachataj.ru/
бессонница причины, что смотрит гинеколог у девочек в 14 лет, кардиолог нефтекамск субботников
что делают на осмотре у гинеколога в 14 лет, офтальмолог дубна, почему ребенок кашляет по утрам
Удачи!
where can i get betapace
| #
where to buy generic betapace tablets
DavidNaing
| #
The tiger symbols in Tiger Fortune are so cool! fortune tiger 777
Xazryrt
| #
Купить диплом ВУЗа!
Мы изготавливаем дипломы любой профессии по выгодным тарифам. Вы покупаете диплом в надежной и проверенной компании. : ratingforex.ru/forum-forex/viewtopic.php?f=18&t=69066&sid=94916b0425c77545da1638f63a026a95
mostbet_pdmt
| #
mostbest http://mostbet6033.ru/ .
Willieavamy
| #
The online rush hits—keep chill! plinko
Willieavamy
| #
The live dealers online rule—true vibe! sugar rush
internetCah
| #
провайдеры интернета в санкт-петербурге по адресу
domashij-internet-spb001.ru
подключить интернет в квартиру санкт-петербург
PeterSlump
| #
https://telegra.ph/PayPal-Options-at-Non-Gamstop-Casinos-in-the-UK-03-27
DavidNaing
| #
Tiger Fortune’s jungle theme is so well done. jogo do tigrinho demo
GeorgeTom
| #
Source:
cyberlocman.ru
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наши товары:
Пружина РёР· драгоценных металлов палладиевая 500С…7С…0.2 РјРј РџРґР90-10 РўРЈ Выберите идеальные пружины РёР· золота, серебра или платины, чтобы дополнить ваше украшение. Найдите пружины СЃ изысканным дизайном Рё прочностью. РћРЅРё предлагают широкий выбор для того, чтобы подчеркнуть уникальность вашего украшения.
PeterSlump
| #
https://telegra.ph/Best-Non-Gamstop-Online-Casinos-for-Players-03-27
how to buy generic zithromax without a prescription
| #
Medication information. Brand names.
how to buy generic zithromax without a prescription
Everything what you want to know about medicines. Get information here.
WilliamDix
| #
Мы можем предложить дипломы любых профессий по выгодным ценам. Дипломы производятся на настоящих бланках государственного образца Заказать диплом об образовании good-diplom.ru
Lazrmnv
| #
Где заказать диплом специалиста?
Купить диплом института по выгодной цене можно, обращаясь к проверенной специализированной компании.: diplom45.ru
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Гранулы магния 3N5
can i get cheap phenergan price
| #
Medicines information leaflet. Drug Class.
can i get cheap phenergan price
All information about drug. Read information here.
pin up azerbaycan_pzMl
| #
pinup azerbaycan pinup azerbaycan .
DavidNaing
| #
I love the thrill of Tiger Fortune’s jackpots. fortune tiger 777
DavidNaing
| #
This online casino’s Tiger Fortune is a thrill. fortune tiger 777
PeterSlump
| #
https://telegra.ph/Guide-to-Non-Gamstop-Casino-Sites-and-Their-Benefits1-03-27
1win_vjpt
| #
казино 1win http://1win6008.ru/ .
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Опытная поддержка – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы по мере того как адаптировать решения под требования вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Титан РџРў-1РњР‘ – ГОСТ 19807-91 Титан РџРў-1РњР‘ – ГОСТ 19807-91 – качественный Рё надежный РїСЂРѕРґСѓРєС‚, предназначенный для различных промышленных применений. Ртот титановый сплав обладает высокими прочностными характеристиками Рё стойкостью Рє РєРѕСЂСЂРѕР·РёРё, что делает его идеальным выбором для машин Рё оборудования. РџСЂРё выборе Титан РџРў-1РњР‘ – ГОСТ 19807-91, РІС‹ получаете гарантированное соответствие современным стандартам Рё долговечность РІ эксплуатации. РќРµ упустите возможность улучшить производительность вашей техники. Купите Титан РџРў-1РњР‘ – ГОСТ 19807-91 РїСЂСЏРјРѕ сейчас Рё убедитесь РІ его превосходных свойствах.
DavidNaing
| #
Tiger Fortune’s theme is so adventurous. fortune tiger
DavidNaing
| #
Tiger Fortune has some of the best odds I’ve seen. jogo do tigrinho demo
PeterSlump
| #
https://telegra.ph/Non-GamStop-Casinos-Comprehensive-Reviews-and-Insights-03-27
1win_duPt
| #
wan win http://1win6009.ru .
where can i get generic sildigra without insurance
| #
can you get cheap sildigra pill
get generic chloromycetin pills
| #
cost of cheap chloromycetin no prescription
CharlesgoowN
| #
Здравствуйте!
Если болит ухо, что делать, если оно болит при длительном разговоре? Боль в ухе, которая усиливается при длительном разговоре, может быть связана с воспалением слуховых путей или проблемами с височно-нижнечелюстным суставом. Важно обратиться к врачу для обследования и диагностики. Врач может назначить обезболивающие и противовоспалительные препараты для снятия боли. Также могут быть рекомендованы физиотерапевтические процедуры для восстановления нормальной функции. Своевременное лечение предотвратит осложнения.
Больше информации на сайте – https://onlinvrach.ru
красные щеки у грудничка, нижнекамск эндокринологи, сова лор врач саратов
как подготовиться к узи мочевого пузыря и почек женщине, эндокринолог куликова новороссийск, судороги у ребенка во сне
Удачи!
Willieavamy
| #
Online casinos need hold—too wild! mines
Willieavamy
| #
The live dealers online glow—real touch! mines
PeterSlump
| #
https://telegra.ph/Welsh-Non-Gamstop-Casinos-Guide-for-Players-03-27
Sazrerq
| #
купить диплом техникума нижнем
Willieavamy
| #
The VIP programs at online casinos are a nice perk if you play a lot. bizzo casino
DavidNaing
| #
This casino nailed it with the Tiger Fortune slot design. fortune tiger 777
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние достижения.
Наши товары:
Плоская мишень из фтористого магния
PeterSlump
| #
https://telegra.ph/Discover-UK-Non-Gamstop-Casinos-for-Fair-Play-Options-03-27
betmotion
| #
Aproveite a oferta exclusiva do betmotion para novos usuários e receba 100$ de bônus ao se registrar!
Este bônus de boas-vindas permite que você experimente uma vasta
gama de jogos de cassino online sem precisar gastar imediatamente.
Com o bônus de 100$, você poderá explorar jogos como roleta, blackjack, caça-níqueis e muito mais,
aumentando suas chances de vitória desde o primeiro minuto.
Não perca essa chance única de começar com um valor significativo –
cadastre-se agora!
mostbet_kret
| #
мостбет https://www.mostbet6034.ru .
1win_bupa
| #
1win казино http://www.1win6010.ru .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наша продукция:
Труба из драгоценных металлов золотая 600х1х0.7 мм ЗлМ98 ТУ Гарантированное качество и изысканный дизайн – купите драгоценные трубы для ювелирных украшений, машиностроения и архитектуры. Широкий выбор и высокое качество! Доставка по России.
Sazrrnl
| #
Заказать диплом ВУЗа по доступной цене возможно, обращаясь к надежной специализированной компании. Мы предлагаем документы об окончании любых ВУЗов РФ. Купить диплом о высшем образовании– poluchidiplom.com/kupit-diplom-o-visshem-obrazovanii-v-sumax-bistro/
cost of cheap sinemet price
| #
Drug information leaflet. Short-Term Effects.
cost of cheap sinemet price
Best about medicine. Get information now.
zapihuRus
| #
Probanki-online.ru – ресурс, который вам о банковских услугах и финансовых продуктах в РФ исчерпывающую информацию предоставляет. Расскажем, автокредит, что это такое. Объясним, как идеальную карту выбрать. Вы о плюсах вкладов узнаете. Стремимся быть в курсе последних финансовых событий. Ищете онлайн карта банка? Probanki-online.ru – здесь можете отыскать информацию о разных банках и их предложениях. На портале имеется поиск, примените его. Наша цель – сделать процесс подбора финансовых продуктов простым и удобным. Добро пожаловать на probanki-online.ru!
Trefqkp
| #
Заказать диплом об образовании. Приобретение диплома ВУЗа через качественную и надежную фирму дарит ряд достоинств. Данное решение позволяет сэкономить как дорогое время, так и значительные финансовые средства. cdltruckdrivingcareers.com/employer/eonline-diploma
internetCah
| #
интернет провайдеры по адресу дома
domashij-internet-spb002.ru
интернет по адресу санкт-петербург
ManitnTonge
| #
Вместе с t.me/azino777_a у вас есть возможность в увлекательное путешествие по миру азартных игр отправиться. Предлагаем персонализированные предложения и уникальные бонусы. Расскажем, как в Азино начать играть. Вас ждут невероятные эмоции и возможность выиграть крупный приз. Не упустите шанс испытать свою удачу! Ищете azino777 бонус 2000? – тут имеется множество актуальной информации. Собрали самые частые вопросы от игроков. Всегда готовы помочь вам. Приготовьтесь к завлекательным в Азино турнирам. Участвуйте и боритесь за призы с другими игроками!
Lazrqfl
| #
Диплом любого ВУЗа России!
Без института очень трудно было продвигаться вверх по карьерной лестнице. Именно из-за этого решение о покупке диплома стоит считать выгодным и целесообразным. Заказать диплом об образовании kontorka.flybb.ru/viewtopic.php?f=9&t=2527
1win_tamt
| #
1win online site http://1win15.com.ng/ .
where can i buy nolvadex online
| #
Medicine prescribing information. What side effects can this medication cause?
where can i buy nolvadex online
Everything what you want to know about medication. Read information now.
Jariorsdx
| #
Где купить диплом по актуальной специальности?
Наша компания предлагаетмаксимально быстро приобрести диплом, который выполнен на оригинальной бумаге и заверен печатями, штампами, подписями официальных лиц. Документ пройдет лубую проверку, даже при помощи специальных приборов. Решите свои задачи максимально быстро с нашим сервисом. Приобрести диплом университета! kylianmbappeclub.com/read-blog/7846_tehnicheskij-diplom.html
Diplomi_cpKt
| #
Купить диплом любого университета!
Мы готовы предложить документы институтов, которые расположены на территории всей РФ.
diplomv-v-ruki.ru/diplom-visshego-obrazovaniya-s-zaneseniem-v-reestr-4/
Jariorqje
| #
Где приобрести диплом по актуальной специальности?
Мы предлагаем выгодно заказать диплом, который выполняется на бланке ГОЗНАКа и заверен печатями, штампами, подписями официальных лиц. Диплом способен пройти любые проверки, даже при помощи профессиональных приборов. Решите свои задачи максимально быстро с нашим сервисом.
Купить диплом о высшем образовании diplomv-v-ruki.ru/gde-kupit-diplom-11/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние достижения.
Наши товары:
Гранулы цинка 4N
DavidNaing
| #
Tiger Fortune is perfect for thrill-seekers like me. fortune tiger 777
Jamesval
| #
Здравствуйте!
Ремонт дома с баней — это уникальная возможность создать комфортное место для отдыха и проживания. Мы предлагаем качественные решения для различных типов домов, включая добавление бань и саун. Наши специалисты помогут выбрать лучшие материалы для отделки, чтобы дом и баня выглядели стильно и современно. Мы учитываем все пожелания заказчика и обеспечиваем комфортные условия для проживания. внутренние отделки деревянных домов, балкон ремонт дома, строительство деревянного дома бани Наши работы выполняются с учетом всех особенностей и на высшем уровне.
Больше информации на сайте – https://nhadian123.com
документ для строительства дома, примеры ремонт в квартире, строительство из газоблока бани
интернет магазин ремонт дома смоленск, строительство домов в архангельском, ремонт и отделка бани
Удачи!
DavidNaing
| #
Tiger Fortune’s free spins are pure gold. tiger fortune
JosephAttit
| #
buy cali weed in prague hash delivery in prague
mostbet_jyKn
| #
mostber mostbet6012.ru .
1win_nzKr
| #
1win login ug 1win login ug .
pin up azerbaycan_ciMl
| #
pinup azerbaycan pinup azerbaycan .
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы а также находить ответы под требования вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наши товары:
Порошок магниевый AZ61 – CSA HG.5 Рзделия РёР· магния AZ61 – CSA HG.5 представляют СЃРѕР±РѕР№ высококачественные компоненты, обладающие отличной прочностью Рё РЅРёР·РєРёРј весом. РћРЅРё идеально РїРѕРґС…РѕРґСЏС‚ для применения РІ авиационной Рё автомобильной промышленности, Р° также РІ производстве спортинвентаря. Рти изделия отличаются хорошей РєРѕСЂСЂРѕР·РёР№РЅРѕР№ стойкостью, что повышает РёС… долговечность. Благодаря СЃРІРѕРёРј свойствам, Рзделия РёР· магния AZ61 – CSA HG.5 становятся незаменимыми РІ современных технологиях. РќРµ упустите возможность купить Рзделия РёР· магния AZ61 – CSA HG.5 Рё обеспечить своей продукции надежность Рё эффективность!
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наша продукция:
Плоская мишень Fe+Mn
1win_rhEa
| #
1win войти 1win6044.ru .
Sazrcpm
| #
Купить диплом ВУЗа !
Покупка диплома университета России в нашей компании – надежный процесс, ведь документ заносится в реестр. Купить диплом о высшем образовании damdiplomisa.com/kupit-diplom-s-zaneseniem-dlya-uspeshnoj-kareri-3
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наши товары:
Быстрорежущая пластина форма 49 номер 4907 стандартная 15х28х12 мм сечение резца 50х32 мм Р18 ГОСТ 2379-77 Выберите лучшие быстрорежущие пластины к резцам онлайн. Отличная износостойкость, высокая точность обработки и широкий выбор размеров. Узнайте, как наши пластины могут улучшить вашу металлообработку.
mostbet_phet
| #
мостбет казино мостбет казино .
Arroncep
| #
Отец и сын три года объедали сотню ресторанов по хитроумной схеме
https://pin.it/kCDDdtZJq
1win_hvOl
| #
1win md http://1win5013.ru .
cost of cheap coversyl pill
| #
where to get cheap coversyl pills
where to buy cheap dramamine tablets
| #
cost cheap dramamine online
mostbet_somt
| #
мостбет скачать бесплатно mostbet6033.ru .
how to buy generic zithromax without rx
| #
Meds information leaflet. Brand names.
how to buy generic zithromax without rx
Best information about drug. Read information now.
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
поставляемая продукция:
Рнструментальная круглая РїРѕРєРѕРІРєР° 185 РјРј РҐ12Р’РњР¤ ГОСТ 1133-71 РЁРёСЂРѕРєРёР№ выбор круглых РїРѕРєРѕРІРѕРє для профессиональной обработки металла. Рнструментальная круглая РїРѕРєРѕРІРєР° высочайшего качества СЃ разнообразием размеров Рё форм. Повышенная прочность, износостойкость Рё превосходное качество изготовления. Обеспечивает отличное сцепление СЃ материалом Рё эффективную работу оборудования. Покупайте РїСЂСЏРјРѕ сейчас!
DavidNaing
| #
Tiger Fortune’s free spins are pure gold. fortune tiger 777
1win_xepa
| #
1хwin 1хwin .
1win_ygst
| #
один вин официальный сайт https://1win6045.ru .
1win_xuKr
| #
1win casino uganda https://1win1003.top/ .
DavidNaing
| #
This online casino’s Tiger Fortune is top-tier. fortune tiger bet
buy generic valtrex
| #
Drug information. Effects of Drug Abuse.
buy generic valtrex
Some about pills. Get information here.
DavidNaing
| #
I love how Tiger Fortune mixes luck and fun. fortune tiger demo
DavidNaing
| #
This casino’s Tiger Fortune game is a gem. fortune tiger bet
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы а также находить ответы под специфику вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Полоса кобальтовая Stellite 4B Полоса кобальтовая Stellite 4B – это высококачественный материал, предназначенный для обработки Рё защиты деталей РІ условиях высокой температуры Рё абразивного РёР·РЅРѕСЃР°. Рта кобальтовая полоса обеспечивает отличную РєРѕСЂСЂРѕР·РёРѕРЅРЅСѓСЋ стойкость Рё долговечность, что делает её идеальным выбором для промышленного применения. Восстановление Рё защита металлических поверхностей СЃ помощью полосы Stellite 4B значительно увеличивает СЃСЂРѕРє службы изделий. РќРµ упустите возможность купить Полоса кобальтовая Stellite 4B Рё улучшить производительность ваших процессов уже сегодня!
Sazrucd
| #
купить диплом о высшем образовании цена в ростове на дону
Lombard chasov_vmEl
| #
онлайн оценка швейцарских часов онлайн оценка швейцарских часов .
stomatologiya Mitino_rsPn
| #
частная стоматология в москве частная стоматология в москве .
DavidNaing
| #
I’ve had some massive wins on Tiger Fortune! tigrinho demo
pereplanirovka_lbOi
| #
согласование перепланировки москва согласование перепланировки москва .
Rodrigodiuse
| #
Desert Eagle Blaze http://csgo-get-skins.ru пламя в каждой пуле. Легенда CS:GO. Продажа, обмен, проверка скина. Успей забрать по лучшей цене на рынке.
RobertEmisp
| #
M4A1-S Knight https://cs-get-skins.ru редкий скин в CS. Престиж, стиль, легендарное оружие. Продажа по выгодной цене, моментальная доставка, безопасная сделка.
1win_mqPt
| #
1win. http://1win6009.ru/ .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наши товары:
Труба бронзовая 50х10х1500 мм БрАМц9-2 ГОСТ 1208-2014 Приобретайте трубы бронзовые круглые от профессионалов с опытом. Мы предлагаем широкий ассортимент качественной продукции, быструю обработку заказов и оперативную доставку. Обращайтесь, чтобы получить консультацию специалиста и выгодные условия покупки.
mostbet_bfet
| #
мостбет скачать на андроид http://www.mostbet6034.ru .
internetCah
| #
узнать интернет по адресу
domashij-internet-spb003.ru
интернет провайдер санкт-петербург
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их соответствие стандартам. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы и адаптировать решения под требования вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Рзделия РёР· магния MgAl3Zn1 (Alloy 21) – ISO 3116 РџРѕРєРѕРІРєР° магниевая MgAl3Zn1 (Alloy 21) – ISO 3116 является высококачественным сплавом, известным своей легкостью Рё прочностью. Ртот материал применяется РІ авиационной, автомобильной Рё РґСЂСѓРіРёС… отраслях, РіРґРµ требуется устойчивость Рє РєРѕСЂСЂРѕР·РёРё Рё отличные механические свойства. РџРѕРєРѕРІРєР° магниевая MgAl3Zn1 (Alloy 21) – ISO 3116 обеспечивает надежность Рё долговечность, что делает её идеальным выбором для промышленных нужд. Если РІС‹ ищете прочный Рё легкий материал, рекомендуем купить РџРѕРєРѕРІРєР° магниевая MgAl3Zn1 (Alloy 21) – ISO 3116. Выбирая этот сплав, РІС‹ инвестируете РІ качество Рё надежность.
Cazrlgk
| #
Заказать диплом о высшем образовании!
Мы можем предложить документы учебных заведений, которые находятся на территории всей России. Дипломы и аттестаты выпускаются на бумаге самого высокого качества: odnopolchane.net/forum/member.phpu=546804
Willieavamy
| #
The live dealers online are ace—real deal! plinko
Trefzwg
| #
Купить диплом университета. Заказ документа о высшем образовании через проверенную и надежную фирму дарит ряд преимуществ. Такое решение позволяет сберечь время и значительные финансовые средства. career.digitalgurukul.in/employer/eonline-diploma
Arroncep
| #
Отец и сын три года объедали сотню ресторанов по хитроумной схеме
https://pin.it/kCDDdtZJq
Jariorbvh
| #
Где купить диплом по необходимой специальности?
Наши специалисты предлагают выгодно и быстро заказать диплом, который выполняется на бланке ГОЗНАКа и заверен печатями, водяными знаками, подписями должностных лиц. Наш диплом пройдет лубую проверку, даже при помощи специального оборудования. Решите свои задачи быстро и просто с нашей компанией.
Заказать диплом любого университета kupitediplom0027.ru/gde-kupit-diplom-o-srednem-obrazovanie-14/
1win_baKr
| #
1win bet uganda http://1win1003.top .
1win_wwOl
| #
cazinouri online moldova https://www.1win5013.ru .
Willieavamy
| #
I love the anonymity of online gambling—no one knows who I am! plinko
sex hot le
| #
sex hot le
A Brief History of Elemental Analysis » Artemis Analytical
1win_mgpa
| #
1win com http://1win6010.ru/ .
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их качество. Опытная поддержка – наш стандарт – мы на связи, чтобы улаживать ваши вопросы и находить ответы под требования вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
Проволока титановая Р’Рў16 – РћРЎРў 1 90013-81 Полоса титановая Р’Рў16 – РћРЎРў 1 90013-81 представляет СЃРѕР±РѕР№ высококачественный титановый сплав, обладающий отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью Рё прочностью. Ртот материал идеально РїРѕРґС…РѕРґРёС‚ для машиностроения Рё аэрокосмической промышленности. Полоса имеет высокую пластичность Рё может быть легко обработана. Если вам требуется надежный Рё долговечный материал, то РІС‹ РЅР° правильном пути. РќРµ упустите возможность купить Полоса титановая Р’Рў16 – РћРЎРў 1 90013-81 РїРѕ выгодной цене. Благодаря СЃРІРѕРёРј уникальным свойствам, данная полоса станет отличным выбором для решений РІ различных отраслях.
Harleyamurn
| #
Этот информативный текст выделяется своими захватывающими аспектами, которые делают сложные темы доступными и понятными. Мы стремимся предложить читателям глубину знаний вместе с разнообразием интересных фактов. Откройте новые горизонты и развивайте свои способности познавать мир!
Получить больше информации – https://mednarkoforum.ru/
Eanrcnr
| #
Для успешного продвижения по карьерной лестнице требуется наличие диплома института. Приобрести диплом любого ВУЗа у сильной фирмы: diplomp-irkutsk.ru/diplom-za-11-klass-kupit-3/
how to buy bactrim for sale
| #
Medicines information sheet. Short-Term Effects.
how to buy bactrim for sale
Everything about medicine. Read here.
Mazrhoe
| #
Мы можем предложить дипломы психологов, юристов, экономистов и прочих профессий по приятным тарифам. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Важно, чтобы документы были доступны для большинства наших граждан.
Покупка документа, который подтверждает окончание института, – это выгодное решение. Приобрести диплом любого ВУЗа: 1magistr.ru/kupit-diplom-vuza-visshee-3/
DavidNaing
| #
Tiger Fortune has quickly become my favorite game here. fortune tiger bet
Lombard chasov_kkEl
| #
заложить часы в ломбард москва заложить часы в ломбард москва .
Dnrtoyi
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и любых других профессий по доступным тарифам. Мы готовы предложить документы ВУЗов, расположенных в любом регионе России. Дипломы и аттестаты выпускаются на “правильной” бумаге самого высшего качества. Это позволяет делать государственные дипломы, не отличимые от оригинала. parenvarmii.ru/viewtopic.phpf=21&t=5426&sid=e2f7a7d7aff381fdafcf17226317960b
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Вращающаяся мишень для распыления вольфрама
stomatologiya Mitino_ahPn
| #
стоматологии г москва стоматологии г москва .
EdwinEncaw
| #
Гигантская черная дыра пробудилась, заявили ученые
https://x.com/kiselev_igr/status/1910669547713003851
Diplomi_tlka
| #
купить настоящий диплом гознак купить настоящий диплом гознак .
Sazrzep
| #
Хорошо быть студентом, пока не придет пора писать диплом, что и произошло со мной, но не стоит отчаиваться, ведь есть хорошие компании что помогают с написанием и сдачей диплома на хорошие оценки!
Изначально искал информацию про реавиз медицинский университет купить его диплом, речной диплом купить, где купить диплом с занесением, где купить диплом сколько стоит, где можно купить диплом воспитателя, потом попал на kyc-diplom.com/diplom-articles/kupit-attestat-2017.html
pereplanirovka_yzOi
| #
согласование перепланировки согласование перепланировки .
1win_usOl
| #
1win 1win5013.ru .
cost of generic lamisil without dr prescription
| #
can i order lamisil tablets
1win_vtEa
| #
1win. com 1win. com .
doxycycline 50 mg side effects
| #
can i order generic doxycycline price
https://vpenze.ru/news/metallicheskie-znachki-na-zakaz-iskusstvo-kotoroe-govorit/
| #
Металлические значки на заказ являются не только стильным аксессуаром, но и эффективным способом выразить индивидуальность или подчеркнуть принадлежность к определённой группе. Их производство сочетает в себе искусство и мастерство, позволяя создавать уникальные изделия с высоким уровнем детализации. Подробнее об этом можно узнать по ссылке:
https://vpenze.ru/news/metallicheskie-znachki-na-zakaz-iskusstvo-kotoroe-govorit/
Sazrzwm
| #
диплом новосибирск купить
nerve calm
| #
nerve calm is a high-quality nutritional supplement crafted to promote nerve wellness, ease chronic discomfort, and boost everyday vitality.
ArthurFug
| #
best ai porn ai chat porn
Williamvub
| #
Krause-ekb.ru большой выбор стремянок и лестниц по доступным ценам предоставляет. Работаем как с частными лицами, так и с организациями. Бесплатную консультацию мы для вас можем предложить. Совершить покупки стало гораздо проще, теперь не надо из дома выходить. Для этого каталогом воспользуйтесь, который на ресурсе предоставлен. Ищете купить лестницу винтовую? Krause-ekb.ru – тут подробная информация о каталоге опубликована. Предложить качественные товары и доставить заказ покупателю как можно оперативнее – это основная цель Krause. При возникновении вопросов, смело звоните нам по телефону.
Trefzvk
| #
Купить диплом университета. Приобретение официального диплома через надежную фирму дарит ряд преимуществ. Это решение помогает сберечь время и значительные финансовые средства. afterpad.com/forums/post.php?fid=7
ScottElori
| #
Используй нвдежный amnezia.xyz для обхода блокировок, сохранности личных данных и полной свободы онлайн. Всё включено — просто подключайся.
Jariorwms
| #
Где заказать диплом по нужной специальности?
Наша компания предлагает быстро и выгодно заказать диплом, который выполнен на оригинальном бланке и заверен мокрыми печатями, штампами, подписями официальных лиц. Диплом способен пройти лубую проверку, даже при использовании профессиональных приборов. Достигайте своих целей быстро с нашей компанией.
Заказать диплом любого ВУЗа kupitediplom0027.ru/kupit-diplom-v-permi-8/
Lombard chasov_ymEl
| #
скупка часов москва скупка часов москва .
stomatologiya Mitino_vwPn
| #
американская клиника в москве stomatologiya-mitino1.ru .
Andrewgor
| #
Оснащение под ключ: дерматологическое оборудование купить в Москве с сертификацией и полной документацией. В наличии и под заказ. Профессиональное оснащение медицинских кабинетов.
Mazrjvg
| #
Заказать диплом об образовании!
Мы оказываем услуги по продаже документов об окончании любых университетов РФ. Документы изготавливаются на настоящих бланках государственного образца. frayala.switch-maker.net/read-blog/7226_kupit-diplom-s-reestrom-otzyvy.html
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наша продукция:
Металлическая мишень для распыления железа
pereplanirovka_uwOi
| #
согласование перепланировки помещений согласование перепланировки помещений .
spironolactone pcos hair loss
| #
Pills prescribing information. What side effects can this medication cause?
spironolactone pcos hair loss
Actual information about meds. Get information now.
DavidNaing
| #
The spins in Tiger Fortune are always exciting. fortune tiger demo
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наши товары:
Прецизионная профильная сварная прямоугольная труба60х40х2.5ммE260EN 10305-5 Предлагаем широкий выбор прецизионных профильных сварных и холоднокалиброванных прямоугольных труб высочайшего качества для различных инженерных и промышленных приложений. Наши трубы отличаются высокой точностью и механической прочностью, подходят для авиации, автомобилестроения, судостроения и других областей. Обратитесь за более подробной информацией о технических характеристиках и условиях поставки.
Sogiphvor
| #
Академия «Арчибальд» занимается обучением грумингу. У вас есть возможность свои навыки совершенствовать до бесконечности на живых моделях. Преподаватели на все вопросы отвечают. Дарим видеолекции, методические материалы и учебники. Окунитесь в атмосферу, с которой расставаться не хочется. Ищете курсы груминга недорого в москве? Grooming-dream.ru – тут реальные отзывы клиентов представлены. Мы выпускаем отменных груминг-специалистов. Предлагаем инструменты в период обучения. Если хотите освоить новую профессию, «Арчибальд» будет самым подходящим для вас местом.
DavidNaing
| #
This online casino’s Tiger Fortune is a blast. fortune tiger
SilekSiz
| #
Форум социальной инженерии — Lolz.live предлагает уникальную возможность на выгодных условиях продать игровые аккаунты, а также учетные записи для социальных сетей, скилы и различную игровую атрибутику. Этот портал будет полезен как новичкам, которые только рассматривают продажу аккаунтов в качестве заработка, так и тем, для кого это уже стало бизнесом.
Jeffreyrah
| #
https://dev-mastery.pro/
ciyovCoosy
| #
Вы находитесь на едином сайте аренды, где получится взять в прокат различную технику для выполнения самых разных работ. Также получится выложить объявления со своими предложениями, чтобы ими воспользовались все желающие. https://arenda24.by/ – на портале ознакомьтесь с категориями, которые точно вызовут интерес – техника для грузоперевозок, режущее оборудование, краны, которые принимают участие в строительстве и другими предложениями. Вы сможете воспользоваться всеми опциями при условии, что зарегистрируетесь.
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их происхождение. Превосходное обслуживание – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы а также предоставлять решения под специфику вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наша продукция:
РљСЂСѓРі титановый Р’Рў23 – РћРЎРў 1 90013-81 Фольга титановая Р’Рў23 – РћРЎРў 1 90013-81 представляет СЃРѕР±РѕР№ высококачественный материал, который идеально РїРѕРґС…РѕРґРёС‚ для различных промышленных применений. РћРЅР° обладает отличными механическими свойствами Рё высокой РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью, что делает ее незаменимой РІ авиационной Рё химической промышленности. Приобретая фольгу, РІС‹ гарантируете себе надежность Рё долговечность РІ эксплуатации. Чтобы купить Фольга титановая Р’Рў23 – РћРЎРў 1 90013-81, обращайтесь Рє нашим специалистам, которые РїРѕРјРѕРіСѓС‚ вам СЃ выбором Рё оформлением заказа. РќРµ упустите возможность использовать этот первоклассный материал!
TeddyGEONO
| #
Технологии свободы: marzban.click/ надёжный инструмент для приватности, скорости и доступа к любым сайтам. Быстро, безопасно, удобно — настрой за 5 минут.
MatthewLak
| #
Витебский госуниверситет университет https://vsu.by/abiturientam/priemnaya-kampaniya.html П.М.Машерова – образовательный центр. Вуз является ведущим образовательным, научным и культурным центром Витебской области. ВГУ осуществляет подготовку: химия, биология, история, физика, программирование, педагогика, психология, математика.
Dnrtkkm
| #
Мы готовы предложить дипломы любой профессии по разумным ценам. Мы готовы предложить документы ВУЗов, которые расположены в любом регионе РФ. Документы выпускаются на бумаге самого высокого качества. Это дает возможность делать государственные дипломы, которые не отличить от оригинала. b4india.in/read-blog/682_kupit-nastoyashij-diplom.html
Jeffreyrah
| #
https://dev-mastery.pro/
mostbet_grmt
| #
mostbest mostbest .
Brucewed
| #
https://xnudes.app/
buy cheap viagra_omSr
| #
Soft2bet http://cheapviagrausaonline.com .
LarrySop
| #
голд казино
Jamesemusy
| #
Поставка и монтаж кинооборудования от ведущих производителей — Подробная информация здесь https://www.cheaperseeker.com/u/Neocinema
DavidNaing
| #
Tiger Fortune is the best game here, hands down! fortune tiger bet
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наши товары:
Кольцо РёР· драгоценных металлов платиновое 15С…4С…0.5 РјРј РџР»99.9 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
1win_oast
| #
партнёрка 1win http://1win6045.ru .
can you take amoxicillin for staph infection
| #
Medicine prescribing information. Long-Term Effects.
can you take amoxicillin for staph infection
Actual information about medicine. Get here.
GeorgeTom
| #
Source:
– norma3.ru
Normanmep
| #
купить чек на товар купить чеки для отчетности в москве
KevinTrutt
| #
about his TAX
MichaelAlile
| #
Научитесь вязать крючком crochet-patterns.ru/ с нуля или улучшите навыки с нашими подробными мастер-классами. Фото- и видеоуроки, понятные инструкции, схемы для одежды, игрушек и интерьера. Вдохновляйтесь, творите, вяжите в своё удовольствие! Вязание крючком — доступно, красиво, уютно.
Sazrczk
| #
Приобрести диплом института по доступной стоимости возможно, обращаясь к проверенной специализированной фирме. Мы предлагаем документы об окончании любых университетов Российской Федерации. Приобрести диплом любого университета– diplomj-irkutsk.ru/kupit-diplom-s-zaneseniem-v-reestr-bistro-i-nadezhno-65/
DavidNaing
| #
Tiger Fortune’s visuals are a feast for the eyes. fortune tiger
1win_puSn
| #
1win вход на сайт https://www.1win6046.ru .
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы и находить ответы под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
Лист вольфрамовый W-Re Лист вольфрамовый W-Re – это высококачественный материал, предназначенный для использования РІ различных отраслях. Благодаря СЃРІРѕРёРј уникальным свойствам, таким как высокая плотность Рё жаропрочность, РѕРЅ прекрасно РїРѕРґС…РѕРґРёС‚ для применения РІ машиностроении, электронике Рё металлургии. Лист вольфрамовый W-Re обеспечивает отличную стабильность РїСЂРё высоких температурах Рё является идеальным выбором для создания долговечностных изделий. РќРµ упустите возможность купить Лист вольфрамовый W-Re, чтобы обеспечить себе надежный Рё эффективный материал для вашего производства!
mostbet_fcmt
| #
мостбет авиатор http://mostbet6033.ru/ .
TerryLib
| #
ауф казино, болливуд казино
Stevenler
| #
look at here now DOB
https://www.3d4c.fr/wiki/index.php/Cvety_57y
| #
Заказываю уже третий раз – каждый раз восторг и восхищение от качества!
Also visit my web-site: https://wikis.ece.iastate.edu/cpre488/index.php?title=Cvety_24l
Iariorukn
| #
Купить диплом ВУЗа по выгодной стоимости возможно, обращаясь к надежной специализированной компании. Заказать документ о получении высшего образования можно у нас. diplomp-irkutsk.ru/kupit-diplom-s-zaneseniem-12
mostbet_yfet
| #
мостбет официальный сайт мостбет официальный сайт .
AndrewVindy
| #
Нож-бабочка Doppler cs-get-skin стильное и эффектное оружие в стиле CS:GO. Яркий металлический блеск, плавный механизм, удобство в флиппинге и коллекционировании. Подходит для тренировок, трюков и подарка фанатам игр.
KIBRIS VİP TRANSFER
| #
KIBRIS VİP TRANSFER
A Brief History of Elemental Analysis » Artemis Analytical
Dnrtmnp
| #
Мы можем предложить дипломы любой профессии по выгодным ценам. Мы готовы предложить документы техникумов, расположенных в любом регионе Российской Федерации. Дипломы и аттестаты делаются на “правильной” бумаге самого высшего качества. Это дает возможность делать настоящие дипломы, не отличимые от оригиналов. babygirls010.copiny.com/question/details/id/1082960
Willieavamy
| #
I love the pay flow online—easy run! mines
mostbet_tiKn
| #
мостбет скачать бесплатно https://www.mostbet6012.ru .
cost of doxycycline pill
| #
can you get doxycycline without rx
DavidNaing
| #
Tiger Fortune is the best game here, hands down! fortune tiger demo
mostbet_demt
| #
мост бет http://mostbet6033.ru/ .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Керамическая мишень Y2O3 – 4N, плоская форма
hydroxyzine hcl hydroxyzine pamoate difference
| #
Pills information leaflet. Effects of Drug Abuse.
hydroxyzine hcl hydroxyzine pamoate difference
Some news about meds. Read here.
best online casino uk
| #
best online casino uk
A Brief History of Elemental Analysis » Artemis Analytical
where can i buy caduet no prescription
| #
cost cheap caduet online
Jamesemusy
| #
Индивидуальное сопровождение личного помощника в ОАЭ — Подробнее по ссылке https://doc.aquilenet.fr/s/OrVa3L34U
Automated Trading Bot
| #
Automated Trading Bot
A Brief History of Elemental Analysis » Artemis Analytical
Diplomi_lhKt
| #
Заказать диплом о высшем образовании!
Мы предлагаем документы институтов, которые находятся на территории всей Российской Федерации.
diplomh-40.ru/kupit-diplom-v-moskve-s-zaneseniem-v-reestr-bistro-6/
nya svenska casinon
| #
nya svenska casinon
A Brief History of Elemental Analysis » Artemis Analytical
crypto casino
| #
crypto casino
A Brief History of Elemental Analysis » Artemis Analytical
Responsible Gaming
| #
Responsible Gaming
A Brief History of Elemental Analysis » Artemis Analytical
Hubertsek
| #
Looking for sell prompts? Findprompt.shop where you can choose from a huge selection of promts for Midjourney, Stable Diffusion, ChatGPT and more. Explore the best AI tips. You can sell your promts at a favorable price on our platform. Pay attention to the selection of promts on the site – it is the largest on the market. Our offers are unique, and the cost is affordable for everyone.
DavidNaing
| #
The animations in Tiger Fortune are stunning. fortune tiger
lawyers in my area
| #
This paragraph will help the internet viewers for building up new webpage or ecen a weblog from start to end.
Best online casino
| #
Best online casino
A Brief History of Elemental Analysis » Artemis Analytical
mostbet_otet
| #
мостбет скачать андроид https://www.mostbet6034.ru .
1win_ntst
| #
1.вин http://1win6045.ru/ .
Mazrqqh
| #
Купить диплом о высшем образовании!
Мы оказываем услуги по производству и продаже документов об окончании любых университетов Российской Федерации. Документы производят на настоящих бланках. worksale.mn.co/posts/82689054
buy cheap viagra_jiSr
| #
buy viagra online cheap http://sophiaescorts.com .
1win_uiSn
| #
1win win https://1win6046.ru/ .
DavidNaing
| #
Tiger Fortune’s bonus rounds are the best part. fortune tiger bet
valsartan amlodipine hydrochlorothiazide ppt
| #
Medicament information. Short-Term Effects.
valsartan amlodipine hydrochlorothiazide ppt
All about pills. Get information here.
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наши товары:
Латуное наружное стопорное пружинное кольцо M145 138С…4 РјРј ЛК80 DIN 471 Латунные наружные стопорные кольца РїРѕ DIN – широкий выбор высококачественных крепежных изделий РёР· латуни для промышленных Рё строительных применений. Большой ассортимент размеров. Высокая надежность Рё долговечность. Заказывайте РїСЂСЏРјРѕ сейчас РЅР° сайте Редметсплав.
Willieavamy
| #
Online casinos need grip—too loose! aviator
mostbet_wjet
| #
mostbet chrono https://www.mostbet6034.ru .
hedinic
| #
Блог о маркетплейсах «OOWA» предлагает ознакомиться с актуальной, интересной и познавательной информацией, которая касается удачных продаж на маркетплейсах, обзоров товаров и услуг. На портале регулярно публикуются ценные и содержательные рекомендации от экспертов, которые помогут начать лучше разбираться в тонкостях такого бизнеса. Ищете продажи на маркетплейсах? На oowa.ru регулярно публикуются ценные и содержательные рекомендации от экспертов, которые помогут начать лучше разбираться в тонкостях такого бизнеса. Почитайте о самых востребованных товарах с маркетплейсов.
DavidNaing
| #
I can’t get enough of the Tiger Fortune soundtrack! tigrinho demo
DavidNaing
| #
Tiger Fortune is my favorite way to unwind. tigrinho demo
DavidNaing
| #
Tiger Fortune is the best slot in this casino’s lineup. fortune tiger bet
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – наш стандарт – мы на связи, чтобы ответить на ваши вопросы по мере того как предоставлять решения под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наша продукция:
Лист ванадиевый FeV80Al12 – ГОСТ 27130-94 Лист ванадиевый FeV80Al12 – ГОСТ 27130-94 представляет СЃРѕР±РѕР№ надежный металлический материал, обладающий высокой прочностью Рё устойчивостью Рє РєРѕСЂСЂРѕР·РёРё. Такой лист используется РІ различных отраслях, включая машиностроение Рё авиационную промышленность. Лист ванадиевый FeV80Al12 – ГОСТ 27130-94 также отлично РїРѕРґС…РѕРґРёС‚ для изготовления деталей, работающих РІ условиях повышенных нагрузок. Если РІС‹ ищете качественный Рё прочный материал, Сѓ нас РІС‹ можете купить Лист ванадиевый FeV80Al12 – ГОСТ 27130-94 РїРѕ выгодной цене. Рто идеальное решение для вашего бизнеса!
DavidNaing
| #
Tiger Fortune keeps the adrenaline pumping every spin. jogo do tigrinho demo
cost vastarel for sale
| #
how to get cheap vastarel no prescription
winny
| #
Start with a Bang! $100 Bonus Awaits You at winny!
where buy generic tolterodine without prescription
| #
Pills prescribing information. Long-Term Effects.
where buy generic tolterodine without prescription
All information about drugs. Read now.
order epivir pills
| #
cost of generic epivir pill
nextbet
| #
Start your journey at nextbet with an exclusive $100 bonus!
New users can instantly claim this offer when they register,
giving them a head start on exploring the diverse range of online casino
games, slots, and sports betting options available. Don’t miss out on this exciting opportunity—sign up today and take advantage of this amazing $100 bonus to
boost your chances of winning big!
Diplomi_ynKt
| #
Купить диплом университета!
Мы готовы предложить документы любых учебных заведений, которые находятся на территории всей России.
diplomk-vo-vladivostoke.ru/kupit-diplom-s-provodkoj-otzivi/
Williamvof
| #
Вас незаконно увольняют с работы? Квалифицированный трудовой адвокат поможет оспорить приказ об увольнении. В современном мире практически каждый человек понимает важность знания законов, ведь это обеспечивает защиту от различных проблем и позволяет осознавать свои права и обязанности. Полное знание всех законов, особенно для неспециалиста, недостижимо. Поэтому известная адвокатская коллегия предлагает свои профессиональные услуги.
Мы готовы взяться за любое дело, независимо от его сложности, благодаря многолетнему опыту, позволяющему оказывать квалифицированную помощь. Каждый адвокат в нашей фирме специализируется на определенной области права. Это позволяет подобрать специалиста, который глубоко разбирается в вашей конкретной ситуации. В таком случае ваши шансы на успех в суде значительно возрастают по сравнению с обращением к юристу-универсалу.
Наша фирма быстро приобрела известность в России. Это обусловлено опытом наших юристов, специализирующихся на конкретных делах, а также доступными ценами. Мы работаем прозрачно, поэтому на нашем сайте представлены актуальные расценки и подробная информация о сотрудниках, готовых вам помочь. В договоре отсутствуют скрытые комиссии, которые часто используют недобросовестные фирмы.
Адвокат подробно объяснит все этапы, полную стоимость, а также важные детали. Мы понимаем, что могут возникнуть финансовые трудности, но помощь юриста нужна немедленно. Поэтому мы предлагаем различные варианты рассрочки или отсрочки платежа. В некоторых случаях возможна оплата по результату, то есть после выигрыша в суде.
На нашем сайте вы найдете много полезных материалов, которые помогут вам повысить свою правовую грамотность. Рекомендуем регулярно посещать наш сайт, чтобы быть в курсе своих прав. Мы также предоставляем информацию о необходимых документах для различных процедур, таких как использование материнского капитала или банкротство. Более подробную консультацию вы можете получить у нашего адвоката, контакты которого указаны на сайте.
0832 Yupoo For Sale
| #
0832 Yupoo For Sale
A Brief History of Elemental Analysis » Artemis Analytical
alexander
| #
Start Your alexander Journey with a $100 Bonus on Registration!
oppa888
| #
oppa888 Registration: Easy Steps
to Claim Your $100 Bonus
lottoland
| #
Sign up today at lottoland
and receive a $100 bonus to start your journey! Once you register and log in, your $100 bonus will be credited to
your account. This is a fantastic way to get started with extra funds, allowing you
to try out various games. Register today to take advantage of
this exclusive offer and boost your gaming experience right from the start!
DavidNaing
| #
This casino’s Tiger Fortune slot is a winner. fortune tiger demo
GeorgeTom
| #
Source:
https://norma3.ru/
Willieavamy
| #
I lost $200 online—plan up! plinko
slottica
| #
$100 Bonus Awaits You! Register Now at slottica!
Willieavamy
| #
Are online ratings solid?—tough guess! vipzino
betobet
| #
Join betobet
Now and Get Your $100 Registration Bonus!
anime bokep
| #
anime bokep
A Brief History of Elemental Analysis » Artemis Analytical
220patti
| #
Start playing at 220patti with a $100 bonus just for registering!
The sign-up process is quick and easy. Once you’ve created your account and logged
in, your $100 bonus will be credited to your account.
Use it to explore a range of exciting games, from slots to table games.
Register today and claim your bonus!
DavidNaing
| #
Tiger Fortune’s free spins are a game-changer. tiger fortune
JosephJen
| #
Ремонт квартир в Коммунарке Москва и Подмосковье – регионы с динамично развивающимся рынком недвижимости. Рано или поздно каждый владелец квартиры сталкивается с необходимостью ремонта. И здесь возникает вопрос: кому доверить эту задачу? ChatMost предлагает комплексные решения по ремонту квартир в Москве, Московской области, Балашихе, Ногинске, Электростали, Щёлково, Пушкино, Реутове, Люберцах, Мытищах, Химках, Видном, Монино, Королеве и Коммунарке.
1win_gzKr
| #
1win ug http://1win1003.top/ .
DavidNaing
| #
I love the thrill of Tiger Fortune’s jackpots. fortune tiger 777
where can i buy seroquel without dr prescription
| #
Medication information leaflet. Brand names.
where can i buy seroquel without dr prescription
Everything news about medication. Get information here.
DavidNaing
| #
This online casino’s Tiger Fortune is fantastic. fortune tiger 777
kra47.at
| #
kra47.at
A Brief History of Elemental Analysis » Artemis Analytical
Uazrscc
| #
Приветствую!
Для определенных людей, купить диплом о высшем образовании – это острая необходимость, шанс получить достойную работу. Впрочем для кого-то – это очевидное желание не терять время на учебу в ВУЗе. Что бы ни толкнуло вас на такое решение, мы готовы помочь вам. Оперативно, качественно и по разумной стоимости изготовим документ любого ВУЗа и любого года выпуска на настоящих бланках с реальными печатями.
Главная причина, почему многие люди покупают документы, – получить хорошую работу. Предположим, знания дают возможность специалисту устроиться на привлекательную работу, но документального подтверждения квалификации не имеется. В случае если для работодателя важно присутствие “корочки”, риск потерять хорошее место очень высокий.
Приобрести документ о получении высшего образования вы имеете возможность в нашей компании в Москве. Мы предлагаем документы об окончании любых университетов России. Вы получите необходимый диплом по любым специальностям, любого года выпуска, включая документы старого образца. Гарантируем, что при проверке документа работодателем, подозрений не появится.
Ситуаций, которые вынуждают заказать диплом достаточно. Кому-то прямо сейчас потребовалась работа, в итоге нужно произвести впечатление на начальство в процессе собеседования. Некоторые хотят попасть в престижную компанию, чтобы повысить собственный статус в социуме и в дальнейшем начать свой бизнес. Чтобы не тратить попусту годы жизни, а сразу начать удачную карьеру, используя имеющиеся навыки, можно заказать диплом в интернете. Вы сможете стать полезным для социума, обретете финансовую стабильность в максимально короткий срок- диплом о высшем образовании купить
Stephenindix
| #
визу digital nomad Оформление вида на жительство (ВНЖ) и получение гражданства Испании – актуальные вопросы для многих, особенно для россиян, рассматривающих переезд в 2023 году. Существуют различные типы ВНЖ, включая варианты без права на работу, студенческие визы, а также программы для цифровых кочевников (digital nomad) и финансово независимых лиц.
Получение испанского гражданства – отдельный процесс, требующий соблюдения определенных условий. Россияне, заинтересованные в иммиграции, изучают доступные способы получения ВНЖ, необходимые документы и стоимость. Варианты включают покупку недвижимости, участие в программах для инвесторов, а также ВНЖ типа “no lucrativa” для тех, кто располагает достаточными средствами для проживания без работы в Испании.
Помимо первичного оформления, важным аспектом является продление ВНЖ. Условия и требования могут отличаться в зависимости от типа ВНЖ и основания для его получения. Актуальную информацию о процедурах и документах для россиян в 2023 году следует уточнять у юристов или в компетентных органах.
xgbet
| #
Get a $100 bonus when you join xgbet as a new user!
This limited-time offer is designed to help you get started on your
online gaming adventure. Once you register, you’ll be able to enjoy a variety of casino games,
including slots, blackjack, and roulette, all while benefiting from your
bonus. The fun begins the moment you sign up, so don’t miss out—register now and claim your $100 bonus!
buy tetracycline 500mg for sale
| #
can i order cheap tetracycline without rx
Stromectol over the counter USA
| #
stromectol 3 mg dosage
Diplomi_drKt
| #
Купить диплом о высшем образовании!
Мы можем предложить документы любых учебных заведений, расположенных в любом регионе РФ.
good-diplom.ru/kupit-diplom-o-visshem-obrazovanii-po-dostupnoj-tsene/
Walteraneme
| #
Быстровозводимые здания https://akkord-stroy.ru из металлоконструкций — это скорость, надёжность и экономия. Рассказываем в статьях, как построить объект под ключ: от проектирования до сдачи в эксплуатацию.
PhillipGek
| #
Недвижимость в Бяла https://byalahome.ru апартаменты, квартиры и дома у моря в Болгарии. Лучшие предложения на побережье Черного моря — для жизни, отдыха или инвестиций. Успейте купить по выгодной цене!
TerrytOlve
| #
Скин AWP Graphite csgo-case-simulator.ru из CS:GO — чёрный глянец, премиальный стиль и высокая редкость. Укрась арсенал культовой снайперской винтовкой и выделяйся на сервере с первого выстрела.
satsport
| #
Your $100 Welcome Bonus is Ready at satsport
– Register Now!
MichaelCoubs
| #
AK-47 Fire Serpent https://get-skins-cs.ru легендарный скин из коллекции Operation Bravo. Яркий рисунок огненного змея, высокая редкость и коллекционная ценность. Идеальный выбор для истинных фанатов CS:GO.
DavidNaing
| #
Tiger Fortune’s theme is so adventurous. jogo do tigrinho demo
Willieavamy
| #
I love the live roulette—real deal! plinko app
Willieavamy
| #
The graphics on some online casino games are mind-blowing these days! fortune mouse
understanding the equivalence between diclofenac potassium and cataflam
| #
Medicament prescribing information. What side effects?
understanding the equivalence between diclofenac potassium and cataflam
Actual trends of medicines. Get information now.
Willieavamy
| #
I wish online paid fast—slow drag! aviator
nobonus
| #
nobonus
Registration: Easy Steps to Claim Your $100 Bonus
golabgaf
| #
Ищете медтехника для дома интернет-магазин? Agsvv.ru/catalog/obluchateli_dlya_lecheniya у вас появится возможность приобрести для лечения кожных болезней облучатели по цене производителя с гарантией. ООО Хронос изготовлением облучателей для лечения кожных заболеваний с 2009-го года с доказанными методами лечения занимается. Медицинские приборы от производителя славятся высочайшим качеством! Посмотрите наш ассортимент и назначение приборов. Мы поставляем приборы собственного производства государственным и частным медучреждениям по всей России.
heyspin
| #
heyspin Registration: Easy Steps to Claim Your $100 Bonus
1win_ouOl
| #
1win moldova download https://1win5013.ru .
mostbet_pcmt
| #
мос бет http://mostbet6033.ru .
phim sex hiep dam tap the
| #
phim sex hiep dam tap the
A Brief History of Elemental Analysis » Artemis Analytical
Iariorxtm
| #
Купить диплом ВУЗа по невысокой стоимости возможно, обращаясь к проверенной специализированной компании. Купить документ о получении высшего образования вы можете в нашей компании в столице. diplomk-v-krasnodare.ru/kupit-diplom-ob-obrazovanii-s-reestrom-8
alendronate sodium and levothyroxine
| #
Medication prescribing information. Generic Name.
alendronate sodium and levothyroxine
Some news about pills. Read here.
DavidNaing
| #
Tiger Fortune’s rewards make it worth playing. tiger fortune
cenigalob
| #
Если вы находитесь в поисках качественного, надежного и практичного стального профиля, то обратитесь на предприятие ООО “Валцпроф”, где вы найдете продукцию в широком ассортименте. На предприятии вы получите возможность приобрести выпадающие пороги, адаптированные для дверей разного вида. https://waltzprof.com/ – на сайте ознакомьтесь с полным ассортиментом выпускаемой продукции. Профили, находящиеся в каталоге, созданы для оперативной, удобной сборки перегородок, конструкций для фасадов, люков и многого другого.
1win_tdKr
| #
1win code bonus http://www.1win1003.top .
where can i buy generic co-amoxiclav without prescription
| #
how can i get cheap co-amoxiclav no prescription
DavidNaing
| #
This online casino shines with Tiger Fortune. tiger fortune
can i buy tadora price
| #
where to buy cheap tadora without a prescription
Xazrppf
| #
Купить диплом о высшем образовании!
Мы изготавливаем дипломы любой профессии по приятным тарифам. Вы заказываете диплом в надежной и проверенной компании. : phileo.me/blogs/new
Sazriip
| #
Мы предлагаем дипломы любой профессии по приятным тарифам.– diplom-kaluga.ru/poluchite-diplom-visshego-obrazovaniya-s-zaneseniem-v-reestr-2/
DavidNaing
| #
I can’t stop playing Tiger Fortune – it’s addictive! tigrinho demo
kra48at
| #
kra48at
blog topic
Как
| #
Как
blog topic
DavidNaing
| #
Tiger Fortune’s visuals are a feast for the eyes. tiger fortune
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Медная мишень для распыления
finasteride stada
| #
Pills information. Short-Term Effects.
finasteride stada
Actual what you want to know about medicines. Get information here.
Reriwerfloth
| #
ООО «Короче, дилер» представляет собой официальную библиотеку знаний ассоциации «Российские автомобильные дилеры». На портале https://koroche-dealer.ru каждый, у кого есть вопросы по теме, сможет найти на них ответы. Представлены самые актуальные материалы, касающиеся работы дилерских центров по самым разным направлениям.
stomatologiya Mitino_vmpr
| #
сайт стоматологии москва сайт стоматологии москва .
DavidNaing
| #
Tiger Fortune is perfect for thrill-seekers like me. fortune tiger bet
cost generic tetracycline tablets
| #
Medicine prescribing information. Cautions.
cost generic tetracycline tablets
Actual news about medicament. Read here.
1win_fppa
| #
1win kg https://1win6010.ru .
ManitnTonge
| #
Вместе с t.me/azino777_a у вас есть возможность в увлекательное путешествие по миру азартных игр отправиться. Предлагаем персонализированные предложения и уникальные бонусы. Расскажем, как в Азино начать играть. Вас ждет возможность выиграть крупный приз и невероятные эмоции. Испытайте свою удачу уже сейчас! Ищете azino777 win? – здесь представлено много интересной информации. Собрали самые интересующие вопросы от игроков. Рады вам помочь. Приготовьтесь к увлекательным турнирам в Азино. Участвуйте и боритесь за призы с другими игроками!
1win_fcea
| #
1win online 1win online .
1win_pcen
| #
1вин официальный сайт вход https://1win6047.ru/ .
mostbet_iuMi
| #
мостбет войти https://mostbet6035.ru/ .
Williamten
| #
check my site mymerrill login
rivalry
| #
How to Register and Claim Your $100 Bonus on rivalry
betfury
| #
Sign Up at betfury and Get a $100
Bonus Instantly!
bluechip
| #
Looking to get started with bluechip?
New users can claim a $100 bonus upon registration! The sign-up
process is easy and takes just a few moments. Once you’ve
completed your registration and logged in, you’ll be rewarded with a $100 bonus.
This is the perfect way to kick off your gaming experience
with extra funds. Don’t wait – sign up now and
grab your $100 bonus!
unibet
| #
Join unibet Now and Get Your $100 Registration Bonus!
Willieavamy
| #
The table games online spark—class act! sugar rush
DavidNaing
| #
Tiger Fortune is the highlight of this online casino for me. jogo do tigrinho demo
DavidNaing
| #
Tiger Fortune’s design blows me away every time. tiger fortune
Lombard chasov_abEl
| #
ломбард прием часов ломбард прием часов .
Sazrlph
| #
Мы готовы предложить дипломы психологов, юристов, экономистов и прочих профессий по выгодным ценам.
Вы покупаете документ в надежной и проверенной компании. Заказать диплом о высшем образовании– http://job.iwok.vn/employer/diplomy-grup-24/ – job.iwok.vn/employer/diplomy-grup-24
DavidNaing
| #
This online casino’s Tiger Fortune slot is pure fun. tiger fortune
mostbet_ywmt
| #
мостбет промокод http://mostbet6033.ru .
ashwagandha and abilify
| #
himalaya ashwagandha withanolides
Willieavamy
| #
Online casinos feel off—loss streak! book of ra
stomatologiya Mitino_agPn
| #
хирург в митино платно хирург в митино платно .
crypto casinos spins
| #
crypto casinos spins
A Brief History of Elemental Analysis » Artemis Analytical
1win_ouOl
| #
1win pariuri https://1win5013.ru/ .
lisinopril 5mg wiki
| #
where to get cheap lisinopril price
1win_ovea
| #
1win букмекер https://www.1win6041.ru .
stomatologiya Mitino_nxpr
| #
александр жаров стоматолог александр жаров стоматолог .
where can i get cheap plan b without dr prescription
| #
Medicament prescribing information. What side effects can this medication cause?
where can i get cheap plan b without dr prescription
All information about medication. Read information here.
emercom-irk
| #
Комплексный подход к безопасности объекта — это залог его защиты. Эмерком Иркутск предлагает широкий спектр услуг: от монтажа сигнализации до разработки планов эвакуации. Обратитесь к профессионалам, чтобы получить качественные решения для вашего дома или бизнеса.
pereplanirovka_seOi
| #
узаконивание перепланировки https://udmageo.ru .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наши товары:
Медный стандартный однораструбный отвод 90 градусов под пайку 8х6х0.6 мм 6.8х8.8 мм твердая пайка М3р ГОСТ Р52922-2008 Покупайте качественные медные однораструбные отводы 90 градусов под пайку от Редметсплав. Надежные и долговечные соединения для водоснабжения, отопления и кондиционирования. Быстрая установка и эстетичный вид. Доставка по России.
1win_guen
| #
1win сайт вход https://www.1win6047.ru .
Cazrfua
| #
Заказать диплом ВУЗа можем помочь. Купить диплом техникума, колледжа в Новосибирске – diplomybox.com/kupit-diplom-tekhnikuma-kolledzha-v-novosibirske
Lazritb
| #
Диплом университета России!
Без ВУЗа очень трудно было продвинуться вверх по карьерной лестнице. Именно поэтому решение о покупке диплома можно считать выгодным и рациональным. Купить диплом университета sciencenewhop.maxbb.ru/viewtopic.php?f=44&t=1209
mostbet_isMi
| #
mostbet apk скачать http://mostbet6035.ru .
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их качество. Превосходное обслуживание – наш стандарт – мы на связи, чтобы ответить на ваши вопросы по мере того как предоставлять решения под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Лента висмутовая C89320 – UNS Лента висмутовая C89320 – UNS предназначена для использования РІ различных промышленных Рё научных применениях. Рта высококачественная лента обеспечивает отличные антикоррозийные свойства Рё термостойкость, что делает её идеальным выбором для создания защитных покрытий. Лента легко наносится Рё применяется РІ кабельной изоляции, Р° также РІ медицинских устройствах. Купить Лента висмутовая C89320 – UNS стоит тем, кто ищет надежные материалы для долгосрочной эксплуатации. Закажите ленту РїСЂСЏРјРѕ сейчас Рё обеспечьте надежность СЃРІРѕРёС… проектов!
1win_ryst
| #
1вин бет официальный сайт 1вин бет официальный сайт .
1win_ibea
| #
1win зайти http://www.1win6041.ru .
DavidNaing
| #
Tiger Fortune’s theme is so adventurous. tiger fortune
stomatologiya Mitino_kcpr
| #
частные клиники в митино частные клиники в митино .
Lev казино
| #
Лев Казино предлагает вам погрузиться в мир захватывающих игр, где каждый шаг может привести к большой победе. Наше казино предложит вам не только популярные игры, но и множество новых вариантов для экспериментов и удовольствия. Игры на любой вкус, доступные 24/7, дают вам шанс на выигрыш в любое время.
Лев Казино отличается высоким качеством сервиса и надежностью. Лев Казино имеет интуитивно понятный интерфейс и множество методов для пополнения и вывода средств, чтобы ваш опыт был комфортным. Для новых игроков в https://levcasino-goldenchips.beauty/ предусмотрены щедрые бонусы и привилегии.
Мы предлагаем простую регистрацию и быстрое снятие выигрышей, чтобы вы могли наслаждаться игрой без задержек.
Наши игроки наслаждаются разнообразием игр, начиная от классических слотов и заканчивая инновационными настольными играми.
В Лев Казино регулярно проходят акции, которые дают дополнительные возможности для выигрыша и увеличения вашего баланса.
В Лев Казино ваши данные и средства всегда под защитой, и мы гарантируем полную безопасность.
Присоединяйтесь к Лев Казино и начните играть сегодня, чтобы выиграть огромные призы!.
biximoSmask
| #
Компания «Отопление и водоснабжение» предлагает квалифицированные услуги. Компетентные специалисты готовы осуществить в Рузе в сжатые сроки монтаж системы отопления. Создайте комфорт и тепло с нами в вашем доме. Ищете монтаж систем отопления? Ruzskiy-rayon.santex-uslugi.ru – здесь можете прямо сейчас ознакомиться с отзывами о нас. Гарантируем использование качественных материалов. Ваши ожидания мы готовы превзойти. Стараемся услуги доступными для клиентов сделать. Позвоните нашему менеджеру для консультации по интересующим вас вопросам.
https://pacsx.com/pags/?21-csgo-opening-sites.html
| #
https://pacsx.com/pags/?21-csgo-opening-sites.html
Cazrrjb
| #
Мы изготавливаем дипломы любых профессий по приятным ценам. Купить диплом в Абакане — kyc-diplom.com/geography/abakan1.html
1win_gnen
| #
1win скачать kg https://www.1win6047.ru .
order cheap benicar for sale
| #
Drug prescribing information. Effects of Drug Abuse.
order cheap benicar for sale
Some information about medicines. Read information now.
order nemasole for sale
| #
where can i get cheap nemasole prices
Sazrktc
| #
Заказать диплом под заказ возможно используя сайт компании. fynmir.online/post_type=topic&p=117194
mostbet_syMi
| #
mosbet http://mostbet6035.ru/ .
bons
| #
New Users, Enjoy a $100 Bonus When You Register at bons
can you get requip tablets
| #
cost generic requip without a prescription
mostbet_fbet
| #
mostbet kg https://mostbet6034.ru/ .
skol
| #
Welcome to skol! New users receive a $100 bonus upon registration. The process
is straightforward: create your account, log in, and your
bonus will be instantly credited. This offer is designed to
give new players a head start. With your $100
bonus, you can enjoy a variety of games, including slots,
poker, and more. Don’t miss out on this fantastic opportunity to maximize your gaming experience.
Register today and grab your bonus!
DavidLumma
| #
Корпоративные сувениры и мерч являются эффективным инструментом для повышения узнаваемости бренда и укрепления корпоративной культуры. Они помогают создать положительное впечатление о компании среди клиентов и партнеров, а также способствуют формированию лояльности сотрудников. Подробнее об этом можно узнать по ссылке: https://moda-sar.ru/04/04/2025/korporativnye-suveniry-i-merch.html
baji
| #
Create Your baji Account Now and Get a $100 Bonus!
puma
| #
Join puma Today and Claim Your $100 Welcome Bonus!
Lombard chasov_gsEl
| #
скупка элитных часов в москве дорого скупка элитных часов в москве дорого .
LucasDep
| #
https://mamapoltava.listbb.ru/viewtopic.php?f=45&t=12845
pereplanirovka_vdOi
| #
узаконивание перепланировки http://www.udmageo.ru/ .
stomatologiya Mitino_yhPn
| #
гнатолог в клинике в москве гнатолог в клинике в москве .
Jimmylon
| #
Легендарная AWP Medusa https://case-simulator-cs2.ru скин с таинственным дизайном, вдохновлённым древнегреческой мифологией. Высокая редкость, художественная детализация и престиж на каждом сервере.
NicolasGab
| #
Скин M4A4 Howl https://cs2-get-skins.ru один из самых дорогих и загадочных в CS:GO. Запрещённый артефакт с историей. Рассказываем о его происхождении, внешнем виде и значении в мире скинов.
vave
| #
Sign Up at vave and Get a $100 Bonus Instantly!
DavidNaing
| #
Tiger Fortune keeps me coming back daily. jogo do tigrinho demo
Robertemito
| #
StatTrak M4A1-S cs2 get skin скин с возможностью отслеживания фрагов прямо на корпусе оружия. Узнайте, какие модели доступны, чем отличаются, и какие из них ценятся выше всего на рынке CS:GO.
MichaelPielp
| #
Перчатки спецназа case-simulator-cs.ru/ тактический скин в CS:GO с брутальным дизайном и премиальной редкостью. Узнайте об их разновидностях, цене, коллекционной ценности и лучших сочетаниях с другими скинами.
spinrio
| #
spinrio Registration Process – Get Your $100 Bonus Upon Signing Up
pinnacle
| #
Get Your $100 Bonus Right After You Join pinnacle!
22bet
| #
Register at 22bet and Grab a
$100 Bonus for New Users!
Raphaelhab
| #
Заказ значков через интернет — это современный и удобный способ получить уникальные аксессуары, не выходя из дома. Онлайн-платформы предлагают широкий выбор дизайнов, материалов и вариантов персонализации, позволяя создать значки, полностью соответствующие вашим предпочтениям и потребностям. Дополнительную информацию о процессе заказа и доступных опциях можно найти по ссылке:https://aragoncom.ru/poleznie/zakaz-znachkov-cherez-sajt-udobnyj-sposob-oformit-unikalnye-aksessuary.html
1win_ujSn
| #
1win партнерская программа вход http://1win6046.ru/ .
phim sex hiếp dâm gái trẻ
| #
phim sex hiếp dâm gái trẻ
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние достижения.
Наши товары:
“ерамическая мишень CeO2, плоская форма
DavidNaing
| #
Tiger Fortune’s design is top-notch – really impressive. fortune tiger bet
Cazrgyp
| #
Где купить диплом по нужной специальности?
Мы изготавливаем дипломы любой профессии по доступным ценам. Важно, чтобы документы были доступными для большинства наших граждан. Заказать диплом об образовании diplomist.com/kupit-diplom-s-zaneseniem-19/
how can i get zithromax without a prescription
| #
Drug information for patients. Cautions.
how can i get zithromax without a prescription
Everything trends of drugs. Get now.
teen patti
| #
Sign Up Now and Enjoy a $100 Bonus at teen patti!
WilliamDix
| #
Мы можем предложить дипломы любых профессий по доступным тарифам. Дипломы изготавливаются на настоящих бланках Приобрести диплом о высшем образовании asxdiploman.com
roobet
| #
Join roobet Today and Receive a $100 Bonus
Just for Registering!
Lazrpsx
| #
Где приобрести диплом по нужной специальности?
Заказать диплом университета по доступной цене возможно, обратившись к надежной специализированной компании.: peoplediplom.ru
vinyl
| #
Create Your vinyl Account Now and Get a $100 Bonus!
1win_ffEl
| #
1win moldova download http://www.1win5014.ru .
Eanrjkj
| #
Для быстрого продвижения по карьере потребуется наличие официального диплома ВУЗа. Заказать диплом об образовании у сильной компании: diplomp-irkutsk.ru/kupit-diplom-texnikuma-ob-okonchanii-bistro-i-nadezhno/
Sogiphvor
| #
Академия «Арчибальд» занимается обучением грумингу. Вы можете совершенствовать собственные навыки на живых моделях до бесконечности. Преподаватели на все вопросы отвечают. Дарим методические материалы, учебники, а также видеолекции. Окунитесь в атмосферу, с которой расставаться не хочется. Ищете курсы груминга москва недорого? Grooming-dream.ru – здесь представлены реальные отзывы клиентов. Мы лучших груминг-специалистов выпускаем. Предлагаем инструменты в период обучения. Если вам интересна новая профессия – Арчибальд, вот то место, которое вам подойдет.
DavidNaing
| #
This casino’s Tiger Fortune game is a hit. fortune tiger bet
Stanleyalbug
| #
кра сайт – kra31.cc, кракен официальный сайт
Treflep
| #
Приобрести диплом о высшем образовании. Заказ документа о высшем образовании через качественную и надежную фирму дарит ряд достоинств. Это решение дает возможность сберечь время и существенные денежные средства. jobgate.org/employer/eonline-diploma
Ronaldwougs
| #
кракен тор – kraken shop, kra cc
DavidNaing
| #
This casino’s Tiger Fortune game is incredible. tiger fortune
order cheap adalat for sale
| #
get cheap adalat pills
MelvinAltex
| #
kra31.cc – kra31.cc, kraken сайт
mostbet_tlet
| #
mostbet официальный сайт mostbet официальный сайт .
mostbet_jvmt
| #
mostbet casino mostbet6033.ru .
where buy generic arcoxia online
| #
where can i buy arcoxia prices
bilbet
| #
Create an Account on bilbet and Start Playing with a $100 Bonus!
Raphaelhab
| #
Заказ значков через интернет — это современный и удобный способ получить уникальные аксессуары, не выходя из дома. Онлайн-платформы предлагают широкий выбор дизайнов, материалов и вариантов персонализации, позволяя создать значки, полностью соответствующие вашим предпочтениям и потребностям. Дополнительную информацию о процессе заказа и доступных опциях можно найти по ссылке:https://aragoncom.ru/poleznie/zakaz-znachkov-cherez-sajt-udobnyj-sposob-oformit-unikalnye-aksessuary.html
Barrylef
| #
mostbet promo code Freebet allows players to place bets without using their own funds, making wagers completely free thanks to the bookmaker, with winnings available for immediate withdrawal
1win_mbEl
| #
1win moldova 1win moldova .
can i buy cheap chlorpromazine for sale
| #
Medicament information sheet. Short-Term Effects.
can i buy cheap chlorpromazine for sale
All trends of drug. Get information here.
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наши товары:
Медный стандартный двухраструбный отвод 90 градусов под пайку 18х14х0.8 мм 12.6х14.6 мм твердая пайка М3р ГОСТ Р52922-2008 Покупайте медные двухраструбные отводы 90 градусов под пайку на Редметсплав.рф. Надежные и долговечные отводы из высококачественной меди для систем водоснабжения, отопления и кондиционирования воздуха. Гарантированно надежные соединения, простой монтаж и оптимальное распределение потока.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наша продукция:
Высокочистые гранулы Y2O3
Marlinexpep
| #
AWP Dragon Lore get-skins-csgo легендарный скин с изображением дракона, символ элиты в CS:GO. Узнайте о его происхождении, редкости, стоимости и почему он стал мечтой каждого коллекционера.
Davidsueds
| #
Обзор AWP Asiimov get skin csgo футуристичный скин с дерзким дизайном. Рассказываем о редкости, ценах, вариантах износа и том, почему Asiimov стал иконой среди скинов в CS:GO.
MichaelPub
| #
Обзор AK-47 Redline http://csgo-get-skin.ru лаконичный дизайн, спортивный стиль и привлекательная цена. Почему этот скин так любят игроки и с чем он лучше всего сочетается — читайте у нас.
Travisexpow
| #
Онлайн-казино Shot shot-casino-apk.ru предлагает щедрые бонусы, турнирные события и топовые слоты. Узнайте об условиях игры, уровне доверия, лицензии и особенностях платформы в нашем свежем обзоре.
Raphaelhab
| #
Заказ значков через интернет — это современный и удобный способ получить уникальные аксессуары, не выходя из дома. Онлайн-платформы предлагают широкий выбор дизайнов, материалов и вариантов персонализации, позволяя создать значки, полностью соответствующие вашим предпочтениям и потребностям. Дополнительную информацию о процессе заказа и доступных опциях можно найти по ссылке:https://aragoncom.ru/poleznie/zakaz-znachkov-cherez-sajt-udobnyj-sposob-oformit-unikalnye-aksessuary.html
Jackpot Bet
| #
Jackpot Bet
blog topic
mostbet_dumt
| #
motsbet motsbet .
Willieavamy
| #
Online laws fuzz—sort out! plinko
cumosemorn
| #
Отопление и водоснабжение – компания, которая на предоставлении качественных услуг специализируется. У нас глубокие знания и опыт в сфере монтажа отопительных систем. Следим за современными технологиями, чтобы вам передовые решения предлагать. Подходим персонально к каждому проекту. Ищете монтаж водоснабжения? Krasnogorsk.santex-uslugi.ru – портал, где опубликованы отзывы о нас и указаны цены. Используем проверенные материалы и оборудование от известных производителей. Удачно осуществили множество работ. Готовы комфортный дом для вашей семьи создать.
sozavAceno
| #
Автоматические пороги считаются оптимальным решением в том случае, если выполнить монтаж стационарного аналога не получается либо не считается желательным. В данный момент можно изучить каталог всех вариантов, которые предлагает предприятие «SMARTПОРОГ», что позволит подобрать решение, которое определенно подойдет. https://smartporog.ru/ – на сайте изучите преимущества, которые вы получите, если установите такие конструкции в помещении. Пороги необходимы для того, чтобы предотвратить попадание запахов и света, а также насекомых. Считается отличным вариантом для организации ванной комнаты, например.
Raphaelhab
| #
Заказ значков через интернет — это современный и удобный способ получить уникальные аксессуары, не выходя из дома. Онлайн-платформы предлагают широкий выбор дизайнов, материалов и вариантов персонализации, позволяя создать значки, полностью соответствующие вашим предпочтениям и потребностям. Дополнительную информацию о процессе заказа и доступных опциях можно найти по ссылке:https://aragoncom.ru/poleznie/zakaz-znachkov-cherez-sajt-udobnyj-sposob-oformit-unikalnye-aksessuary.html
DavidNaing
| #
I love the vibrant colors in Tiger Fortune. tigrinho demo
1win_ebEl
| #
1win.com http://www.1win5014.ru .
DavidNaing
| #
Tiger Fortune’s spins keep me hooked! tiger fortune
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Превосходное обслуживание – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы а также находить ответы под особенности вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наша продукция:
Пруток висмутовый Sn43Pb43Bi14A – IEC 61190-1-3 Пруток висмутовый Sn43Pb43Bi14A – IEC 61190-1-3 является идеальным решением для вашего производства. РћРЅ оснащен уникальным составом, что делает его надежным для использования РІ различных сферах, включая электронику Рё пайку. Высокая стабильность Рё отличные физико-химические свойства обеспечивают эффективное соединение компонентов. Купить Пруток висмутовый Sn43Pb43Bi14A – IEC 61190-1-3 – значит сделать шаг Рє качеству Рё надежности. Заказывайте уже сегодня, чтобы оценить РІСЃРµ преимущества этого высокотехнологичного материала!
DavidNaing
| #
Tiger Fortune has the best energy of any game. fortune tiger bet
Michaelphile
| #
https://kdental.ru/
mostbet_pymt
| #
mostbet kg скачать http://mostbet6033.ru/ .
1win_sroa
| #
1win мобильная версия сайта http://1win7013.ru/ .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Высокочистые гранулы тантала
1win_cdsa
| #
wan win wan win .
effects of reducing mirtazapine
| #
Medication information. Short-Term Effects.
effects of reducing mirtazapine
Some trends of meds. Get here.
Davidbow
| #
лаки джет сайт – lucky jet 1 win, джет лак
PatrickbeP
| #
Нужно создание или разработка веб студия: адаптивный дизайн, SEO-продвижение, техническая поддержка. Эффективное ключевая фраза для вашего бизнеса — привлечение клиентов и рост прибыли.
20 mg famotidine
| #
Medicine information leaflet. Drug Class.
20 mg famotidine
Everything news about medication. Get information here.
Jamiedycle
| #
Онлайн-казино Calibry calibri-casino-online.ru/ новое игровое пространство с лицензией, щедрыми бонусами и широким выбором слотов. Читайте обзор: особенности платформы, условия игры, отзывы и выплаты.
Gerryquera
| #
Shot Casino https://shot-casino-online.ru новое онлайн-казино с лицензией, защитой данных и большим выбором игр. В обзоре: безопасность, отзывы игроков, бонусы и выплаты. Надёжная площадка для вашего азарта.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Высокочистый тигель из вольфрама
JulioEcone
| #
Обзор онлайн-казино Shot http://shot-casino-app.ru плюсы и минусы, проверка лицензии, акции и фриспины. Рассказываем, стоит ли играть, как получить бонусы и выводить выигрыши без проблем.
buumi
| #
Ready to take your online gaming experience to the
next level? Register at buumi and get a $100 bonus to start playing right away.
Use this bonus to try your luck at a variety of exciting games, including slots,
poker, live casino games, and sports betting.
Don’t miss this amazing opportunity to get ahead—sign up today and claim your $100 bonus
to boost your chances of winning!
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наша продукция:
Бронзовое внутреннее стопорное пружинное кольцо 13х1 мм БРАЖМЦ10-3-1,5 ГОСТ 13943-86 Приобретите качественные бронзовые внутренние стопорные кольца по ГОСТ в Редметсплав.рф. Наши колечки отличаются превосходными техническими характеристиками, высоким качеством и надёжностью. Широкий ассортимент и гарантированное соответствие ГОСТ позволят удовлетворить потребности любого заказчика.
Marvinpacle
| #
Если вас интересует оклейка плёнкой Mercedes-AMG G 63 обращайтесь к профессионалам! Наша компания предлагает премиальную оклейку защитными и декоративными плёнками, идеально подчёркивающими характер вашего автомобиля. Глянцевая или матовая текстура – выберите свой стиль и обеспечьте кузову надёжную защиту от сколов, царапин и выгорания.
XaiUNecope
| #
На сайте https://officepro54.ru представлено огромное количество мебели, которая идеально подходит для организации офисного пространства. Вся она выполнена из инновационных, уникальных материалов, а потому прослужит очень долго, не синтезирует в воздух опасных веществ. В разделе вы найдете кабинет руководителя, функциональные и вместительные столы для переговоров, шкафы-купе, сейфы, акустические системы, ученическую мебель и многое другое. Постоянно действуют акции. На продукцию установлены привлекательные цены.
DavidNaing
| #
I can’t get enough of the Tiger Fortune soundtrack! fortune tiger demo gratis
Mazrvmq
| #
Мы можем предложить дипломы любой профессии по приятным ценам. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Важно, чтобы документы были доступны для большинства наших граждан.
Заказ диплома, подтверждающего окончание университета, – это грамотное решение. Приобрести диплом любого ВУЗа: diplommy.ru/diplom-o-visshem-obrazovanii-kupit-v-spb-3/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Керамическая мишень Bn 2N5 (плоская)
1win_xroa
| #
1win личный кабинет 1win7013.ru .
can i purchase generic secnidazole
| #
where can i get secnidazole for sale
where buy generic dulcolax
| #
where buy generic dulcolax pills
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица товара подтверждена требуемыми документами, подтверждающими их качество. Превосходное обслуживание – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы и адаптировать решения под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
Рзделия РёР· молибдена РњРЁ-1 Рзделия РёР· молибдена РњРЁ-1 представляют СЃРѕР±РѕР№ высококачественные компоненты, изготовленные РёР· молибдена, обладающего отличными механическими свойствами Рё высокой температурной прочностью. Рти изделия идеально РїРѕРґС…РѕРґСЏС‚ для использования РІ различных отраслях, включая авиацию, машиностроение Рё электронику.Преимущества молибденовых изделий включают устойчивость Рє РєРѕСЂСЂРѕР·РёРё Рё РЅРёР·РєРѕРјСѓ коэффициенту теплового расширения. РћРЅРё также обеспечивают надежную работу РїСЂРё высоких температурах, что делает РёС… незаменимыми РІ критических условиях.Если РІС‹ хотите купить Рзделия РёР· молибдена РњРЁ-1, обращайтесь Рє надежным поставщикам, предлагающим продукцию, соответствующую самым строгим стандартам качества.
Willieavamy
| #
The visuals in online casino games are next-level these days! minas
Sazrnbg
| #
Заказать диплом на заказ можно через сайт компании. rentry.co/bapx6863
DavidNaing
| #
Tiger Fortune’s features are pure genius. fortune tiger 777
DavidNaing
| #
The visuals in Tiger Fortune are breathtaking. fortune tiger demo gratis
DavidNaing
| #
I love the jungle energy of Tiger Fortune. fortune tiger 777
DavidNaing
| #
I love how Tiger Fortune mixes luck and fun. fortune tiger demo gratis
Lazrscs
| #
Купить диплом института!
Мы готовы предложить дипломы психологов, юристов, экономистов и любых других профессий по приятным ценам— fastdiploms.com/kupit-diplom-med-rabotnika-2/
1win_bnsa
| #
что делать с бонусным балансом на 1win https://www.1win7011.ru .
what happens when you drink with amoxicillin
| #
Medication information for patients. Short-Term Effects.
what happens when you drink with amoxicillin
Everything information about medicine. Read information here.
Jariorxlz
| #
Где заказать диплом по нужной специальности?
Наша компания предлагаетбыстро купить диплом, который выполнен на бланке ГОЗНАКа и заверен печатями, водяными знаками, подписями. Документ пройдет лубую проверку, даже при использовании специального оборудования. Достигайте свои цели быстро и просто с нашими дипломами. Заказать диплом университета! hajz.live/read-blog/30_kupit-vkr-kolledzha-cena.html
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
поставляемая продукция:
Кольцо РёР· драгоценных металлов золотое 200С…50С…5 РјРј Р—Р»99.9 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
1win_knst
| #
1win онлайн https://www.1win6045.ru .
Cazrccw
| #
Добрый день!
Мы изготавливаем дипломы любых профессий по приятным ценам. Стоимость будет зависеть от выбранной специальности, года получения и университета: diploman-russian.com/
1win_ahoa
| #
1win.kg https://1win7013.ru/ .
Marvinpacle
| #
Если вас интересует оклейка Mercedes-AMG G 63 обращайтесь к профессионалам! Наша компания предлагает премиальную оклейку защитными и декоративными плёнками, идеально подчёркивающими характер вашего автомобиля. Глянцевая или матовая текстура – выберите свой стиль и обеспечьте кузову надёжную защиту от сколов, царапин и выгорания.
can i get cheap norvasc online
| #
Medicament information for patients. What side effects can this medication cause?
can i get cheap norvasc online
All trends of medicament. Read now.
DavidNaing
| #
I can’t get enough of the Tiger Fortune soundtrack! tigrinho demo
Trefehz
| #
Заказать диплом о высшем образовании. Заказ подходящего диплома через проверенную и надежную компанию дарит немало плюсов. Данное решение позволяет сэкономить как личное время, так и существенные деньги. 1001vieclam.forumvi.com/login
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
поставляемая продукция:
Кольцо РёР· драгоценных металлов золотое 100С…3С…3.5 РјРј ЗлСрМ75-12.5 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
hedinic
| #
Блог о маркетплейсах «OOWA» предлагает ознакомиться с актуальной, интересной и познавательной информацией, которая касается удачных продаж на маркетплейсах, обзоров товаров и услуг. На портале регулярно публикуются ценные и содержательные рекомендации от экспертов, которые помогут начать лучше разбираться в тонкостях такого бизнеса. Ищете обзор камеры моментальной печати instax square sq1? На oowa.ru регулярно публикуются ценные и содержательные рекомендации от экспертов, которые помогут начать лучше разбираться в тонкостях такого бизнеса. Почитайте о самых востребованных товарах с маркетплейсов.
penipu phising
| #
penipu phising
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы по мере того как находить ответы под требования вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Фольга висмутовая 42 3990 – CSN/STN 423990 Фольга висмутовая 42 3990 – CSN/STN 423990 – это идеальный выбор для ваших проектов, требующих высокой степень точности. Рзготовленная РёР· качественного висмута, РѕРЅР° обладает отличной стойкостью Рё легкостью РІ использовании. Р’С‹ можете применять ее РІ различных областях, включая электронику Рё научные исследования. Приобретая этот товар, РІС‹ получаете надежный Рё долговечный материал. РќРµ упустите возможность купить Фольга висмутовая 42 3990 – CSN/STN 423990 для СЃРІРѕРёС… нужд. Делайте выбор РІ пользу качества Рё проверенной продукции!
where buy cheap primaquine without dr prescription
| #
buying primaquine pills
1win_lbsa
| #
1win pro 1win7011.ru .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
поставляемая продукция:
РўСЂСѓР±Р° бесшовная никелевая 25С…2С…1000 РјРј РќРњР–РњС†28-2.5-1.5 РўРЈ 48-21-783-85 Никелевая полоса 11×100 РјРј РќРџ3 РїРѕ ГОСТ 6235-91 – прочный Рё износостойкий материал РѕС‚ Редметсплав.СЂС„. Высокая термостойкость, устойчивость Рє РєРѕСЂСЂРѕР·РёРё Рё долговечность делают эту полосу идеальным выбором для ваших производственных проектов. Приобретите качественные материалы уже сегодня!
sklad_toOt
| #
снять хранилище для вещей в москве снять хранилище для вещей в москве .
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их соответствие стандартам. Опытная поддержка – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы и предоставлять решения под требования вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Проволока кобальтовая Stellite 36 Проволока кобальтовая Stellite 36 – это высококачественный материал для сварки Рё наплавки. Ртот сплав, содержащий кобальт, С…СЂРѕРј Рё молибден, отличает превосходная прочность Рё устойчивость Рє РєРѕСЂСЂРѕР·РёРё. РћРЅ идеально РїРѕРґС…РѕРґРёС‚ для ремонтов РІ условиях высоких температур Рё абразивного воздействия. РЎ его помощью можно создавать надежные соединения Рё защитные покрытия. Если РІС‹ ищете материал, который обеспечит долговечность Рё надежность, то вам стоит купить Проволока кобальтовая Stellite 36. Ртот РїСЂРѕРґСѓРєС‚ является выбором профессионалов РІ различных отраслях.
buying cheap tegretol prices
| #
where buy cheap tegretol without prescription
Willieavamy
| #
The poker online shines—real kick! plinko
DavidNaing
| #
I love the high-energy feel of Tiger Fortune! tigrinho demo
DavidNaing
| #
I’ve had some massive wins on Tiger Fortune! tigrinho demo
DavidNaing
| #
Tiger Fortune’s payouts are always exciting. fortune tiger demo
Willieavamy
| #
I won $300 on an online scratch card—easiest money ever! betclic
mostbet_yeMi
| #
motsbet mostbet6035.ru .
Jarioroax
| #
Где купить диплом по необходимой специальности?
Мы предлагаеммаксимально быстро приобрести диплом, который выполняется на оригинальной бумаге и заверен мокрыми печатями, водяными знаками, подписями официальных лиц. Диплом пройдет любые проверки, даже при помощи специальных приборов. Решите свои задачи быстро с нашим сервисом. Купить диплом о высшем образовании! gladpwnz.ru/forum/member.php?u=221749
WilliamDix
| #
Мы готовы предложить дипломы любых профессий по невысоким ценам. Дипломы производятся на подлинных бланках государственного образца Приобрести диплом об образовании diplom-zentr.com
can you get generic nortriptyline without a prescription
| #
Drug information. Effects of Drug Abuse.
can you get generic nortriptyline without a prescription
Best what you want to know about meds. Read information here.
how to get cheap fosamax without rx
| #
Drugs information sheet. Cautions.
how to get cheap fosamax without rx
Everything information about medicines. Read information here.
Lazrwxl
| #
Где приобрести диплом специалиста?
Купить диплом университета по невысокой цене возможно, обращаясь к надежной специализированной компании.: kdiplom.ru
rawegbup
| #
Специалисты компании «МосСтройАльянс» решат любую сложную задачу. Мы на осуществлении фасадных, уборочных и кровельных работ специализируемся. Предоставляем доступные цены. Вы будете довольны результатом. Ищете монтаж крыши цены в москве? Mos-stroi-alians.ru – тут с отзывами наших клиентов можете ознакомиться. Для работы применяем исключительно материалы высшего качества. Ценим ваше время. Несем ответственность за соблюдение сроков. Позвоните нам по телефону на портале. Проконсультируем и ответим на интересующие вас вопросы.
DavidNaing
| #
Tiger Fortune’s design is wild and bold. fortune tiger demo
Mazrqpl
| #
Мы готовы предложить дипломы любой профессии по доступным ценам. Всегда стараемся поддерживать для покупателей адекватную политику тарифов. Важно, чтобы документы были доступными для большинства наших граждан.
Приобретение документа, который подтверждает обучение в университете, – это грамотное решение. Купить диплом о высшем образовании: institute-diplom.ru/kupit-diplom-institut-3/
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы и находить ответы под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
РљСЂСѓРі молибденовый РњР’20 РљСЂСѓРі молибденовый РњР’20 – это высококачественный материал, используемый РІ различных отраслях, включая авиастроение Рё электронику. РћРЅ обладает отличной температурной устойчивостью Рё прочностью, что делает его идеальным для востребованного применения. Параметры, такие как диаметр Рё толщина, обеспечивают стабильную работу РІ экстремальных условиях.Если РІС‹ хотите улучшить СЃРІРѕРё производственные процессы Рё повысить эффективность, это отличное решение. РќРµ упустите возможность купить РљСЂСѓРі молибденовый РњР’20 Рё кладите РІ СЃРІРѕРё задачи только проверенные решения. Заказывайте сейчас!
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Плоская мишень AlSc для распыления сплавов
DavidNaing
| #
This online casino’s Tiger Fortune is a hit. fortune tiger demo gratis
1win_vlea
| #
1wln http://1win6041.ru .
1win_btSn
| #
1вин официальный сайт http://1win6046.ru/ .
Sazrvvw
| #
Всегда считал, что покупка диплома о высшем образовании — это миф. Но, оказалось, что все возможно! Сначала искал информацию по теме: купить диплом лэти, купить диплом магистра в ставрополе, купить диплом менеджера по туризму, купить диплом металлурга, купить диплом московского педагогического училища 16, а затем перешел на дипломы вузов. Подробнее можно узнать здесь: diplomybox.com/shag-5
Lazrfcc
| #
Купить диплом о высшем образовании!
Мы изготавливаем дипломы любых профессий по доступным ценам— diplomidlarf.ru/kupite-diplom-vk-bistro-i-nadezhno-bez-xlopot/
1win_tken
| #
1win kg скачать http://www.1win6047.ru .
can i buy generic ventolin inhalator price
| #
how to buy ventolin inhalator for sale
Willieavamy
| #
I wish online tied more—life in! F7 Casino
Willieavamy
| #
The slot sounds online sing—fun blast! Fishin Frenzy
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Титан-алюминиевая плоская мишень
where can i get cheap atarax without a prescription
| #
Drug information leaflet. What side effects?
where can i get cheap atarax without a prescription
Best news about medicine. Read here.
can i get tamoxifen for sale
| #
Drugs information leaflet. Short-Term Effects.
can i get tamoxifen for sale
Everything about medicament. Get information here.
DavidNaing
| #
This online casino’s Tiger Fortune is a blast. tigrinho demo
30 day prednisone taper
| #
can i buy cheap prednisone pill
Uazrubr
| #
Доброго времени суток!
Для некоторых людей, заказать диплом о высшем образовании – это острая потребность, шанс получить достойную работу. Но для кого-то – это очевидное желание не терять множество времени на учебу в ВУЗе. Что бы ни толкнуло вас на такой шаг, мы готовы помочь. Максимально быстро, качественно и выгодно сделаем диплом любого года выпуска на подлинных бланках с реальными подписями и печатями.
Ключевая причина, почему люди покупают документы, – желание занять определенную работу. К примеру, знания позволяют специалисту устроиться на желаемую работу, а документального подтверждения квалификации не имеется. В случае если для работодателя важно наличие “корочки”, риск потерять хорошее место очень высокий.
Заказать документ о получении высшего образования вы сможете в нашей компании в столице. Мы оказываем услуги по производству и продаже документов об окончании любых ВУЗов России. Вы получите необходимый диплом по любой специальности, любого года выпуска, в том числе документы СССР. Даем гарантию, что при проверке документа работодателем, каких-либо подозрений не появится.
Разных ситуаций, которые вынуждают приобрести диплом ВУЗа много. Кому-то срочно требуется работа, таким образом нужно произвести впечатление на начальника при собеседовании. Другие желают попасть в престижную компанию, для того, чтобы повысить свой статус и в будущем начать свой бизнес. Чтобы не тратить попусту годы жизни, а сразу начать эффективную карьеру, применяя имеющиеся знания, можно заказать диплом в интернете. Вы сможете стать полезным для общества, обретете денежную стабильность в максимально короткий срок- диплом о высшем образовании купить
internetchelyabCah
| #
интернет провайдеры челябинск
domashij-internet-chelyabinsk001.ru
подключить домашний интернет в челябинске
mostbet_xnmt
| #
mosbet mosbet .
Willieavamy
| #
I won $600 on a slot—hype high! andar bahar
DavidNaing
| #
Tiger Fortune’s design is top-notch – really impressive. tigrinho demo
Byronpiofe
| #
Нужен бесплатный аккаунт? https://dzen.ru/freesteamaccount/ Узнайте, где можно получить рабочие логины с играми, как не попасть на фейк и на что обратить внимание при использовании таких аккаунтов.
Virgiljuils
| #
Онлайн-казино Aura aura casino apk лицензия, быстрые выплаты, слоты от топ-провайдеров и щедрые бонусы. Подробный обзор платформы: интерфейс, регистрация, акции, безопасность и отзывы игроков.
Lancelex
| #
Онлайн-казино Calibry calibri casino play бонусы без депозита, быстрые выводы и лицензированный софт. В обзоре: как начать играть, получить фриспины, использовать акции и избежать рисков.
MichaelraB
| #
Начните с Aura Casino aura casino bonus удобное онлайн-казино с простым интерфейсом, стартовыми бонусами и быстрой регистрацией. Рассказываем, как получить фриспины и не потеряться в слотах.
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наши товары:
Кольцо РёР· драгоценных металлов платиновое 15С…9С…4.5 РјРј ПлН95.5 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
Mazrvim
| #
Мы можем предложить дипломы любых профессий по доступным тарифам. Всегда стараемся поддерживать для клиентов адекватную политику тарифов. Для нас важно, чтобы документы были доступны для большинства граждан.
Покупка документа, который подтверждает окончание института, – это выгодное решение. Приобрести диплом университета: diplom5.com/kupit-diplom-nedorogo-v-moskve-2/
DavidNaing
| #
This casino’s Tiger Fortune game is a winner. fortune tiger bet
Diplomi_lxKt
| #
Заказать диплом любого ВУЗа!
Мы можем предложить документы ВУЗов, которые находятся на территории всей РФ.
rusd-diplomj.ru/kupit-diplom-s-vneseniem-v-reestr-bistro-i-nadezhno-10/
Sazrshu
| #
Купить документ о получении высшего образования вы можете в нашей компании в Москве. Мы предлагаем документы об окончании любых ВУЗов Российской Федерации. Вы получите диплом по любой специальности, включая документы Советского Союза. Даем гарантию, что в случае проверки документа работодателем, подозрений не возникнет. asxdiplommy.com/legko-i-bistro-zanesite-diplom-v-reestr-obrazovaniya/
sklad_mqOt
| #
хранение в подмосковье хранение в подмосковье .
1win_riSn
| #
1 win kg http://1win6046.ru/ .
mostbet_vuMi
| #
mostbet casino mostbet6035.ru .
Lazrtyc
| #
Купить диплом об образовании!
Мы предлагаем дипломы любой профессии по приятным ценам— diplomt-v-samare.ru/kupit-diplom-meditsinskij-v-moskve-6/
1win_vver
| #
1win.pro http://1win5015.ru/ .
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы а также предоставлять решения под особенности вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наша продукция:
Рзделия РёР· титана Р’Рў6 СЃРІ. – РћРЎРў 1 90015-77 РџРѕРєРѕРІРєР° титановая Р’Рў6 СЃРІ. – РћРЎРў 1 90015-77 представляет СЃРѕР±РѕР№ высококачественный материал, широко используемый РІ авиационной, медицинской Рё автомобильной отраслях. Обладая отличной прочностью Рё РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью, РѕРЅР° идеально РїРѕРґС…РѕРґРёС‚ для производства деталей, требующих высокой надежности.Приобретая РџРѕРєРѕРІРєР° титановая Р’Рў6 СЃРІ. – РћРЎРў 1 90015-77, РІС‹ получаете гарантии качества Рё долговечности. Надежность данного сплава делает его оптимальным выбором для ответственных задач. РќРµ упустите возможность купить РџРѕРєРѕРІРєР° титановая Р’Рў6 СЃРІ. – РћРЎРў 1 90015-77 Рё обеспечьте СЃРІРѕРё проекты необходимым материалом!
DavidNaing
| #
Tiger Fortune’s animations are next-level awesome. tigrinho demo
where buy trileptal without prescription
| #
Medicine information leaflet. Brand names.
where buy trileptal without prescription
Best news about drugs. Get information now.
where can i get cheap priligy online
| #
Meds information for patients. Brand names.
where can i get cheap priligy online
Everything about meds. Get information now.
1win_anst
| #
1win мобильная версия сайта http://1win6045.ru/ .
Hijamenetly
| #
На сайте https://pilmi.ru вы найдете огромное количество фильмов самых разных жанров, включая комедии, мелодрамы, драмы, романтические. Для того чтобы подыскать наиболее подходящий вариант, нужно воспользоваться специальным каталогом. Вашему вниманию представлены и ожидаемые любопытные новинки этого года, которые произведут эффект. Все это можно просматривать в любое время и на различных устройствах. Прямо сейчас воспользуйтесь возможностью посмотреть азиатское кино. Используйте поиск для облегчения выбора.
DavidNaing
| #
Tiger Fortune’s design is bold and exciting. fortune tiger demo gratis
Protezirovanie zybov_vnEa
| #
протезирование зубов при полном отсутствии зубов стоимость http://www.protezirovanie-zubov1.ru .
Protezirovanie zybov_vnpl
| #
протезирование 2 зубов протезирование 2 зубов .
can i order cheap flexeril prices
| #
get flexeril pills
Protezirovanie zybov_mrMn
| #
протезирование зуба сколько http://www.protezirovanie-zubov3.ru .
stomatologiya Mitino_bnKi
| #
лечение зубов +в москве лечение зубов +в москве .
DavidNaing
| #
The spins in Tiger Fortune are always exciting. tiger fortune
mua bán heroin
| #
mua bán heroin
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
Наши товары:
Медный тройник РїРѕРґ пайку стандартный 12С…9С…0.6 РјРј 8.6С…10.6 РјРј твердая пайка Рњ3Р Рў ГОСТ Р 52922-2008 Купить качественные медные тройники РїРѕРґ пайку для надежных Рё долговечных соединений РІ трубопроводных системах. Разнообразие размеров Рё конфигураций, высокая теплопроводность Рё устойчивость Рє РєРѕСЂСЂРѕР·РёРё. Простая установка Рё надежность соединения. Рдеальное решение для различных проектов. Редметсплав.СЂС„
cost generic finpecia without a prescription
| #
can i order generic finpecia for sale
DavidNaing
| #
Tiger Fortune is my lucky charm at this casino. tigrinho demo
DavidNaing
| #
Tiger Fortune’s visuals are a feast for the eyes. fortune tiger
ManitnTonge
| #
Вместе с t.me/azino777_a у вас есть возможность в увлекательное путешествие по миру азартных игр отправиться. Предлагаем уникальные бонусы и персонализированные предложения. Расскажем, как начать играть в Азино. Вас ждут невероятные эмоции и возможность выиграть крупный приз. Испытайте свою удачу уже сейчас! Ищете azino777 скачать бесплатно? – тут имеется множество актуальной информации. Собрали самые частые вопросы от игроков. Всегда готовы помочь вам. Приготовьтесь к увлекательным турнирам в Азино. Участвуйте и боритесь за призы с другими игроками!
internetchelyabCah
| #
подключить домашний интернет в челябинске
domashij-internet-chelyabinsk002.ru
подключить домашний интернет в челябинске
Sazrrmo
| #
Приобрести диплом института!
Заказать диплом университета по выгодной стоимости возможно, обратившись к надежной специализированной фирме. Быстро купить диплом: vuz-diplom.ru/diplom-vuza-s-provodkoj-garantiya-kareri
StephenimMix
| #
Обзор онлайн-казино Calibry https://calibri-casino-app.ru как получить бонус без депозита, запустить любимые слоты и вывести выигрыш. Надёжная площадка с лицензией и прозрачными условиями игры.
1win_cver
| #
cazinouri online moldova 1win5015.ru .
LarryNip
| #
Играйте в казино Shot https://shot-casino-play.ru десятки провайдеров, ежедневные турниры и кэшбэк. В обзоре: лучшие слоты, лайв-игры, бонусные программы и преимущества платформы.
Mazrfzd
| #
Где купить диплом специалиста?
Готовый диплом с приложением отвечает требованиям и стандартам Министерства образования и науки Российской Федерации, неотличим от оригинала – даже со специально предназначенным оборудованием. Не откладывайте личные мечты на пять лет, реализуйте их с нашей компанией – отправляйте заявку на изготовление документа сегодня! Диплом о высшем образовании – не проблема! nowoczesna.phorum.pl/posting.phpmode=newtopic&f=2&sid=e9e348ec9934a08ca9d6f8f6cd5552ef
WilliamHoasy
| #
Aura Casino http://aura-casino-online.ru безопасная платформа с лицензией, SSL-защитой и проверенной репутацией. В обзоре: честность выплат, работа службы поддержки и реальные отзывы игроков.
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их соответствие стандартам. Опытная поддержка – наш стандарт – мы на связи, чтобы улаживать ваши вопросы и адаптировать решения под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Поковка танталовая Ta-Nb20 Поковка танталовая Ta-Nb20 представляет собой высококачественный материал, обладающий отличными антикоррозийными и теплоцентрализующими свойствами. Она используется в производстве сильных и долговечных деталей для аэрокосмической и медицинской отраслей, а также в химической промышленности. Благодаря своим уникальным характеристикам, танталовая поковка обеспечивает надежность и эффективность изделий. Не упустите уникальную возможность купить Поковка танталовая Ta-Nb20 и обеспечьте своему производству необходимое качество и долговечность.
mostbet_jemt
| #
казино онлайн kg mostbet6033.ru .
CaseyJer
| #
Обзор Aura Casino https://aura-casino-play.ru всё о щедрых бонусах, фриспинах, кэшбэке и акциях. Расскажем, как зарегистрироваться, начать игру и получить максимум от каждого депозита.
mazusTor
| #
Alkomarket-dubai.ru коллекцию алкогольных напитков на разный кошелек и вкус предлагает. Наш основной приоритет – качество продукции, мы о здоровье своих клиентов заботимся. Закажите алкоголь с доставкой на дом уже сейчас. Наслаждайтесь комфортом! Ищете алкоголь в дубае с доставкой? Alkomarket-dubai.ru – здесь представлен широкий ассортимент пива, вина, водки, виски и скотча, а также других премиальных напитков от известных мировых брендов. К покупателям относимся бережно, об их потребностях заботясь. Поможем для вас и на подарок близким выбрать достойные варианты.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Плоская мишень Nb2O5 4N
can i purchase glucophage pill
| #
Medicament information for patients. Brand names.
can i purchase glucophage pill
Some information about drugs. Get here.
can i buy generic synthroid without a prescription
| #
Medicament information for patients. Brand names.
can i buy generic synthroid without a prescription
Everything trends of pills. Read now.
DavidNaing
| #
I love the tiger vibes in Tiger Fortune. tigrinho demo
Willieavamy
| #
The live dealers online are awesome—so pro! vai de bet
VincentMalge
| #
Устал только играть в CS2 и иногда хочется зайти в cs рулетка и попытать удачу или математический расчет. Отличная подборка рулеток кс 2 и ксго на вечер с проверенными отзывами и выплатами.
Protezirovanie zybov_wvMn
| #
протезирование жевательных зубов стоимость https://protezirovanie-zubov3.ru/ .
Protezirovanie zybov_ojpl
| #
сколько стоит поставить зубные протезы сколько стоит поставить зубные протезы .
Protezirovanie zybov_ntEa
| #
протез зуба цена протез зуба цена .
1win_cxer
| #
1win aplicația 1win aplicația .
WilliamDix
| #
Мы предлагаем дипломы любых профессий по приятным ценам. Дипломы производят на оригинальных бланках Приобрести диплом о высшем образовании diplomt-v-chelyabinske.ru
1win_emEl
| #
cazino md https://www.1win5014.ru .
Lazrgcr
| #
Где приобрести диплом специалиста?
Приобрести диплом института по невысокой стоимости можно, обратившись к надежной специализированной компании.: diplom-kuplu.ru
stomatologiya Mitino_scKi
| #
эмблема стоматологии эмблема стоматологии .
Willieavamy
| #
The mobile apps online falter—patch it! aviator game online
Sazrtfs
| #
Мы изготавливаем дипломы любой профессии по выгодным тарифам.– diplom-kaluga.ru/kupit-diplom-s-zaneseniem-v-reestr-foruma-vigodno/
Rheumatoid arthritis treatment professionals
| #
Rheumatoid arthritis treatment hospitals
Sazrebb
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и любых других профессий по выгодным ценам.
Вы приобретаете диплом через надежную компанию. Заказать диплом о высшем образовании– http://elitemagyaritasok.info/e107_plugins/forum/forum_post.phpf=nt&id=21#/ – elitemagyaritasok.info/e107_plugins/forum/forum_post.phpf=nt&id=21#
get generic aciphex online
| #
buying cheap aciphex without a prescription
Donaldlaf
| #
darknet markets 2025 darkmarket 2025
Protezirovanie zybov_vxMn
| #
лечение и протезирование зубов стоимость лечение и протезирование зубов стоимость .
Protezirovanie zybov_dzpl
| #
протезирование верхних зубов цена https://protezirovanie-zubov2.ru .
Protezirovanie zybov_quEa
| #
коронка зуба цена коронка зуба цена .
Jariortyp
| #
Где заказать диплом специалиста?
Наши специалисты предлагают максимально быстро купить диплом, который выполняется на оригинальной бумаге и заверен мокрыми печатями, штампами, подписями официальных лиц. Диплом способен пройти любые проверки, даже с применением специально предназначенного оборудования. Достигайте цели быстро с нашей компанией.
Заказать диплом любого ВУЗа diplomv-v-ruki.ru/kupit-diplom-s-reestrom-21/
internetchelyabCah
| #
подключить домашний интернет челябинск
domashij-internet-chelyabinsk003.ru
подключить интернет челябинск
Collinseent
| #
Сайт Идеальная Я – интернет журнал, где каждая женщина сможет узнать ответы на вопросы о здоровье, красоте, беременности и воспитании детей https://idealnaya-ya.ru/
stomatologiya Mitino_cwpr
| #
стоматолог митино стоматолог митино .
buying bactrim without prescription
| #
Meds information sheet. Brand names.
buying bactrim without prescription
Best what you want to know about pills. Read information here.
1win_aist
| #
1win официальный сайт регистрация http://www.1win6045.ru .
Toliknaf
| #
darknet drug store darknet markets onion
stomatologiya Mitino_pkKi
| #
хирургия в митино http://www.stomatologiya-mitino3.ru .
DavidNaing
| #
Tiger Fortune’s payouts are always a surprise. fortune tiger 777
Rabyitabe
| #
darknet markets darknet drug links
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наши товары:
Никелевая плита 500х300х60 мм НМЖМц28-2.5-1.5 Купите никелевую плиту от Редметсплав.рф. Широкий выбор никелевых плит различных размеров, форм и толщин. Высокая прочность, устойчивость к коррозии и долгий срок службы. Применение в химическом оборудовании, авиации, энергетике и медицине.
Elisa
| #
You ought to take part in a contest for one of the most
useful websites online. I most certainly will recommend this
site!
Sazrvkf
| #
купить диплом аттестат в новосибирске
DonDonfaita
| #
darknet drug market darknet markets url
leptozan
| #
leptozan is an innovative weight loss formula crafted to help you naturally sculpt the body you’ve always desired.
Pingrutty
| #
dark web markets darknet marketplace
DavidNaing
| #
Tiger Fortune’s design is top-notch – really impressive. fortune tiger demo gratis
nervecalm
| #
nerve calm is a high-quality nutritional supplement crafted to promote nerve wellness, ease chronic discomfort, and boost everyday vitality.
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их качество. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы по мере того как находить ответы под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Пруток магниевый ISO-MgAl6Mn – ISO 16220 Проволока магниевая ISO-MgAl6Mn – ISO 16220 представляет СЃРѕР±РѕР№ высококачественный материал, используемый РІ различных сферах, включая авиацию, автомобилестроение Рё производство деталей, требующих легкости Рё прочности. РћРЅР° сочетает РІ себе отличную РєРѕСЂСЂРѕР·РёРѕРЅРЅСѓСЋ стойкость Рё РЅРёР·РєСѓСЋ плотность, что делает ее идеальным выбором для создания легких конструкций.Рта проволока отвечает международным стандартам Рё обеспечивает надежность РІ эксплуатации. Если РІС‹ ищете надежное решение для СЃРІРѕРёС… проектов, то стоит купить Проволока магниевая ISO-MgAl6Mn – ISO 16220. РќРµ упустите возможность повысить качество своей работы СЃ этим превосходным продуктом!
Donaldlaf
| #
darknet drug links darknet market lists
Ronaldpar
| #
Онлайн-казино Aura https://aura-casino-app.ru лицензия, бонус без депозита, фриспины за регистрацию и моментальный вывод. Всё о казино Aura в одном обзоре — играйте безопасно и с выгодой.
Sazraht
| #
Студенческая жизнь прекрасна, пока не приходит время писать диплом, как это случилось со мной. Не стоит отчаиваться, ведь существуют компании, которые помогают с написанием и защитой диплома на высокие оценки!
Сначала я искал информацию по теме: купить диплом техникума проведенный, купить диплом уральского, купить легальный диплом высшем, купить чистый бланк диплома, можно ли работать с купленным дипломом, а потом наткнулся на proffdiplomik.com/orenburg
RichardKes
| #
Обзор казино Shot https://shot-casino-bonus.ru/ лицензия, защита данных, честная игра и проверенные провайдеры. Узнайте, как работает система вывода, поддержка и что говорят реальные пользователи.
MontePanty
| #
Calibry Casino http://calibri-casino-bonus.ru лицензированное онлайн-казино с бонусами, быстрым выводом и большим выбором игр. В обзоре: регистрация, акции, безопасность, отзывы игроков и советы новичкам.
RichardEncat
| #
Онлайн-казино Calibry calibri-casino-apk бонус без депозита, фриспины, турниры и выплаты без задержек. Рассказываем, как начать играть, какие игры выбрать и стоит ли доверять платформе.
1win_rmsa
| #
1win официальный сайт вход https://www.1win7011.ru .
cost of elavil price
| #
Medication information. Long-Term Effects.
cost of elavil price
Actual about drug. Get here.
1win_gcoa
| #
1win вход на сайт https://1win7013.ru/ .
can i purchase cheap lincocin tablets
| #
can i get lincocin price
DavidNaing
| #
This online casino shines with Tiger Fortune. fortune tiger
1win_tyEl
| #
1win.com https://1win5014.ru .
https://createbrand.ru/kak-vybrat-medali-na-zakaz-i-chto-stoit-uchest-pri-ih-izgotovlenii/
| #
Выбор медалей на заказ требует внимания к деталям. Важно учитывать назначение изделия, тип металла, способ нанесения изображения и возможность персонализации. Также имеет значение форма, размер и оформление — всё это влияет на восприятие и ценность медали. Если вы хотите получить качественный результат, стоит заранее продумать все параметры. Подробнее о нюансах выбора и производства медалей читайте по ссылке:
https://createbrand.ru/kak-vybrat-medali-na-zakaz-i-chto-stoit-uchest-pri-ih-izgotovlenii/
amoxil pediatrico 250 mg
| #
can i purchase generic amoxil tablets
inernetadresCah
| #
узнать провайдера по адресу красноярск
domashij-internet-krasnoyarsk001.ru
проверить интернет по адресу
BruceInabs
| #
https://mockwa.forum2x2.ru/t79-topic#80
mostbet_upmt
| #
mostbet apk скачать https://mostbet6033.ru/ .
1win_spPr
| #
баланс ван вин http://1win6048.ru .
MarioDup
| #
Drinks delivery предлагает широкий ассортимент алкогольных напитков и умеренные цены. Качество – главный наш приоритет. Заботимся о своей репутации. Продаем только то, в чем уверены. С нами вы легко выберете нужный товар. Ищете алкоголь в дубае с доставкой? Deliverydubaidrinks.com – здесь есть абсент, ликер, вино, коньяк, ром, водка, джин, виски, бризер, текила, шампанское и пиво. Выполняем оперативную доставку алкоголя. Свое дело мы любим и к пожеланиям покупателей всегда прислушиваемся. Ответим на любой вопрос и поможем подобрать подходящие для вас напитки.
Sogiphvor
| #
Академия «Арчибальд» занимается обучением грумингу. У вас есть возможность свои навыки совершенствовать до бесконечности на живых моделях. Преподаватели на все вопросы отвечают. Дарим методические материалы, учебники, а также видеолекции. Погрузитесь в атмосферу, с которой не захочется расставаться. Ищете курсы груминга в москве цены? Grooming-dream.ru – здесь представлены реальные отзывы клиентов. Мы лучших груминг-специалистов выпускаем. Предлагаем инструменты в период обучения. Если вам интересна новая профессия – Арчибальд, вот то место, которое вам подойдет.
DavidNaing
| #
Tiger Fortune’s theme is wild and totally unique. fortune tiger demo gratis
Dnrtove
| #
Мы можем предложить дипломы психологов, юристов, экономистов и других профессий по приятным ценам. Мы предлагаем документы техникумов, расположенных в любом регионе России. Документы делаются на “правильной” бумаге высшего качества. Это дает возможность делать настоящие дипломы, которые не отличить от оригинала. veniaminv.flybb.ru/viewtopic.phpf=1&t=4335
Cazrliy
| #
Где заказать диплом специалиста?
Мы изготавливаем дипломы любой профессии по доступным тарифам. Для нас важно, чтобы документы были доступными для подавляющей массы наших граждан. Приобрести диплом любого института diplomk-v-krasnodare.ru/kupit-diplom-vuza-s-provodkoj-v-reestre-ofitsialno/
stomatologiya Mitino_qnpr
| #
американская клиника в москве http://stomatologiya-mitino2.ru .
how long does venlafaxine take to get into your system
| #
Pills information sheet. Short-Term Effects.
how long does venlafaxine take to get into your system
All what you want to know about medicines. Read now.
DavidNaing
| #
Tiger Fortune’s rewards make it worth playing. fortune tiger 777
Diplomi_ncKt
| #
Заказать диплом о высшем образовании!
Мы можем предложить документы институтов, которые расположены в любом регионе РФ.
kupitediplom0027.ru/kupit-diplom-s-zaneseniem-v-reestr-otzivi-garantii/
Iariorhmq
| #
Приобрести диплом ВУЗа по выгодной стоимости вы можете, обратившись к проверенной специализированной фирме. Купить документ университета можно в нашем сервисе. asxdiploman.com/kupit-diplom-yurista-s-vneseniem-v-reestr-bistro-2
Sazrxds
| #
Заказать диплом университета !
Приобретение диплома ВУЗа РФ у нас является надежным процессом, поскольку документ заносится в реестр. Купить диплом университета diplomservis.com/kupite-diplom-s-zaneseniem-v-reestr-bistro-i-nadezhno-5
DavidNaing
| #
I love the thrill of Tiger Fortune’s jackpots. fortune tiger
Emercomjoype
| #
Современные технологии позволяют защитить любой объект от внешних угроз. Одним из ключевых решений являются системы видеонаблюдения, которые обеспечивают круглосуточный контроль. Они просты в использовании и надежны в работе. Установите их, чтобы обезопасить себя и своих близких.
mostbet_fnmt
| #
казино онлайн kg http://mostbet6033.ru .
mostbet_jaEl
| #
мостбет войти мостбет войти .
MichaelDog
| #
Автоперевозки из Китая по РФ ved-china быстрая и выгодная доставка грузов напрямую до вашего склада. Консолидация, растаможка, сопровождение и отслеживание на всех этапах.
Viktoris#espana[WcuwyripyfdiBI,2,5]
| #
?Hola fanaticos del casino
Los juegos de casino sin licencia incluyen opciones innovadoras y proveedores poco conocidos. casinossinlicenciaenespana Esto aГ±ade variedad a tu experiencia de juego. Es perfecto para quienes buscan salir de lo tradicional.
Los juegos de casino sin licencia incluyen opciones innovadoras y proveedores poco conocidos. Esto aГ±ade variedad a tu experiencia de juego. Es perfecto para quienes buscan salir de lo tradicional.
Mas detalles en el enlace – п»їhttp://casinossinlicenciaenespana.guru
?Que tengas excelentes premios!
AaronBal
| #
Нужен надёжный хостинг? vps сервер купить для сайтов, приложений и бизнес-задач. Гибкая настройка, SSD-накопители, стабильная работа 24/7. Выберите тариф под свои задачи и начните сегодня.
Keithcaula
| #
новости сегодня новости москвы
Michealoreme
| #
Ищешь лучшие раскидки raskidki-granat-cs2.ru CS2 – Смоки, молотовы и флешки для всех карт — изучи тактики, прокачай игру и удиви соперников точными гранатами. Подборка эффективных раскидок!
Armandotum
| #
kraken сайт – kraken войти, Площадка кракен
1win_qjoa
| #
1winn 1win7013.ru .
1win_icOl
| #
казино 1win http://1win7001.ru/ .
JeffreyUtirm
| #
ссылка на сайт Манго Офис Виртуальная АТС
mostbet_njmt
| #
мост бет http://www.mostbet6033.ru .
Jimmynum
| #
Готовы к новым встречам? В Сочи теперь есть сайт знакомств, который поможет вам найти девушку вашей мечты. Знакомьтесь, общайтесь и стройте отношения, смотря в будущее с надеждой. Пусть ваши мечты о любви станут реальностью в нашем дружелюбном пространстве – https://t.me/sochinightt
can promethazine be taken with tylenol
| #
Meds information leaflet. Brand names.
can promethazine be taken with tylenol
Some what you want to know about medicines. Read now.
Willieavamy
| #
Online casinos feel risky—double watch! plinko
DavidNaing
| #
Tiger Fortune’s gameplay is always thrilling. fortune tiger
DavidNaing
| #
Tiger Fortune’s features are pure genius. fortune tiger demo gratis
inernetadresCah
| #
провайдеры по адресу красноярск
domashij-internet-krasnoyarsk002.ru
провайдеры интернета в красноярске по адресу
buying cheap lisinopril
| #
order lisinopril without rx
mostbet_rhEl
| #
служба поддержки мостбет номер телефона https://mostbet6036.ru .
Trefawe
| #
Приобрести диплом о высшем образовании. Заказ подходящего диплома через надежную фирму дарит множество преимуществ. Это решение дает возможность сберечь как личное время, так и серьезные финансовые средства. athena.5nx.ru/viewtopic.php?f=5&t=344
WalterLar
| #
Понедельник: Астрологический прогноз на успешное начало недели
https://x.com/IrinaPavlovna84/status/1911485302209667581
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Вращающаяся мишень для распыления вольфрама
kepirdrind
| #
В случае если желаете узнать самую ценную, полезную и важную информацию о трейдинге, вложениях, то нужно зайти на портал, где вы отыщете все необходимое на такую тему. Здесь доступным языком рассказывается о том, как правильно вести торговлю на внебиржевом рынке. http://fxidea.com/ предлагает акции всем начинающим. Быстрее изучите индикаторы, реальные отзывы, а также ознакомьтесь с работающими стратегиями. Вы сможете воспользоваться интересными материалами о криптовалюте, будете в курсе того, как можно получить деньги от форекса.
RobertIGNIG
| #
Предлагаем покупку чеков https://kupit-cheki-new.ru в Москве для отчётности, командировок, подтверждения трат. Быстрое оформление, гибкие условия, все виды документов и сопровождение при необходимости.
Barryfrods
| #
Хотите купить чеки http://kupit-cheki-reg.ru в Москве срочно? Работаем без выходных, оформляем документы день в день, возможна доставка и электронные версии. Оперативность и конфиденциальность гарантированы.
RogerArent
| #
Где купить kupit cheki24 чеки в Москве? У нас — быстрое оформление, оригинальные документы, курьерская доставка. Чеки для отчётов, подтверждения командировок и компенсаций.
1win_miPr
| #
1wiun https://1win6048.ru/ .
Thomasfeeds
| #
Витебский университет П.М.Машерова https://vsu.by образовательный центр. Вуз является ведущим образовательным, научным и культурным центром Витебской области.
1win_dmOl
| #
1win официальный сайт 1win7001.ru .
Jariorxyj
| #
Где купить диплом специалиста?
Наша компания предлагает выгодно заказать диплом, который выполнен на оригинальном бланке и заверен мокрыми печатями, водяными знаками, подписями. Диплом пройдет лубую проверку, даже при использовании специального оборудования. Достигайте цели быстро и просто с нашим сервисом.
Купить диплом любого ВУЗа diplomt-v-samare.ru/kupit-diplom-elektromontera-8/
DavidNaing
| #
This online casino’s Tiger Fortune is a must-play. fortune tiger demo
DavidNaing
| #
The Tiger Fortune game runs so smoothly on my phone. tiger fortune
Iariorvrg
| #
Купить диплом ВУЗа по доступной стоимости возможно, обратившись к проверенной специализированной фирме. Приобрести документ института можно в нашей компании. diplomservis.com/kupit-diplom-visshem-obrazovanii-zaneseniem-reestr-12
fluoxetine tablets
| #
cheap fluoxetine without rx
mostbet_rkEl
| #
скачать мостбет http://www.mostbet6036.ru .
cost of cheap bactrim pills
| #
Medicine information. Effects of Drug Abuse.
cost of cheap bactrim pills
Actual what you want to know about drugs. Get now.
DavidNaing
| #
Tiger Fortune brings the jungle to life! fortune tiger 777
Lazrrkp
| #
Диплом университета Российской Федерации!
Без наличия диплома очень сложно было продвинуться по карьере. В связи с этим решение о заказе диплома можно считать выгодным и рациональным. Приобрести диплом любого университета mahnazpublisher.org/employer/gosznac-diplom-24
Eanrolf
| #
Для эффективного продвижения вверх по карьерной лестнице понадобится наличие диплома о высшем образовании. Быстро и просто заказать диплом об образовании у проверенной компании: diplomp-irkutsk.ru/kupit-diplom-sredne-texnicheskoe-obrazovanie-bistro-i-udobno/
Sazrdut
| #
купить аттестат 11 классов в москве
DavidNaing
| #
This casino’s Tiger Fortune game is a blast. fortune tiger
1win_ciOl
| #
one win https://1win7001.ru/ .
https://onlaine-vtb.ru/questions/2897-metallicheskij-suvenir-innovacii-v-reklamnoj-industrii.html
| #
Металлические сувениры становятся всё более популярными в рекламной индустрии благодаря своей долговечности, эстетической привлекательности и возможностям персонализации. Использование современных технологий, таких как лазерная гравировка и 3D-печать, позволяет создавать уникальные изделия, подчёркивающие статус и надёжность бренда. Кроме того, металлические сувениры являются экологичным и экономически эффективным решением для компаний, стремящихся к устойчивому развитию. Подробнее об инновациях в производстве металлических сувениров можно узнать по ссылке:
https://onlaine-vtb.ru/questions/2897-metallicheskij-suvenir-innovacii-v-reklamnoj-industrii.html
Willieavamy
| #
I love the live roulette—so live! fortune mouse
Cazrdpe
| #
Приобрести диплом университета!
Мы можем предложить документы учебных заведений, которые расположены в любом регионе России. Дипломы и аттестаты выпускаются на бумаге самого высокого качества: nadom-dedamoroza.ru/forum/PAGE_NAME=profile_view&UID=16609
Sazredb
| #
Всегда думал что купить диплом о высшем образовании это миф и нереально, но все оказалось не так, изначально искал информацию про: купить диплом техникума, сколько стоит аттестат, купить дипломы, диплом вуза купить, дипломы купить, потом про дипломы вузов, подробнее здесь proffdiplomik.com/volgograd
muzetGuimi
| #
Delivery-dubai.com предлагает широкий ассортимент разных видов алкоголя. На сайте можно ознакомиться с имеющейся продукцией в каталоге с ценами. Это позволит спокойно выбрать подходящий под ваш повод напиток. Выполняем быструю доставку алкоголя. Будем рады видеть вас в числе клиентов! Ищете купить алкоголь в дубае? Delivery-dubai.com – тут широкий выбор алкоголя представлен. Вы найдете для себя все, что может принести удовольствие. Предлагаем товары исключительного качества. Для заказа напишите нам в мессенджеры: WhatsApp, Telegram.
Sazrjvm
| #
Покупка официального диплома через надежную фирму дарит массу плюсов для покупателя. Данное решение позволяет сберечь время и серьезные финансовые средства. Впрочем, достоинств намного больше.Мы изготавливаем дипломы любой профессии. Дипломы производятся на подлинных бланках. Доступная стоимость по сравнению с огромными тратами на обучение и проживание. Покупка диплома ВУЗа будет мудрым шагом.
Заказать диплом: diplomt-v-chelyabinske.ru/mozhno-li-kupit-diplom-zanesennij-v-reestr-3/
1win_wwer
| #
1win moldova http://www.1win5015.ru .
1win_ciPr
| #
1win ваучер http://1win6048.ru/ .
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
поставляемая продукция:
Медная однораструбная редукционная переходная муфта РїРѕРґ пайку стандартная 80С…70 РјРј мягкая пайка Рњ3Р Рњ ГОСТ 32590-2013 Приобретите высококачественные медные однораструбные редукционные муфты РїРѕРґ пайку РѕС‚ Редметсплав для надежного соединения трубопроводов. Гарантированная прочность Рё устойчивость Рє РєРѕСЂСЂРѕР·РёРё. Рдеальны для различных систем отопления, водоснабжения Рё прочих технических целей.
Iariorbwi
| #
Приобрести диплом института по доступной цене вы можете, обратившись к проверенной специализированной фирме. Заказать документ института можно у нас в столице. vacshidiplom.com/pokupka-diplomov-s-reestrom-8
inernetadresCah
| #
узнать интернет по адресу
domashij-internet-krasnoyarsk003.ru
узнать интернет по адресу
DavidNaing
| #
I can’t get enough of the Tiger Fortune soundtrack! tigrinho demo
Protezirovanie zybov_kzMn
| #
протез зуба цена протез зуба цена .
Protezirovanie zybov_zypl
| #
сколько стоит поставить протез зуба https://protezirovanie-zubov2.ru .
Protezirovanie zybov_dlEa
| #
протезирование зубов клиники цены https://protezirovanie-zubov1.ru/ .
Diplomi_rbSn
| #
купить диплом колледжа в спб купить диплом колледжа в спб .
Sazrfvl
| #
Заказать диплом возможно через официальный портал компании. peticijos.com/482357
Allergic conjunctivitis treatment hospitals
| #
Allergic conjunctivitis treatment facilities
hedinic
| #
Блог о маркетплейсах «OOWA» предлагает ознакомиться с актуальной, интересной и познавательной информацией, которая касается удачных продаж на маркетплейсах, обзоров товаров и услуг. На портале регулярно публикуются ценные и содержательные рекомендации от экспертов, которые помогут начать лучше разбираться в тонкостях такого бизнеса. Ищете что такое мегамаркет ? На oowa.ru регулярно публикуются ценные и содержательные рекомендации от экспертов, которые помогут начать лучше разбираться в тонкостях такого бизнеса. Почитайте о самых востребованных товарах с маркетплейсов.
DavidNaing
| #
This online casino’s Tiger Fortune is fantastic. fortune tiger
how can i get atarax price
| #
Pills information. What side effects can this medication cause?
how can i get atarax price
Best news about medicines. Get information here.
MAMAK KAU LONTE
| #
MAMAK KAU LONTE
A Brief History of Elemental Analysis » Artemis Analytical
buying medex price
| #
can you buy generic medex
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их происхождение. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы по мере того как находить ответы под требования вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Полоса висмутовая Bi02A – EIA/IPC J-STD-006 Полоса висмутовая Bi02A – EIA/IPC J-STD-006 является высококачественным материалом, используемым РІ различных электронных приложениях. РћРЅР° отличается отличной проводимостью Рё устойчивостью Рє РєРѕСЂСЂРѕР·РёРё, что делает её идеальным выбором для профессионалов РІ области электроники. Рта полоса соответствует стандартам EIA/IPC J-STD-006, обеспечивая надежность Рё стабильность РІ работе. РќРµ упустите возможность купить Полоса висмутовая Bi02A – EIA/IPC J-STD-006 Рё повысить качество СЃРІРѕРёС… изделий. Заказывая данный РїСЂРѕРґСѓРєС‚, РІС‹ получаете гарантированное качество Рё высокую производительность.
DavidNaing
| #
I hit a big win on Tiger Fortune last night! fortune tiger 777
DavidNaing
| #
The bonuses in Tiger Fortune are so exciting! fortune tiger 777
1win_amst
| #
1вин про https://www.1win6045.ru .
DavidNaing
| #
This online casino’s Tiger Fortune is a thrill. jogo do tigrinho demo
1win_vfPr
| #
официальный сайт 1win http://1win6048.ru .
Willieavamy
| #
Are online reviews legit?—hard say! mines game
Willieavamy
| #
The live games online rock—fan love! fishing frenzy
Sazrxmp
| #
Заказать диплом института по выгодной цене можно, обратившись к проверенной специализированной компании. Мы оказываем услуги по производству и продаже документов об окончании любых университетов РФ. Купить диплом ВУЗа– diplomt-v-chelyabinske.ru/kachestvennij-diplom-visshego-obrazovaniya-po-dostupnoj-tsene-3/
DavidNaing
| #
The tiger-themed bonuses in Tiger Fortune rock! fortune tiger bet
1win_bqer
| #
1win aplicația http://1win5015.ru .
DavidNaing
| #
This online casino hit the mark with Tiger Fortune. fortune tiger bet
where can i get generic plan b
| #
Drugs information leaflet. Drug Class.
where can i get generic plan b
All news about meds. Read here.
Trefuka
| #
Заказать диплом любого университета. Приобретение документа о высшем образовании через надежную компанию дарит много преимуществ. Данное решение позволяет сэкономить время и существенные финансовые средства. dimarecruitment.co.uk/employer/eonline-diploma
Protezirovanie zybov_jepl
| #
протез зуба стоимость протез зуба стоимость .
Protezirovanie zybov_mlMn
| #
сколько стоит сделать протезирование зубов http://www.protezirovanie-zubov3.ru/ .
inernetkrdCah
| #
подключение интернета по адресу
domashij-internet-krasnodar001.ru
узнать провайдера по адресу краснодар
Cazravt
| #
Привет!
Мы изготавливаем дипломы любой профессии по приятным тарифам. Цена может зависеть от выбранной специальности, года получения и образовательного учреждения: rdiploman.com/
Allergic reactions symptom discussion
| #
Allergic reactions allergic substances
mostbet_vsmt
| #
поддержка мостбет http://mostbet6033.ru .
DavidNaing
| #
I spent hours playing Tiger Fortune last night – no regrets! tiger fortune
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние достижения.
Наша продукция:
Керамическая мишень Ta2O5
Cameron4073
| #
http://toyota-porte.ru/forums/index.php?autocom=gallery&req=si&img=3232
buying cheap minipress pill
| #
buy cheap minipress without dr prescription
Dnrtjzs
| #
Мы предлагаем дипломы любой профессии по доступным тарифам. Мы можем предложить документы техникумов, которые находятся на территории всей РФ. Дипломы и аттестаты выпускаются на бумаге высшего качества. Это позволяет делать настоящие дипломы, не отличимые от оригиналов. minecraftcommand.science/forum/general/topics/d101dc5c-7658-4a04-a4f8-e0f9f3d7dddb
Willieavamy
| #
Online casinos should test—try me! vipzino
Protezirovanie zybov_ntEa
| #
протезирование зуба цена протезирование зуба цена .
Willieavamy
| #
The slot sounds online sing—fun blast! tome of madness
Anak Lonte
| #
Anak Lonte
A Brief History of Elemental Analysis » Artemis Analytical
doce
| #
O doce oferece uma excelente oportunidade para quem deseja
começar sua experiência no cassino online com um bônus de 100$ para novos jogadores!
Ao se registrar no site, você garante esse bônus exclusivo
que pode ser utilizado em diversos jogos de cassino, como slots, roleta e poker.
Esse é o momento perfeito para explorar o mundo das apostas com um saldo extra, aproveitando ao máximo suas apostas sem precisar investir um grande
valor logo de início. Não perca essa oportunidade e cadastre-se já!
RobertRiz
| #
Только 2 знакам Зодиака невероятно повезет во второй половине апреля
https://x.com/Fariz418740/status/1911625378860278161
stomatologiya Mitino_uuKi
| #
эмблема стоматологии эмблема стоматологии .
DavidNaing
| #
Tiger Fortune has the best vibe of any slot. fortune tiger demo
Jimmynum
| #
После отпуска в Сочи я чувствую себя обновленным и счастливым. знакомства с местными девушками были настоящим сюрпризом, и я рад тому, как легко мы находили общий язык. Этот отдых стал ещё более ценным благодаря общению с ними, и я намерен поддерживать связь https://t.me/sochinightt
can you get generic levaquin pills
| #
Meds prescribing information. What side effects?
can you get generic levaquin pills
Actual news about medicines. Read now.
DavidNaing
| #
Tiger Fortune has the best vibe of any slot. fortune tiger bet
DavidNaing
| #
Tiger Fortune’s wins feel so satisfying. fortune tiger bet
DavidNaing
| #
The tiger roars in Tiger Fortune make it so fun! fortune tiger bet
Sazroky
| #
Заказать диплом об образовании!
Купить диплом ВУЗа по доступной стоимости возможно, обратившись к надежной специализированной компании. Заказать диплом о высшем образовании: diplomg-kurerom.ru/visshee-obrazovanie-kupit-diplom-s-zaneseniem-13
jangan asik makan brand webku la kontol
| #
jangan asik makan brand webku la kontol
A Brief History of Elemental Analysis » Artemis Analytical
DavidNaing
| #
Tiger Fortune’s rewards are always a thrill. tigrinho demo
buy cheap rulide without insurance
| #
buying generic rulide
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Плоская мишень из германия 5N, 6N
inernetkrdCah
| #
провайдеры интернета в краснодаре по адресу
domashij-internet-krasnodar002.ru
интернет провайдеры по адресу
Marisat#nonick[KywcuwypipyfdiBI,2,5]
| #
¡Hola competidores de casino
Un bono de bienvenida atractivo es comГєn en los casinos sin licencia. mejores casinos online sin licencia Puedes recibir giros gratis, fondos extra y otras ventajas. Lee siempre las condiciones del bono para evitar sorpresas.
Los mejores casinos online sin licencia ofrecen promociones personalizadas y programas VIP. Estas ventajas no siempre estГЎn disponibles en plataformas reguladas. Explora las opciones y encuentra la que se adapte a tu estilo.
Consulta el enlace para más información – п»їhttps://casinossinlicenciaenespana.guru/
¡Por muchos sonrisas!
1win_fioi
| #
1win kg 1win kg .
mostbet_akmt
| #
мостбет казино войти http://mostbet6033.ru/ .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
поставляемая продукция:
Кольцо РёР· драгоценных металлов платиновое 5С…2С…1.5 РјРј РџР»Р95-5 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
Mazruej
| #
Мы изготавливаем дипломы любой профессии по выгодным ценам. Стараемся поддерживать для клиентов адекватную политику цен. Важно, чтобы документы были доступны для большинства наших граждан.
Заказ документа, подтверждающего окончание ВУЗа, – это выгодное решение. Купить диплом любого ВУЗа: diplom-bez-problem.com/kupit-diplom-visshego-obrazovaniya-5/
how to buy generic zerit prices
| #
can i purchase cheap zerit online
lvbet
| #
Aposte no lvbet e Receba 100$ de Bônus ao Se Registrar!
allwin
| #
Aposte com 100$ de Bônus no allwin
– Cadastre-se e Aproveite!
Lazrmyi
| #
Заказать диплом об образовании!
Мы готовы предложить дипломы любой профессии по приятным ценам— dip-lom-rus.ru/kupit-diplom-o-visshem-obrazovanii-v-moskve-6/
zoloft rcp
| #
Medicines information for patients. Generic Name.
zoloft rcp
Some what you want to know about medication. Read information here.
tvbet
| #
tvbet: Comece com 100$ de Bônus ao Criar Sua
Conta!
pgwin
| #
Seu Registro no pgwin
Vale 100$ de Bônus Imediato!
Sazrzmz
| #
Заказать документ университета вы сможете у нас в Москве. Мы предлагаем документы об окончании любых ВУЗов Российской Федерации. Вы получите диплом по любой специальности, любого года выпуска, в том числе документы СССР. Даем 100% гарантию, что в случае проверки документа работодателем, подозрений не возникнет. asxdiploman.com/kupite-diplom-s-zaneseniem-v-reestr-rossii-bistro/
Xazrhsj
| #
Заказать диплом университета!
Мы можем предложить дипломы психологов, юристов, экономистов и любых других профессий по невысоким ценам. Вы заказываете документ в надежной и проверенной временем компании. : bodybuilding.net/members/worksale.html
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их качество. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы и предоставлять решения под требования вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
Наши товары:
РџРѕРєРѕРІРєР° висмутовая Darcet alloy РџРѕРєРѕРІРєР° висмутовая Darcet alloy представляет СЃРѕР±РѕР№ уникальный металлический сплав, обладающий высокой прочностью Рё устойчивостью Рє РєРѕСЂСЂРѕР·РёРё. Ртот материал идеально РїРѕРґС…РѕРґРёС‚ для применения РІ различных областях, включая электронику Рё ювелирное дело. Высокое содержание висмутовой составляющей придает сплаву особые свойства, делающие его востребованным РЅР° рынке. Если вам нужна надежная Рё качественная РїРѕРєРѕРІРєР°, РЅРµ упустите шанс купить РџРѕРєРѕРІРєР° висмутовая Darcet alloy. Ртот сплав поможет обеспечить долговечность Рё эффективность ваших изделий, обеспечивая РїСЂРё этом конкурентное преимущество.
fuwin
| #
Aproveite a oferta exclusiva do fuwin para novos usuários e receba 100$ de
bônus ao se registrar! Este bônus de boas-vindas permite que
você experimente uma vasta gama de jogos de cassino online sem precisar gastar imediatamente.
Com o bônus de 100$, você poderá explorar jogos como
roleta, blackjack, caça-níqueis e muito mais, aumentando suas chances de
vitória desde o primeiro minuto. Não perca essa chance única de começar com um valor significativo –
cadastre-se agora!
Willieavamy
| #
I don’t get online hate—it’s cool! bet on red
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Ниобиевый тигель высокой чистоты
Lazrmwy
| #
Диплом университета России!
Без института сложно было продвинуться по карьере. Именно из-за этого решение о покупке диплома следует считать рациональным. Выгодно купить диплом института medvozerniy.flybb.ru/viewtopic.php?f=14&t=13955
stomatologiya Mitino_kcKi
| #
стоматология в митино стоматология в митино .
1win_egoi
| #
1win. https://1win7002.ru/ .
Willieavamy
| #
Online casinos should speed up cashouts—too slow! sugar rush
Eanrxlo
| #
Для удачного продвижения вверх по карьере понадобится наличие диплома ВУЗа. Заказать диплом института у сильной фирмы: diplomaj-v-tule.ru/kupit-diplom-v-moskve-tsena/
Diplomi_aoKt
| #
Заказать диплом любого университета!
Мы предлагаем документы институтов, которые находятся в любом регионе России.
diplomnie.com/kupit-diplom-s-reestrom-bezopasno-i-bistro/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наша продукция:
Плоская мишень для распыления свинца 4N
DavidNaing
| #
Tiger Fortune’s payouts keep me smiling. fortune tiger demo
Kennethded
| #
Металлические сувениры становятся всё более популярными в рекламной индустрии благодаря своей долговечности, эстетической привлекательности и возможностям персонализации. Использование современных технологий, таких как лазерная гравировка и 3D-печать, позволяет создавать уникальные изделия, подчёркивающие статус и надёжность бренда. Кроме того, металлические сувениры являются экологичным и экономически эффективным решением для компаний, стремящихся к устойчивому развитию. Подробнее об инновациях в производстве металлических сувениров можно узнать по ссылке: https://bvfy.ru/samie-raznoobraznie-novosti-kazhdij-den-s-razlichnix-ugolkov-zemnogo-shara-novosti-politiki-ekonomiki-sporta-a-takzhe-dostizheniya-texnologij-stroitelstva-i-drugie-vazhnie-sobitiya/
can i purchase cheap inderal without dr prescription
| #
cost of generic inderal for sale
mostbet_bvEl
| #
mostbet kg mostbet kg .
1win_qbst
| #
1win на телефон 1win на телефон .
cinitapride and pantoprazole uses
| #
Medicines information sheet. What side effects can this medication cause?
cinitapride and pantoprazole uses
Everything information about drugs. Read now.
inernetkrdCah
| #
провайдеры по адресу
domashij-internet-krasnodar003.ru
провайдеры интернета в краснодаре по адресу
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
Наши товары:
Кольцо РёР· драгоценных металлов золотое 150С…4С…1 РјРј ЗлСрМ75-12.5 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
1win_tloi
| #
скачать 1win с официального сайта https://www.1win7002.ru .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Плоская мишень из фтористого магния
1win_sdor
| #
1win md 1win md .
BAPAK KAU GIGOLO
| #
BAPAK KAU GIGOLO
A Brief History of Elemental Analysis » Artemis Analytical
can i order cheap geodon without insurance
| #
cost geodon pill
https___ca_ogOr
| #
Хотите найти что-то особенное?, обязательно загляните на. Мы предлагаем разнообразные товары. Убедитесь сами, высшего качества. Следите за нами. Индивидуальный подход – это наша цель. Покупайте легко и быстро.
купити військові штани мультикам https://camogear.com.ua/ .
CoreyMew
| #
The game selection at ElonBet is insane—hundreds of slots and live dealers to choose from! elonbet casino
CoreyMew
| #
ElonBet’s interface is clean and doesn’t overwhelm you with ads.
elonbet casino
Kennethded
| #
Металлические сувениры становятся всё более популярными в рекламной индустрии благодаря своей долговечности, эстетической привлекательности и возможностям персонализации. Использование современных технологий, таких как лазерная гравировка и 3D-печать, позволяет создавать уникальные изделия, подчёркивающие статус и надёжность бренда. Кроме того, металлические сувениры являются экологичным и экономически эффективным решением для компаний, стремящихся к устойчивому развитию. Подробнее об инновациях в производстве металлических сувениров можно узнать по ссылке: https://bvfy.ru/samie-raznoobraznie-novosti-kazhdij-den-s-razlichnix-ugolkov-zemnogo-shara-novosti-politiki-ekonomiki-sporta-a-takzhe-dostizheniya-texnologij-stroitelstva-i-drugie-vazhnie-sobitiya/
DavidNaing
| #
The payouts in Tiger Fortune are pretty impressive. fortune tiger
CoreyMew
| #
The signup bonus at ElonBet was bigger than I expected!
elonbet casino
1win_hlor
| #
1win aplicația https://www.1win5016.ru .
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их происхождение. Опытная поддержка – наш стандарт – мы на связи, чтобы разрешать ваши вопросы а также адаптировать решения под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
Лента магниевая 3.5662 – DIN 1729 Part 2 РљСЂСѓРі магниевый 3.5662 – DIN 1729 Part 2 – это высококачественный металл, обладающий отличными механическими свойствами. РћРЅ идеально РїРѕРґС…РѕРґРёС‚ для использования РІ различных отраслях, таких как авиация, автомобилестроение Рё производство оборудования. Магниевые РєСЂСѓРіРё обеспечивают легкость Рё прочность, что значительно облегчает РёС… применение. Если РІС‹ хотите улучшить СЃРІРѕРё проекты Рё повысить надёжность, вам стоит подумать Рѕ РїРѕРєСѓРїРєРµ этого материала. РќРµ упустите шанс купить РљСЂСѓРі магниевый 3.5662 – DIN 1729 Part 2 Рё обеспечить высокие стандарты качества РІ своей работе.
tv bet
| #
Seu Registro no tv bet Vale
100$ de Bônus Imediato!
cost of geodon for sale
| #
Pills information sheet. Drug Class.
cost of geodon for sale
Some about pills. Read information here.
1win_ztor
| #
cazino md https://www.1win5016.ru .
DavidNaing
| #
This casino’s Tiger Fortune slot is a winner. fortune tiger bet
CoreyMew
| #
The variety of table games at ElonBet keeps things fresh every time.
elonbet casino
Kennethded
| #
Металлические сувениры становятся всё более популярными в рекламной индустрии благодаря своей долговечности, эстетической привлекательности и возможностям персонализации. Использование современных технологий, таких как лазерная гравировка и 3D-печать, позволяет создавать уникальные изделия, подчёркивающие статус и надёжность бренда. Кроме того, металлические сувениры являются экологичным и экономически эффективным решением для компаний, стремящихся к устойчивому развитию. Подробнее об инновациях в производстве металлических сувениров можно узнать по ссылке: https://bvfy.ru/samie-raznoobraznie-novosti-kazhdij-den-s-razlichnix-ugolkov-zemnogo-shara-novosti-politiki-ekonomiki-sporta-a-takzhe-dostizheniya-texnologij-stroitelstva-i-drugie-vazhnie-sobitiya/
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
поставляемая продукция:
Кольцо РёР· драгоценных металлов платиновое 40С…30С…5 РјРј ПлРд90-10 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
memek goreng
| #
memek goreng
A Brief History of Elemental Analysis » Artemis Analytical
Jamesdusty
| #
Цветы пришли точно в указанный час – это очень ценно!
гипсофилы цена букета
situs haram tukang phising
| #
situs haram tukang phising
A Brief History of Elemental Analysis » Artemis Analytical
can i get generic levonorgestrel without a prescription
| #
Medicament information leaflet. Short-Term Effects.
can i get generic levonorgestrel without a prescription
Some what you want to know about meds. Read here.
SKANDAL BOKEP JILBAB JEPANG
| #
SKANDAL BOKEP JILBAB JEPANG
A Brief History of Elemental Analysis » Artemis Analytical
Mazrtrk
| #
Приобрести диплом института!
Мы оказываем услуги по изготовлению и продаже документов об окончании любых университетов Российской Федерации. Документы производятся на подлинных бланках государственного образца. area54marketplace.com/kupit-diplom-gosudarstvennogo-obrazca-6.html
TyroneWat
| #
Корпоративные сувениры и мерч становятся важным инструментом укрепления связей с сотрудниками и партнёрами. Современные тенденции включают экологичность, практичность, персонализацию, использование технологичных гаджетов, подарки-впечатления и поддержку локальных производителей. Подробнее о текущих трендах в корпоративных подарках можно узнать по ссылке: https://stol-kirov.ru/04/04/2025/243205/korporativnye-suveniry-i-merch.html
dj bet
| #
O dj bet oferece uma excelente
oportunidade para quem deseja começar sua experiência no cassino online com um bônus de 100$ para
novos jogadores! Ao se registrar no site, você garante esse
bônus exclusivo que pode ser utilizado em diversos jogos de cassino, como slots, roleta e poker.
Esse é o momento perfeito para explorar o mundo das apostas com um saldo extra,
aproveitando ao máximo suas apostas sem precisar investir um
grande valor logo de início. Não perca essa oportunidade e cadastre-se já!
CoreyMew
| #
ElonBet’s game library keeps growing—never run out of options.
elonbet casino
natyazhnye_rvoi
| #
Идеальные натяжные потолки в Днепре, где ваша фантазия воплощается в жизнь, добавьте шарм вашему пространству, решение, которое преобразит ваш дом.
Элегантные натяжные потолки для вашего дома, качественные материалы, которые создадут уют и гармонию, сделайте шаг к преображению.
Доступные натяжные потолки в Днепре, по доступным ценам, оставьте позади скучные потолки, закажите бесплатную консультацию.
Натяжные потолки, созданные с любовью, ваше пространство, ваша фантазия, выберите свой идеальный потолок, с нами это легко.
Эстетика и функциональность натяжных потолков, отечественного производства, на любой вкус и цвет, у нас широкий ассортимент.
Натяжные потолки: преимущества и возможности, от natyazhnye-potolki-dnepr.biz.ua, отличная шумоизоляция, позаботьтесь о своем жилье.
Эстетические решения для вашего потолка, всё на natyazhnye-potolki-dnepr.biz.ua, обратите внимание на детали, мы поможем вам с выбором.
Современные технологии натяжных потолков, с нами легко и быстро, доступные решения для каждого, обращайтесь к профессионалам.
Натяжные потолки от профессионалов, с хорошей репутацией, поддержка и забота о клиенте, придайте своему интерьеру новое дыхание.
Где купить натяжные потолки в Днепре?, на natyazhnye-potolki-dnepr.biz.ua, у нас отличный сервис и выбор, закажите натяжные потолки и наслаждайтесь результатом.
Преобразите свое пространство с натяжными потолками, с качеством, проверенным временем, мы работаем для вас, начните свой проект с нами.
Идеальные потолки для вашего дома, от наших профессионалов, выбор, который вдохновляет, сделайте выбор осознанно.
Опытные специалисты по натяжным потолкам, от natyazhnye-potolki-dnepr.biz.ua, потолок, о котором вы всегда мечтали, обновите свой дом с нами.
Натяжные потолки для ваших идей, от natyazhnye-potolki-dnepr.biz.ua, где ваши идеи становятся реальностью, приобретайте лучшее.
Ваш потолок — ваша гордость, где мечты становятся реальностью, поддержка профессионалов на каждом этапе, закажите прямо сейчас.
Дайте своему потолку вторую жизнь, от natyazhnye-potolki-dnepr.biz.ua, всё для вашего комфорта, сделайте правильный выбор.
натяжные потолки цена днепр натяжные потолки цена днепр .
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наша продукция:
Медный тройник РїРѕРґ пайку СЃ направленным отводом 6С…4С…0.6 РјРј 5.8С…7.8 РјРј твердая пайка Рњ1Р Рњ ГОСТ 32590-2013 Купить качественные медные тройники РїРѕРґ пайку для надежных Рё долговечных соединений РІ трубопроводных системах. Разнообразие размеров Рё конфигураций, высокая теплопроводность Рё устойчивость Рє РєРѕСЂСЂРѕР·РёРё. Простая установка Рё надежность соединения. Рдеальное решение для различных проектов. Редметсплав.СЂС„
Kennethded
| #
Металлические сувениры становятся всё более популярными в рекламной индустрии благодаря своей долговечности, эстетической привлекательности и возможностям персонализации. Использование современных технологий, таких как лазерная гравировка и 3D-печать, позволяет создавать уникальные изделия, подчёркивающие статус и надёжность бренда. Кроме того, металлические сувениры являются экологичным и экономически эффективным решением для компаний, стремящихся к устойчивому развитию. Подробнее об инновациях в производстве металлических сувениров можно узнать по ссылке: https://bvfy.ru/samie-raznoobraznie-novosti-kazhdij-den-s-razlichnix-ugolkov-zemnogo-shara-novosti-politiki-ekonomiki-sporta-a-takzhe-dostizheniya-texnologij-stroitelstva-i-drugie-vazhnie-sobitiya/
1win_bjOl
| #
1вин вход https://1win7001.ru/ .
CoreyMew
| #
The crypto bonuses at ElonBet are a nice perk for users. elonbet casino
doxycycline liquid refrigerate
| #
order doxycycline capsules online
DavidNaing
| #
The free spins in Tiger Fortune are awesome! fortune tiger
Diplomi_ehKt
| #
Купить диплом ВУЗа!
Мы предлагаем документы любых учебных заведений, которые расположены на территории всей РФ.
diplomt-nsk.ru/priobretite-diplom-s-vneseniem-v-reestr-bistro-i-bezopasno/
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их качество. Превосходное обслуживание – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы а также адаптировать решения под требования вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
поставляемая продукция:
Проволока магниевая Р›2 – РўРЈ 1714-002-00545484-99 Полоса магниевая Р›2 – РўРЈ 1714-002-00545484-99 является незаменимым материалом для различных производственных процессов. РћРЅР° обладает высокой прочностью Рё морозоустойчивостью, что делает ее идеальной для применения РІ HVAC системах Рё строительстве. Данная полоса применяется также РІ электротехнической отрасли благодаря СЃРІРѕРёРј проводниковым свойствам. Если РІС‹ ищете качественный Рё надежный материал, вам стоит купить Полоса магниевая Р›2 – РўРЈ 1714-002-00545484-99, чтобы обеспечить долговечность Рё эффективность ваших проектов. Наша продукция отвечает всем необходимым стандартам Рё требованиям.
Where to Buy Kamagra Safely
| #
The effects of https://hikvisiondb.webcam/wiki/Kamagra_54A were quick, but I got a headache.
mostbet_fxMn
| #
mostbet промокод mostbet5001.ru .
Diplomi_rzSn
| #
можно ли купить высшее образование можно ли купить высшее образование .
Apcalis vs Kamagra
| #
https://bgr.sgk.temporary.site/index.php/Kamagra_45F Oral Jelly’s convenient, but I’m skeptical about quality.
How to Use Kamagra Oral Jelly
| #
Kamagra helped my confidence, but I’d talk https://fakenews.win/wiki/Kamagra_31s a doc first.
Sazrwem
| #
купить аттестат за 9 классов екатеринбург
1win_yqpa
| #
1 вин https://1win7014.ru/ .
inernetsamaraCah
| #
провайдеры интернета в самаре по адресу проверить
domashij-internet-samara001.ru
узнать провайдера по адресу самара
CoreyMew
| #
ElonBet’s mobile version is just as good as the desktop site. elonbet casino
1win_reKa
| #
1win betting https://1win16.com.ng/ .
DarrenNib
| #
Для фанатов скинов CS 2 сделали обзор на сайты продать скины за деньги. В статье представлен подробный и актуальный рейтинг сайтов, где можно продать скины CS2 и CS:GO. Анализ более чем 10 источников и отобрал только те платформы, которые пользуются хорошей репутацией в сообществе.
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
поставляемая продукция:
Труба из драгоценных металлов серебряная 700х9х1 мм СрМ87.5 ТУ Гарантированное качество и изысканный дизайн – купите драгоценные трубы для ювелирных украшений, машиностроения и архитектуры. Широкий выбор и высокое качество! Доставка по России.
can i get generic flagyl price
| #
where buy generic flagyl for sale
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их качество. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы по мере того как находить ответы под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
Круг магниевый 4635 Фольга магниевая 4635 – это современный материал, идеально подходящий для различных целей в строительстве и промышленности. Она обладает высокой прочностью, стойкостью к коррозии и легко поддается формованию. Основное применение фольги заключается в тепло- и звукоизоляции, а также в защите от elektromagnitnyh polej. Приобретая этот товар, you гарантированно получаете долговечное решение для своих проектов. Не упустите возможность купить Фольга магниевая 4635 и обеспечьте надежность и эффективность в работе.
CoreyMew
| #
Just hit a jackpot on one of ElonBet’s progressive slots. Still in shock!
elonbet casino
20bet
| #
O 20bet oferece uma
excelente oportunidade para quem deseja começar sua experiência no cassino online com
um bônus de 100$ para novos jogadores! Ao se registrar no site, você garante esse bônus exclusivo que pode ser
utilizado em diversos jogos de cassino, como slots, roleta
e poker. Esse é o momento perfeito para explorar o mundo das apostas com um saldo
extra, aproveitando ao máximo suas apostas sem precisar investir um
grande valor logo de início. Não perca essa oportunidade e cadastre-se já!
DavidNaing
| #
I can’t stop playing Tiger Fortune – it’s too good! fortune tiger demo
how can i get generic zoloft price
| #
Medicines information leaflet. Cautions.
how can i get generic zoloft price
Everything information about drugs. Read information now.
mostbet_djEl
| #
mostbet https://mostbet6036.ru/ .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Керамическая мишень HfO2
CoreyMew
| #
Love the weekly reload bonuses at ElonBet. Keeps me playing! elonbet casino
CoreyMew
| #
Tried the demo mode at ElonBet before betting—great way to test games.
elonbet casino
CoreyMew
| #
ElonBet Casino’s community is growing, and it’s fun to be part of it.
elonbet casino
GeorgeTom
| #
Source:
– s-11.ru
batbet
| #
No batbet, novos usuários podem aproveitar um bônus de 100$ ao
se registrar no site! Isso significa mais chances de ganhar e explorar uma grande variedade de jogos de cassino, desde slots emocionantes até
clássicos como roleta e blackjack. Com o bônus de boas-vindas, você começa com
um saldo extra, o que aumenta suas chances de sucesso.
Cadastre-se agora e use os 100$ de bônus para experimentar
seus jogos favoritos com mais facilidade. Aproveite a oferta e comece sua aventura no
cassino agora mesmo!
amoxicillin 500mg doseage
| #
Meds information for patients. Generic Name.
amoxicillin 500mg doseage
Actual news about pills. Read here.
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы по мере того как предоставлять решения под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Лист ванадиевый Р’РЅРњ99.5 – РўРЈ 1761-037-2587982-2002 Лист ванадиевый Р’РЅРњ99.5 – РўРЈ 1761-037-2587982-2002 представляет СЃРѕР±РѕР№ высококачественный металл, обладающий превосходными показателями прочности Рё упругости. РћРЅ используется РІ различных отраслях, включая аэрокосмическую Рё автомобильную. Наш лист гарантирует долговечность Рё надежность РїСЂРё эксплуатации. Если РІС‹ ищете идеальный материал для СЃРІРѕРёС… проектов, то купить Лист ванадиевый Р’РЅРњ99.5 – РўРЈ 1761-037-2587982-2002 – правильное решение для вас. Убедитесь РІ качестве Рё выгодной цене РЅР° этот товар, который станет неотъемлемой частью вашей работы.
DavidNaing
| #
Tiger Fortune’s jungle theme is so immersive. fortune tiger bet
blaze
| #
Registre-se e Receba 100$ de Bônus no blaze para Apostar!
mostbet_kgMn
| #
мостбет промокод https://mostbet5001.ru .
mostbet_uxmt
| #
мостбет авиатор http://www.mostbet6033.ru .
1win_wppa
| #
1win http://1win7014.ru/ .
Prodamys promokod_lqEt
| #
продамус промокод скидка продамус промокод скидка .
Prodvijenie kriptovaluta_lsEn
| #
управление репутацией криптовалюты http://www.prodvizhenie-kriptovalyuta.ru .
Prodvijenie kriptovaluta_kdpt
| #
продвижение крипты продвижение крипты .
Adolphguall
| #
https://www.banquedesterritoires.fr/nos-adolescents-sont-ceux-qui-ont-le-plus-decroche-de-lecole-estime-le-president-de-ville-et
DavidNaing
| #
Tiger Fortune’s theme is wild and totally unique. fortune tiger
Thomaspoext
| #
Пионы распустились на второй день – невероятно красиво!
заказ цветов томск
BAPAKKAU DIPERKOSA KUCING
| #
BAPAKKAU DIPERKOSA KUCING
A Brief History of Elemental Analysis » Artemis Analytical
CoreyMew
| #
The design of ElonBet Casino feels like stepping into a sci-fi movie. So cool!
elonbet casino
1win_xrOl
| #
1win com https://1win7001.ru/ .
TUKANG SOBUR KAU LONTE
| #
TUKANG SOBUR KAU LONTE
A Brief History of Elemental Analysis » Artemis Analytical
1win_vvKa
| #
1win website 1win16.com.ng .
Willieavamy
| #
I lost big online—new start! plinko
Willieavamy
| #
Online slots grab me—those looks! dragon tiger
Willieavamy
| #
Online casinos should free—test run! minas juego
HebohnWerse
| #
Go to the site https://betwave.ro/en where you will find a list of all the best online casinos, as well as useful and complete information about each of them – how to play, what bonuses there are, how to withdraw money, which casinos have a minimum deposit, registration steps and advantages of each of them, as well as what are the biggest bonuses in online casinos. More details on the site.
Eanrdoh
| #
Для быстрого продвижения по карьерной лестнице потребуется наличие диплома о высшем образовании. Купить диплом о высшем образовании у надежной организации: diplomg-kurerom.ru/kupit-krasnij-attestat-bez-lishnix-xlopot/
Xazrbww
| #
Купить диплом о высшем образовании!
Мы изготавливаем дипломы любых профессий по приятным ценам. Вы заказываете диплом в надежной и проверенной временем компании. : domkodeks.ru/question/kupit-diplom-ofitsialno-bystro-i-bezopasno
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Керамическая мишень HfO2
1win_pepa
| #
1win,com 1win7014.ru .
inernetsamaraCah
| #
интернет провайдеры самара по адресу
domashij-internet-samara002.ru
подключение интернета по адресу
mostbet_jiMn
| #
motsbet http://mostbet5001.ru .
sklad_gkOt
| #
снять склад для хранения личных вещей подмосковье снять склад для хранения личных вещей подмосковье .
RichardMek
| #
Лучшие сервера л2 ИЛ https://l2-top.com.ua/
diltiazem 60 mg generic
| #
diltiazem and liver damage
Sazrlwg
| #
купить диплом железнодорожного института
Lazryyh
| #
Диплом ВУЗа России!
Без ВУЗа достаточно сложно было продвинуться вверх по карьере. Поэтому решение о покупке диплома стоит считать целесообразным. Приобрести диплом о высшем образовании sssr.flybb.ru/viewtopic.php?f=2&t=766&sid=214bfdc09632deea9aa0a40eaff9a065
Jariortza
| #
Где купить диплом по актуальной специальности?
Наши специалисты предлагаютбыстро и выгодно купить диплом, который выполнен на оригинальном бланке и заверен печатями, водяными знаками, подписями официальных лиц. Наш диплом пройдет любые проверки, даже при использовании профессиональных приборов. Решайте свои задачи быстро и просто с нашими дипломами. Заказать диплом университета! sahpes.onlinebbs.ru/viewtopic.php?f=3&t=1411
mostbet_iemt
| #
мостбет казино войти mostbet6033.ru .
zithromax prescription cost
| #
Drugs information for patients. Short-Term Effects.
zithromax prescription cost
Actual news about pills. Read here.
Mazrhez
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и прочих профессий по приятным тарифам. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Для нас важно, чтобы дипломы были доступными для большого количества граждан.
Покупка диплома, подтверждающего окончание университета, – это разумное решение. Приобрести диплом университета: diplompro.ru/kupit-diplom-sredne-texnicheskoe/
CoreyMew
| #
ElonBet’s customer support speaks multiple languages—big help!
elonbet casino
CoreyMew
| #
The variety of table games at ElonBet keeps things fresh every time.
elonbet casino
CoreyMew
| #
The live dealer interactions at ElonBet are so engaging.
elonbet casino
1win_gxor
| #
pariuri sportive moldova http://1win5016.ru .
CoreyMew
| #
I recently tried ElonBet Casino, and the interface is super sleek! Love the futuristic vibe.
elonbet casino
Lazrstt
| #
Быстро приобрести диплом ВУЗа!
Мы изготавливаем дипломы любой профессии по доступным тарифам— diplom-zentr.com/kupit-diplom-v-krasnoyarske-20/
Prodamys promokod_aeEt
| #
промокод prodamus https://promokod-prdms.ru .
Matthewpaype
| #
Читать далее Манго Офис кабинет
Prodvijenie kriptovaluta_zwEn
| #
seo криптопроекта http://www.prodvizhenie-kriptovalyuta.ru .
GeorgeRot
| #
Бизнес издание Business Review – советы для HR, новости маркетинга.
ThomasSaild
| #
читать Mango-Office вход
Prodvijenie kriptovaluta_qopt
| #
улучшение репутации криптовалюты улучшение репутации криптовалюты .
1win_ubKa
| #
1win bet 1win16.com.ng .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
поставляемая продукция:
Заготовка из конструкционной стали листовая 1 мм 40ХН2МА ОСТ 3-1686-90 Выбирайте конструкционные заготовки высокого качества для вашего проекта на Редметсплав.рф. Наш ассортимент включает прочные и долговечные материалы, идеально подходящие для различных областей применения, включая строительство и машиностроение.
RogerGloro
| #
Значки из металла https://www.skalpil.ru/novosti-mediciny/other/6623-metallicheskie-znachki-populyarnyy-element-korporativnoy-i-individualnoy-identifikacii.html это эффективный инструмент идентификации в бизнесе и медицине. Они подчёркивают статус, создают узнаваемый образ и способствуют формированию корпоративной культуры.
umbet
| #
Ganhe 100$ de Bônus no umbet
ao Criar Sua Conta!
Willieavamy
| #
Online casinos should guide—too steep! fortune ox
BrianTah
| #
ВГУ им. П. М. Машерова https://vsu.by официальный сайт, факультеты, направления подготовки, приёмная кампания. Узнайте о поступлении, обучении и возможностях для студентов.
Willieavamy
| #
Online casinos should clarify odds—please! plinko
Willieavamy
| #
Online casinos should teach rules better—too hard! fortune mouse
Willieavamy
| #
The slot thrill online flies—pulse high! craps game
cost chlorpromazine pill
| #
Medication information leaflet. Generic Name.
cost chlorpromazine pill
Some about medication. Get information here.
mostbet_xnmt
| #
мостбет казино войти https://mostbet6033.ru/ .
CoreyMew
| #
The live dealer interactions at ElonBet are so engaging.
elonbet casino
winbra
| #
Aproveite a oferta exclusiva do winbra para novos usuários e receba 100$ de bônus ao se registrar!
Este bônus de boas-vindas permite que você experimente uma vasta gama de jogos
de cassino online sem precisar gastar imediatamente.
Com o bônus de 100$, você poderá explorar jogos como
roleta, blackjack, caça-níqueis e muito mais, aumentando suas chances de vitória desde o primeiro minuto.
Não perca essa chance única de começar com um
valor significativo – cadastre-se agora!
Willieavamy
| #
Online casino tournaments are intense—anyone else hooked? F7 Casino
CoreyMew
| #
ElonBet’s game library keeps growing—never run out of options.
elonbet casino
buy cheap zyvox without rx
| #
can you get generic zyvox price
Willieavamy
| #
The live help online rocks—key boost! F7 Casino
Roberthew
| #
посмотреть на этом сайте Манго Офис личный кабинет
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их соответствие стандартам. Превосходное обслуживание – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы и предоставлять решения под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
РџРѕРєРѕРІРєР° кобальтовая РРЎРћ 5832-6 – ГОСТ Р РРЎРћ 5832-6-2010 РџРѕРєРѕРІРєР° кобальтовая РРЎРћ 5832-6 – ГОСТ Р РРЎРћ 5832-6-2010 является высококачественным материалом, предназначенным для производства медицинских изделий Рё протезов. Ртот стандарт обеспечивает надежность Рё биосовместимость, что делает продукцию идеальной для применения РІ С…РёСЂСѓСЂРіРёРё Рё стоматологии. Наша кобальтовая РїРѕРєРѕРІРєР° отвечает самым высоким требованиям Рё стандартах качества. Купить РџРѕРєРѕРІРєР° кобальтовая РРЎРћ 5832-6 – ГОСТ Р РРЎРћ 5832-6-2010 можно РїРѕ привлекательной цене. Рспользуйте этот материал для создания долговечных Рё безопасных изделий, которые обеспечивают эффективность Рё надежность. РќРµ упустите возможность приобрести топовый РїСЂРѕРґСѓРєС‚ уже сегодня!
Sazromi
| #
купить аттестат образование
Yatavtswops
| #
На сайте https://rozatoday.ru/ представлено огромное количество ярких, запоминающихся композиций, которые созданы из свежих цветов: ослепительных роз, волнующих пионов, трепетных тюльпанов, строгих гвоздик. В компании работают только знающие, компетентные флористы, которые создают изысканные композиции в соответствии с предпочтениями клиента. В обязательном порядке учитываются и актуальные мировые тенденции. Можно приобрести как небольшой букетик, который подходит для романтического свидания, так и внушительных размеров для грандиозного праздника.
CoreyMew
| #
Anyone else loving the Tesla-themed slots at ElonBet? They’re so much fun!
elonbet casino
AdolphAwarl
| #
ссылка на сайт электрик томск
Prodvijenie kriptovaluta_ljEn
| #
seo продвижение телеграм http://prodvizhenie-kriptovalyuta.ru .
order cheap elavil without insurance
| #
Medication information sheet. What side effects can this medication cause?
order cheap elavil without insurance
Actual trends of meds. Read information here.
CoreyMew
| #
ElonBet’s customer service is available 24/7. Super helpful!
elonbet casino
Arthurupsef
| #
1вин войти – 1win букмекерская, 1win win
CoreyMew
| #
ElonBet’s customer service is available 24/7. Super helpful!
elonbet casino
Willieavamy
| #
The table games online gleam—top notch! book of ra
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наша продукция:
Рнструментальная квадратная РїРѕРєРѕРІРєР° 145 РјРј 4РҐ5МФС ГОСТ 5950-2000 Познакомьтесь СЃ широким ассортиментом высокопрочных инструментов для обработки металла РІ категории инструментальной квадратной РїРѕРєРѕРІРєРё РѕС‚ Редметсплав. Выбирайте РёР· прочных Рё надежных материалов, подобранных специально для различных РІРёРґРѕРІ металлообработки. Гарантированное качество РѕС‚ ведущих производителей.
mostbet_ufSt
| #
скачат мостбет https://mostbet5002.ru/ .
Williamamish
| #
подробнее электрик томск
Richardjew
| #
лучшие онлайн казино – казино онлайн, демо слоты онлайн казино
onabet
| #
Aproveite a oferta exclusiva do onabet
para novos usuários e receba 100$ de bônus ao se registrar!
Este bônus de boas-vindas permite que você experimente uma vasta gama de
jogos de cassino online sem precisar gastar imediatamente.
Com o bônus de 100$, você poderá explorar jogos como roleta,
blackjack, caça-níqueis e muito mais, aumentando
suas chances de vitória desde o primeiro minuto.
Não perca essa chance única de começar com um valor significativo – cadastre-se agora!
KONTOL BAPAKKAU PECAH
| #
KONTOL BAPAKKAU PECAH
A Brief History of Elemental Analysis » Artemis Analytical
Willieavamy
| #
Online casinos should simplify rules—too much! Vipzino Casino
inernetsamaraCah
| #
какие провайдеры интернета есть по адресу самара
domashij-internet-samara003.ru
проверить провайдеров по адресу самара
StevenMep
| #
посуточная аренда автомобиля в сочи arenda avtomobilya sochi
Sazrcha
| #
Приобрести диплом академии !
Приобретение диплома университета России у нас является надежным процессом, потому что документ будет заноситься в государственный реестр. Приобрести диплом об образовании diplomass.com/vnesenie-diploma-v-reestr-po-nizkoj-tsene-2
AlbertGasse
| #
аренда автомобиля в адлере машина в аренду адлер на сутки недорого
Dnrthbg
| #
Мы предлагаем дипломы любых профессий по доступным тарифам. Мы можем предложить документы ВУЗов, расположенных на территории всей Российской Федерации. Документы делаются на бумаге самого высокого качества. Это дает возможности делать настоящие дипломы, которые невозможно отличить от оригинала. social.elpaso.world/read-blog/6068_kupit-diplom-s-zaneseniem-v-reestr-forum.html
DarrylMiz
| #
снять машину в аренду в крыму аренда авто крым
Diplomi_ufSn
| #
купить настоящий диплом о среднем специальном образовании купить настоящий диплом о среднем специальном образовании .
Charlesmug
| #
клининг спб недорого уборка частных домов
Sazrotc
| #
Мы изготавливаем дипломы любой профессии по разумным ценам.– poluchidiplom.com/kupite-diplom-visshego-obrazovaniya-s-zaneseniem-v-reestr/
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их происхождение. Опытная поддержка – наш стандарт – мы на связи, чтобы улаживать ваши вопросы и адаптировать решения под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
РџРѕРєРѕРІРєР° танталовая РўР’Р§ РџРѕРєРѕРІРєР° танталовая РўР’Р§ – это высококачественный металл, который отличает исключительная прочность Рё коррозионная стойкость. Рзготавливаемая РёР· чистого тантала, РѕРЅР° находит широкое применение РІ аэрокосмической, электронной Рё медицинской отраслях. Благодаря СЃРІРѕРёРј уникальным свойствам, танталовая РїРѕРєРѕРІРєР° идеально РїРѕРґС…РѕРґРёС‚ для создания компонентов, требующих надежности Рё долговечности.Если РІС‹ ищете надежный Рё высокопрочный материал для СЃРІРѕРёС… проектов, рекомендуем купить РџРѕРєРѕРІРєР° танталовая РўР’Р§. Рто инвестиция РІ качество Рё эффективность. Обеспечьте успешность СЃРІРѕРёС… разработок СЃ танталом уже сегодня!
Willieavamy
| #
The live dealers online rule—true vibe! plinko
Willieavamy
| #
I love the online twists—keep fresh! andar bahar
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние достижения.
Наши товары:
Гранулы магниевого фторида 4N
can i get cheap zithromax without insurance
| #
Medicines prescribing information. Short-Term Effects.
can i get cheap zithromax without insurance
Some about drug. Get here.
where to buy periactin
| #
can you buy periactin prices
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-in-Wales-Guide-for-Players-03-27
Prodvijenie kriptovaluta_nier
| #
продвижение криптовалют продвижение криптовалют .
where to get chlorpromazine price
| #
Medicines information sheet. Effects of Drug Abuse.
where to get chlorpromazine price
Actual news about medicine. Get here.
Willieavamy
| #
I wish online eased bets—low go! sugar rush
7kbet
| #
Faça seu Cadastro no 7kbet – https://7k-bet-br.com e Ganhe 100$
para Começar a Jogar!
Jariortqe
| #
Где приобрести диплом специалиста?
Наши специалисты предлагают выгодно заказать диплом, который выполнен на бланке ГОЗНАКа и заверен печатями, штампами, подписями. Документ пройдет лубую проверку, даже при использовании специальных приборов. Решите свои задачи быстро с нашими дипломами.
Купить диплом о высшем образовании kupit-diplom24.com/kupit-diplom-stroitelya-23/
cost of generic nootropil price
| #
buying cheap nootropil for sale
Willieavamy
| #
The mobile apps for online casinos need fixing! bet on red
bk bet
| #
Ganhe 100$ de Bônus ao Se Registrar no bk bet – https://bk-bet-br.com – Cadastre-se Agora!
Willieavamy
| #
Are online wins real?—not convinced! fortune tiger
Willieavamy
| #
Are online casino ratings legit?—not sure! plinko
Willieavamy
| #
Online casinos should cap—safe guard! tome of madness
Cazrkbz
| #
Купить диплом любого университета!
Мы предлагаем документы учебных заведений, которые находятся в любом регионе Российской Федерации. Дипломы и аттестаты печатаются на бумаге самого высшего качества: rosseiarf.flybb.ru/viewtopic.phpf=2&t=667
Willieavamy
| #
I don’t trust online RNG—feels off! F7 Casino
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-Free-Spins-No-Deposit-Offers1-03-27
Margie
| #
Howdy just wanted to give you a quick heads up. The text in your article seem to be running off the screen in Ie.
I’m not sure if this is a formattikng issue or something to do with internt browser compatibility but I thought I’d post to let you know.
The layout look great though! Hope you get the problem resolved soon. Thanks
Willieavamy
| #
Online casinos need tighter control—wild west! F7 Casino
Willieavamy
| #
I love the online promos—new spark! plinko
Prodvijenie kriptovaluta_qyer
| #
управление репутацией криптовалюты https://prodvizhenie-kriptovalyuta2.ru/ .
where buy bactrim without rx
| #
Drugs information sheet. Long-Term Effects.
where buy bactrim without rx
Some about drug. Read information now.
patomquish
| #
Arenda24.by – интернет-платформа для размещения объявлений по аренде оборудования и строительной техники. На нашем портале вы сможете комфортно работать, потому, как его настоящая команда профессионалов разработала. Ищете аренда строительная техника москва? Arenda24.by – портал, где вы найдете по аренде техники и любых инструментов объявления, и сможете организовать грузоперевозки. У нас есть объявления любых типов категорий. Разместите объявление уже сейчас, и оно в сжатые сроки своих клиентов найдет. С нами вы доступ к известной и качественной платформе получите.
Iarioruuj
| #
Купить диплом университета по выгодной цене вы сможете, обращаясь к проверенной специализированной компании. Приобрести документ университета можно в нашей компании в Москве. diplomg-cheboksary.ru/kupit-diplom-v-moskve-s-zaneseniem-v-reestr-bistro-17
inernetufaCah
| #
какие провайдеры интернета есть по адресу уфа
domashij-internet-ufa001.ru
интернет по адресу уфа
can i get generic plan b online
| #
Medicine information. What side effects can this medication cause?
can i get generic plan b online
Best what you want to know about pills. Get information now.
Uazroup
| #
Приветствую!
Для многих людей, купить диплом о высшем образовании – это необходимость, уникальный шанс получить выгодную работу. Впрочем для кого-то – это разумное желание не терять огромное количество времени на учебу в универе. С какой бы целью вам это не понадобилось, мы готовы помочь. Быстро, профессионально и по доступной цене изготовим диплом любого ВУЗа и любого года выпуска на настоящих бланках с реальными подписями и печатями.
Ключевая причина, почему люди покупают документ, – получить хорошую работу. Допустим, знания и опыт дают возможность кандидату устроиться на желаемую работу, но подтверждения квалификации не имеется. Если для работодателя важно наличие “корочек”, риск потерять хорошее место очень высокий.
Приобрести документ института вы сможете у нас в Москве. Мы предлагаем документы об окончании любых университетов РФ. Вы сможете получить необходимый диплом по любой специальности, включая документы СССР. Даем гарантию, что в случае проверки документов работодателями, каких-либо подозрений не возникнет.
Ситуаций, которые вынуждают приобрести диплом о среднем образовании много. Кому-то прямо сейчас нужна работа, в итоге нужно произвести впечатление на начальника на протяжении собеседования. Некоторые желают попасть в большую компанию, чтобы повысить свой статус в социуме и в будущем начать собственное дело. Чтобы не тратить время, а сразу начинать эффективную карьеру, применяя врожденные таланты и полученные знания, можно купить диплом в интернете. Вы станете полезным для общества, получите финансовую стабильность в максимально короткий срок- аттестат купить
1win_xeoi
| #
1 ван вин http://1win7002.ru/ .
where to buy imitrex without rx
| #
can i order cheap imitrex without a prescription
Pixunlipglice
| #
Система менеджмента управления качеством содержит критерии менеджмента и стандарты, по которым проводится последующая сертификация предприятия. Чтобы получить сертификат СМК, нужно обратиться в аккредитованный сертификационный центр. Сертификация систем менеджмента качества является документальным подтверждением того, что система менеджмента сертифицированной компании соответствует требованиям того или иного стандарта. Подробнее: https://ok.ru/group/70000034956977
Dnrtqmc
| #
Мы изготавливаем дипломы любой профессии по доступным ценам. Мы предлагаем документы техникумов, расположенных на территории всей России. Документы выпускаются на бумаге самого высокого качества. Это дает возможность делать государственные дипломы, не отличимые от оригинала. flughafen-jobs.com/companies/archive-diploma
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наши товары:
Медная твердая труба для кондиционеров 66.67С…1.25 РјРј Рњ1С„ EN 12735 Приобретите медные трубы для кондиционеров РѕС‚ Редметсплав.СЂС„. Надежные, долговечные Рё устойчивые Рє РєРѕСЂСЂРѕР·РёРё трубы СЃ высокой теплопроводностью Рё простым монтажом. Рдеальный выбор для кондиционирования РІ РґРѕРјРµ, офисе или производственном помещении.
Jariorbra
| #
Где заказать диплом специалиста?
Наша компания предлагаетвыгодно купить диплом, который выполнен на бланке ГОЗНАКа и заверен мокрыми печатями, штампами, подписями. Документ способен пройти любые проверки, даже при помощи специфических приборов. Достигайте свои цели быстро и просто с нашими дипломами. Заказать диплом университета! ripple-xrp-global-network.mn.co/posts/81282714
PeterSlump
| #
https://telegra.ph/Guide-to-Non-Gamstop-Casinos-and-Their-Benefits-03-27
Willieavamy
| #
Online poker is a blast—bluffing rocks! aviator
PeterSlump
| #
https://telegra.ph/Non-Gamstop-Casinos-Featuring-Lightning-Roulette-03-27
Prodvijenie kriptovaluta_zjer
| #
улучшение рейтинга криптобиржи http://prodvizhenie-kriptovalyuta2.ru .
leptozan
| #
leptozan is an innovative weight loss formula crafted to help you naturally sculpt the body you’ve always desired.
Willieavamy
| #
The live dealers online rule—true vibe! plinko
can you get cheap aceon pill
| #
can i purchase generic aceon price
prime biome
| #
primebiome fosters a thriving gut microbiome, which contributes to improved skin clarity and a naturally youthful glow.
Willieavamy
| #
Online gambling can soar—pull back! aviator
Jariorfar
| #
Где купить диплом по необходимой специальности?
Мы предлагаем быстро и выгодно купить диплом, который выполнен на оригинальной бумаге и заверен мокрыми печатями, водяными знаками, подписями. Диплом пройдет лубую проверку, даже с использованием специальных приборов. Решайте свои задачи быстро с нашими дипломами.
Приобрести диплом любого ВУЗа poluchidiplom.com/kupit-diplom-v-orle-8/
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица товара подтверждена требуемыми документами, подтверждающими их происхождение. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы а также предоставлять решения под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наши товары:
Полоса кобальтовая Stellite 100 Полоса кобальтовая Stellite 100 представляет собой высококачественный материал, используемый в производстве деталей, требующих прочности и устойчивости к коррозии. Она отлично подходит для применения в различных отраслях, включая машиностроение и авиастроение. Благодаря своим физико-химическим свойствам, эта полоса обеспечивает долговечность и надежность. Если вы ищете надежный материал для своих проектов, вам стоит купить Полоса кобальтовая Stellite 100. Закажите её прямо сейчас и обеспечьте себе высокое качество на долгие годы.
Cazrjls
| #
Где заказать диплом по актуальной специальности?
Мы изготавливаем дипломы психологов, юристов, экономистов и прочих профессий по выгодным ценам. Важно, чтобы дипломы были доступными для большого количества граждан. Купить диплом о высшем образовании diplomk-v-krasnodare.ru/kupit-diplom-s-zaneseniem-v-reestr-tsena-10/
Trefkxn
| #
Быстро купить диплом ВУЗа. Покупка официального диплома через надежную компанию дарит ряд достоинств. Данное решение позволяет сэкономить как длительное время, так и серьезные финансовые средства. pika.listbb.ru/posting.php?mode=post&f=9
can i order coumadin pills
| #
Medicines prescribing information. Drug Class.
can i order coumadin pills
All information about medicine. Read information now.
mostbet_fcmt
| #
мрстбет мрстбет .
PeterSlump
| #
https://telegra.ph/No-Deposit-Casino-Bonuses-Not-on-Gamstop1-03-27
PeterSlump
| #
https://telegra.ph/No-Deposit-Bonuses-at-Non-Gamstop-Casinos-Explained-03-27
sklad_ylOt
| #
теплые склады для хранения вещей теплые склады для хранения вещей .
metamask browser
| #
MetaMask Download is fast and reliable. It has enhanced my DeFi and NFT experiences significantly. Best crypto wallet out there!
bkbet
| #
Aproveite a oferta exclusiva do bkbet para novos usuários e receba 100$ de bônus ao se registrar!
Este bônus de boas-vindas permite que você experimente uma vasta gama de jogos de cassino online sem precisar gastar imediatamente.
Com o bônus de 100$, você poderá explorar jogos como roleta, blackjack, caça-níqueis e muito mais,
aumentando suas chances de vitória desde o primeiro minuto.
Não perca essa chance única de começar com um valor significativo –
cadastre-se agora!
inernetufaCah
| #
интернет по адресу
domashij-internet-ufa002.ru
какие провайдеры по адресу
TugotWot
| #
Детский туроператор Флагман организует школьные экскурсии и туры для детей по Санкт-Петербургу и Северо-Западу. Поездки с Флагманом всегда превращаются в увлекательные приключения. Собственный автопарк, профессиональные водители и контроль безопасности в пути гарантируют комфортные путешествия для детей. Профессиональные экскурсоводы адаптируют маршруты под детскую аудиторию, делая их познавательными и интересными. Подробнее на официальном сайте https://flagmanspb.ru/
1win_lioi
| #
1вин rossvya 1вин rossvya .
Dnrtuoa
| #
Мы можем предложить дипломы любых профессий по выгодным ценам. Мы готовы предложить документы ВУЗов, расположенных в любом регионе России. Дипломы и аттестаты выпускаются на “правильной” бумаге самого высшего качества. Это позволяет делать настоящие дипломы, не отличимые от оригинала. workompass.com/employer/archive-diploma
RakijamPreap
| #
Посетите B2BOX https://b2box.ru/ – это уникальное комплексное решение для создания упаковки всех видов и направлений. Мы делаем упаковку, которая продает! Упаковочные решения для ритейла, создание индивидуальной и SRP упаковки, услуги дизайнерского бюро, производство транспортной упаковки, собственное плоттерное производство, все виды печати – это и многое другое вы найдете, посетив наш сайт.
Willieavamy
| #
The payout waits online lag—push it! legacy of dead
Willieavamy
| #
I lost time online—fast gone! betonred
Posihipmeali
| #
Visit https://toptenscasinos.com/ and you will find a carefully selected list of the top 10 online casinos, games and strategies. Our goal is to provide informative, unbiased and entertaining content to help players make informed decisions in the world of online gaming. Helping players understand the games, rules and strategies in a simple and fun way.
how can i get generic celexa without a prescription
| #
where can i buy generic celexa pill
tadacip without prescription
| #
Meds information for patients. What side effects can this medication cause?
tadacip without prescription
Some news about pills. Get here.
mostbet_lqSt
| #
mostbets http://mostbet5002.ru .
PeterSlump
| #
https://telegra.ph/Explore-Non-Gamstop-Live-Casinos-for-Real-Gaming-Fun-03-27
PeterSlump
| #
https://telegra.ph/Explore-Non-GamStop-Casinos-Available-in-the-UK-03-27
Willieavamy
| #
The slot rush online pulls—watch it! blue wizard
Jarioreyu
| #
Где купить диплом по необходимой специальности?
Мы предлагаем максимально быстро купить диплом, который выполняется на оригинальном бланке и заверен печатями, водяными знаками, подписями. Диплом способен пройти лубую проверку, даже с применением специально предназначенного оборудования. Достигайте цели быстро и просто с нашей компанией.
Заказать диплом любого университета poluchidiplom.com/kupit-diplom-universiteta-10/
Sazrziz
| #
Мы можем предложить дипломы любой профессии по приятным тарифам.
Вы покупаете диплом через надежную и проверенную компанию. Заказать диплом университета– http://mop.5nx.ru/viewtopic.phpf=38&t=1789/ – mop.5nx.ru/viewtopic.phpf=38&t=1789
buy cheap stromectol pill
| #
ivermectin cost in usa stromectolinfo3
can you get generic paxil without a prescription
| #
Medicines information for patients. Effects of Drug Abuse.
can you get generic paxil without a prescription
Some trends of meds. Get here.
WilliamDix
| #
Мы изготавливаем дипломы любой профессии по выгодным тарифам. Дипломы производят на настоящих бланках Заказать диплом о высшем образовании diplomc-v-ufe.ru
BillyCew
| #
аренда автомобиля минеральные воды arenda-avto-mineralnye-vody.ru/
Lazrzuq
| #
Где заказать диплом специалиста?
Заказать диплом института по невысокой цене можно, обращаясь к надежной специализированной компании.: 1magistr.ru
OliverLen
| #
аренда автомобиля в москве аренда автомобиля в москве и области
JasonPaw
| #
аренда авто в калининграде в аэропорту аренда авто храброво
mostbet_vymt
| #
mostbet скачать mostbet скачать .
Willieavamy
| #
Online casino ads push—soften it! plinko
1win_ghpa
| #
win 1 http://www.1win7014.ru .
Iariorekr
| #
Заказать диплом института по невысокой цене возможно, обращаясь к надежной специализированной компании. Приобрести документ о получении высшего образования можно у нас в столице. diplom4you.com/kupit-diplom-o-srednem-obrazovanii-dlya-reestra
Willieavamy
| #
Are online reviews fake?—hard to tell! plinko
1win_sqPr
| #
1 win вход https://1win6048.ru .
RobertLoork
| #
обеспыливание стен цена cleaning-top24.ru
where can i get phenytoin
| #
Medicine information for patients. Cautions.
where can i get phenytoin
All about medicament. Get now.
Sazruzz
| #
Заказать диплом под заказ в столице возможно используя сайт компании. aijobsphilippines.com/employer/frees-diplom
Willieavamy
| #
Online casinos feel chancy—watch out! plinko
inernetufaCah
| #
интернет провайдеры уфа по адресу
domashij-internet-ufa003.ru
интернет по адресу уфа
where can i get accutane price
| #
can i purchase generic accutane pill
AndreDap
| #
Букет шикарный! Все цветы свежие.
цветы
Sazrowc
| #
Купить диплом института по доступной цене возможно, обращаясь к проверенной специализированной фирме. Мы оказываем услуги по производству и продаже документов об окончании любых ВУЗов РФ. Заказать диплом о высшем образовании– diplomg-cheboksary.ru/kupit-diplom-v-reestre-legko-i-bistro/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Плоская мишень NiV
Willieavamy
| #
I don’t trust online data—shaky! vipzino
Sazruas
| #
Мы изготавливаем дипломы любой профессии по приятным ценам.– damdiplomisa.com/kupit-diplom-s-zaneseniem-v-reestr-instituta-bistro/
технический план здания
| #
технический план здания
moverbet
| #
O moverbet oferece uma excelente oportunidade
para quem deseja começar sua experiência no cassino online com um bônus de 100$
para novos jogadores! Ao se registrar no site, você garante esse bônus exclusivo
que pode ser utilizado em diversos jogos de cassino,
como slots, roleta e poker. Esse é o momento perfeito para
explorar o mundo das apostas com um saldo extra, aproveitando ao máximo
suas apostas sem precisar investir um grande valor logo de início.
Não perca essa oportunidade e cadastre-se já!
where to buy cheap tetracycline without rx
| #
cost generic tetracycline for sale
mostbet_ilMn
| #
motsbet http://mostbet5001.ru .
Prodvijenie kriptovaluta_kppt
| #
orm криптопроекта http://prodvizhenie-kriptovalyuta1.ru/ .
Prodamys promokod_teEt
| #
продамус промокод скидка на подключение продамус промокод скидка на подключение .
mostbet_hsSt
| #
mostbet промокод https://www.mostbet5002.ru .
1win_wsSi
| #
1win.kg https://1win7003.ru/ .
1win_muKa
| #
1win login nigeria https://1win16.com.ng .
afbouwen van mirtazapine
| #
Pills information. What side effects can this medication cause?
afbouwen van mirtazapine
All about drugs. Read now.
Vlarizapex
| #
Иногда всё, что нужно — правильный сайт. mikro-zaim-online.ru — надёжный помощник в мире микрофинансов. Здесь вы найдете список МФО с моментальной выдачей, займами без отказа и возможностью получить до 30 дней без процентов. Подборка постоянно обновляется. Начните с уверенного шага.
Каждая микрофинансовая организация в подборке зарегистрирована в реестре ЦБ РФ и работает по закону №151-ФЗ. Деньги переводятся мгновенно после одобрения. Проверка минимальна, отказов — практически нет. Всё по закону: официально, безопасно, с чёткими условиями и прозрачными ставками. Просто, как должно быть.
Sazrric
| #
Приобретение документа о высшем образовании через надежную фирму дарит ряд преимуществ. Такое решение помогает сэкономить как личное время, так и значительные финансовые средства. Тем не менее, только на этом выгоды не ограничиваются, плюсов гораздо больше.Мы можем предложить дипломы любой профессии. Дипломы производятся на подлинных бланках государственного образца. Доступная цена сравнительно с крупными тратами на обучение и проживание в чужом городе. Покупка диплома об образовании из российского института является разумным шагом.
Приобрести диплом о высшем образовании: poluchidiplom.com/diplom-s-zaneseniem-v-reestr-4/
"oppna ett binance-konto
| #
I don’t think the title of your article matches the content lol. Just kidding, mainly because I had some doubts after reading the article.
Willieavamy
| #
I lost time online—swift pass! jackpotraider
Willieavamy
| #
Online casinos should free—try it! plinko app
1win_rbpi
| #
1вин сайт https://www.1win7015.ru .
1win_djpa
| #
адин вин адин вин .
Mazrrpe
| #
Купить диплом об образовании!
Мы предлагаем документы об окончании любых ВУЗов России. Документы производятся на фирменных бланках. itkr.com.ua/forum/viewtopic.php?t=27755
1win_hxSi
| #
1winn https://1win7003.ru .
CharlesViods
| #
Наша компания оказывает услуги в сфере кадастровых работ и проектирования перепланировок. Выполняем полный комплекс кадастровых работ – от межевания до подготовки технических планов. Также разрабатываем проекты перепланировки жилых помещений в соответствии с действующими нормами и требованиями. Гарантируем точность, юридическую чистоту и соблюдение сроков на каждом этапе: проект перепланировки цена
Willieavamy
| #
The rewards online shine—good perk! plinko
can i purchase cozaar without rx
| #
cost of cozaar online
internetchelyabCah
| #
домашний интернет
domashij-internet-chelyabinsk001.ru
подключить домашний интернет челябинск
Trefdyz
| #
Приобрести диплом об образовании. Приобретение документа о высшем образовании через качественную и надежную компанию дарит много достоинств. Данное решение помогает сэкономить время и существенные деньги. ieo-worktravel.com/employer/eonline-diploma
1win_ahpi
| #
1вин приложение https://www.1win7015.ru .
1win_xiSi
| #
1win сайт вход 1win сайт вход .
RedV5
| #
Полное руководство по путешествию от А до Я: http://roeliepost.com/2018/03/european-ombudsman-to-look-into-treatment-of-whistleblower-roelie-post/
solicitor law firms
| #
This is nicely put. !
AlphaV5
| #
Как войти в тему путешествия? https://wecanprevent20.org/avtobusnye-ekskursii-iz-spb.html
1win_lapi
| #
1 вин официальный сайт вход http://1win7015.ru .
Voboypoild
| #
Visit https://wpstore.pro/ where you will find premium WordPress themes and plugins. We have collected the best solutions for WordPress in one place. Official products from developers. We have the most affordable prices, original products, unlimited use, lifetime updates and much more. More details on the site.
order generic asacol without insurance
| #
where can i buy cheap asacol without rx
Prodamys promokod_doEt
| #
промокод на продамус скидка подключение promokod-prdms.ru .
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наши товары:
Лента никелевая 1.4С…15 РјРј РќРџ2РГОСТ 2170-73 Познакомьтесь СЃ высококачественной никелевой лентой РѕС‚ Редметсплав.СЂС„. РЁРёСЂРѕРєРёР№ выбор размеров Рё толщин. Прочность, устойчивость Рє высоким температурам. Применение РІ различных отраслях. Рдеально РїРѕРґС…РѕРґРёС‚ для электротехники, химической промышленности Рё медицинской техники.
Prodvijenie kriptovaluta_bept
| #
повышение доверия криптовалюты повышение доверия криптовалюты .
can i buy rizatriptan without dr prescription
| #
Medicament information for patients. Cautions.
can i buy rizatriptan without dr prescription
Best information about drug. Read now.
Claytonclurl
| #
Официальный веб-сайт казино Рояль предлагает располагающую обстановку для азартных игроков. С его элегантным дизайном и высококачественным выбором игр, Рояль является превосходным местом для тех, кто обожает качественное азартное развлечение. Исследуйте многочисленные игровых автоматов и настольных игр на официальном веб-сайте https://royalcasino-slots.com.
Казино Рояль предоставляет высокий уровень клиентов и поддержку. Внимательная команда поддержки в наличии круглосуточно, настроена откликнуться на любые возникающие вопросы и урегулировать проблемы, гарантируя непрерывную игру и удовлетворение.
CyberOverlord
| #
Нужны практические советы по путешествию. http://www.projectorlampsworld.com/activnosti
Willieavamy
| #
I’m off online—shady risk! plinko
Toliknaf
| #
darknet markets url darkmarket link
mostbet_iymt
| #
мостбет казино http://mostbet6033.ru .
Thunder_Guardian
| #
Столкнулся с проблемой путешествием. Решение здесь: https://myaoc.org/razvlecheniya.html
Willieavamy
| #
The online flow is wild—stay sharp! marvel casino
OmegaPioneer
| #
Эксперты рекомендуют эти ресурсы https://webosarena.com/aktivnyy-otdyh-v-sankt-peterburge.html по путешествию.
Rabyitabe
| #
darkmarkets darknet websites
Cazrkxa
| #
Где приобрести диплом по актуальной специальности?
Мы изготавливаем дипломы любых профессий по приятным тарифам. Важно, чтобы документы были доступными для большого количества граждан. Приобрести диплом о высшем образовании diplom-top.ru/diplom-v-reestre-garantiya-podlinnosti-i-uspexa/
Willieavamy
| #
Online casinos need stricter regulations—too many shady ones! doradobet
mostbet_fyMn
| #
aviator mostbet https://www.mostbet5001.ru .
GeorgeTom
| #
Source:
– snowlands.ru
Pingrutty
| #
dark market 2025 darknet links
AlphaV6
| #
Путешествие для стартапов: https://blockhouseraces.com/aktivnyy-otdyh-v-spb.html
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Опытная поддержка – наш стандарт – мы на связи, чтобы улаживать ваши вопросы по мере того как адаптировать решения под особенности вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
РќРёРѕР±РёР№ РќР±РџР“-3 РќРёРѕР±РёР№ РќР±РџР“-3 – уникальный материал, обладающий высокими антикоррозийными свойствами Рё отличной термостойкостью. Ртот металл идеально РїРѕРґС…РѕРґРёС‚ для применения РІ химической промышленности, производстве авиадеталей Рё РІ электронике. Благодаря своей прочности Рё легкости, РЅРёРѕР±РёР№ широко используется РІ производстве специальных сплавов Рё катализаторов. Если РІС‹ ищете надежный источник этого уникального материала, рекомендуем купить РќРёРѕР±РёР№ РќР±РџР“-3. Доступный Рё высококачественный, РѕРЅ обеспечит вам отличные результаты РІ ваших проектах.
Uazrqdc
| #
Доброго времени суток!
Для многих людей, купить диплом ВУЗа – это острая необходимость, уникальный шанс получить выгодную работу. Но для кого-то – это понятное желание не терять множество времени на учебу в институте. Что бы ни толкнуло вас на это решение, мы готовы помочь вам. Оперативно, профессионально и выгодно сделаем документ любого года выпуска на подлинных бланках со всеми печатями.
Основная причина, почему многие покупают документы, – получить хорошую должность. Например, знания и опыт позволяют специалисту устроиться на работу, но документального подтверждения квалификации не имеется. Когда для работодателя важно наличие “корочек”, риск потерять вакантное место очень высокий.
Приобрести документ о получении высшего образования вы имеете возможность у нас в столице. Мы оказываем услуги по продаже документов об окончании любых ВУЗов РФ. Вы получите необходимый диплом по любой специальности, включая документы образца СССР. Даем 100% гарантию, что при проверке документов работодателем, каких-либо подозрений не возникнет.
Разных ситуаций, которые вынуждают заказать диплом немало. Кому-то прямо сейчас потребовалась работа, и нужно произвести особое впечатление на руководителя при собеседовании. Другие планируют устроиться в серьезную компанию, чтобы повысить собственный статус в обществе и в дальнейшем начать свой бизнес. Чтобы не терять драгоценное время, а сразу начинать успешную карьеру, применяя имеющиеся навыки, можно приобрести диплом в онлайне. Вы сможете быть полезным для общества, обретете финансовую стабильность максимально быстро и просто- диплом купить о высшем образовании
Willieavamy
| #
I love the live roulette—real deal! plinko
DonDonfaita
| #
darknet site darknet drug store
Willieavamy
| #
Online casinos can hook—edge it! plinko
PhantomOverlord
| #
Использование на практике путешествия: https://pregmind.com/audiogid
Willieavamy
| #
I love the online chats—great crew! Dragon Hatch
Sazrjyj
| #
Заказать диплом об образовании!
Купить диплом ВУЗа по невысокой цене возможно, обращаясь к проверенной специализированной компании. Приобрести диплом о высшем образовании: diplomc-v-ufe.ru/kupit-diplom-s-zaneseniem-v-reestr-instituta-9
Sazraoc
| #
Мы готовы предложить дипломы любых профессий по приятным ценам.
Вы приобретаете документ в надежной и проверенной временем компании. Заказать диплом института– http://t98223u0.beget.tech/2025/04/03/kupit-podlinnyy-diplom-dostupno-i-prosto.html/ – t98223u0.beget.tech/2025/04/03/kupit-podlinnyy-diplom-dostupno-i-prosto.html
Dextershory
| #
Онлайн-казино 7k — это современная платформа для азартных развлечений с большим выбором игр. Здесь можно найти известные игровые автоматы, классические игры и игры в реальном времени с опытными дилерами. Дизайн удобный и простой в освоении, что делает игру удобной и увлекательной. На 7k740.casino/ru/ предусмотрены щедрые призы для любителей азартных игр. В целом, платформа предоставляет неповторимый опыт для каждого игрока.
https://proektmedia-stat.ams3.digitaloceanspaces.com/2019/01/bob_king_main_image.jpg
| #
https://proektmedia-stat.ams3.digitaloceanspaces.com/2019/01/bob_king_main_image.jpg
Hunter5978
| #
Где тренироваться путешествию? https://www.800ta.org/audio-gid-po-ermitazhu.html
Willieavamy
| #
Online bonuses mask—dig in! gates of olympus
Donaldlaf
| #
darknet sites dark market url
1win_svKi
| #
portofele electronice casino https://1win5024.ru/ .
Steel118
| #
Смешные случаи в путешествии: https://brinasings.com/oruzheina-palata.html.
how to buy cheap benemid online
| #
where can i buy generic benemid tablets
can you get cheap seroquel
| #
Drug information for patients. Cautions.
can you get cheap seroquel
Actual information about medicament. Read information here.
mostbet_ltmt
| #
mostbet игры http://mostbet6033.ru/ .
Sazrttv
| #
Заказать документ о получении высшего образования вы сможете в нашей компании в столице. Мы оказываем услуги по производству и продаже документов об окончании любых университетов Российской Федерации. Вы получите необходимый диплом по любым специальностям, любого года выпуска, включая документы Советского Союза. Гарантируем, что при проверке документа работодателями, никаких подозрений не возникнет. freediplom.com/kupit-diplom-s-reestrom-bistro-i-legko/
internetchelyabCah
| #
подключить домашний интернет челябинск
domashij-internet-chelyabinsk002.ru
домашний интернет
Prodvijenie kriptovaluta_rtEn
| #
seo криптовалюты seo криптовалюты .
RedarnsuenI
| #
Посетите сайт Мир Часов https://mir-watch.ru/ – это скупка часов в Москве. Вы сможете сдать часы в ломбард по выгодной стоимости. Занимаемся выкупом (скупкой) швейцарских часов и других известных брендов. Выкупаем не только элитные часы, но и часовые аксессуары: виндеры, коробки, шкатулки, ремни, ювелирные украшения и аксессуары с бриллиантовыми камнями.
Toliknaf
| #
dark market list dark websites
Hunter3504
| #
Где учиться https://www.hymnscript.com/bilety-v-pushkinskij-muzej.html путешествию?
Reaper9587
| #
Какой http://www.projectorlampsworld.com/russkii-musei.html вариант предпочесть в путешествии?
Pingrutty
| #
dark web markets darknet market
Willieavamy
| #
Online casinos can be trouble—set boundaries! minas
Mazrnro
| #
Где заказать диплом по необходимой специальности?
Готовый диплом с нужными печатями и подписями 100% отвечает запросам и стандартам Министерства образования и науки России, неотличим от оригинала – даже со специально предназначенным оборудованием. Не откладывайте свои мечты на несколько лет, реализуйте их с нашей компанией – отправляйте заявку на диплом прямо сейчас! Диплом о среднем специальном образовании – не проблема! district-jobs.com/profile/vbfgeorge36787
Rabyitabe
| #
dark web markets dark web marketplaces
can prednisone affect your taste
| #
prednisone without prescription canadian pharmacy
Pioneer8897
| #
Как сэкономить на путешествии: https://webosarena.com/bilety-v-stroganovskiy-dvorec.html.
Omega_Byte
| #
https://stuttgart-isst.com/bilety-v-tretyakovskuyu-galereyu/ Последние обновления по путешествию.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Гранулы оксида алюминия 4N
DonDonfaita
| #
darknet websites dark web market
mostbet_usmt
| #
скачать mostbet mostbet6033.ru .
Willieavamy
| #
Online gambling laws are so confusing—can I even play legally? bac bo
Willieavamy
| #
The slot rush online pulls—watch it! aviator
Spark7987
| #
ТОП-10 ресурсов по путешествию: https://ragdollresearch.org/bilet-v-tretiakovku.html
Willieavamy
| #
Online blackjack rocks—fun twist! plinko
Willieavamy
| #
Online casinos should ease—too much! plinko
Larryfut
| #
Имплантация зубов — это надежный способ вернуть здоровую улыбку, не затрагивая соседние зубы, особенно актуально при полной или частичной адентии https://www.vevioz.com/read-blog/327658
Walker9555
| #
Мемы про путешествие: https://www.socalfreenet.org/bilety-na-kreiser-aurora/
Donaldlaf
| #
darkmarket list darknet market links
GhostMaster
| #
Подборка ресурсов по путешествию: https://myaoc.org/kreml.html
https://proektmedia-stat.ams3.digitaloceanspaces.com/2018/10/sosed-putina-boris-listov.png
| #
https://proektmedia-stat.ams3.digitaloceanspaces.com/2018/10/sosed-putina-boris-listov.png
Willieavamy
| #
The mobile apps online falter—patch it! razor returns
buying cheap zithromax price
| #
Meds information. Cautions.
buying cheap zithromax price
Best what you want to know about medicament. Read here.
JupudtCow
| #
На сайте http://oknaksa.ru закажите расчет технического задания, чтобы воспользоваться такой важной, полезной услугой, как остекление. На монтажные работы, а также сами конструкции предоставляются гарантии, вы сможете воспользоваться бесплатным обслуживанием. А все работы выполняются точно по ГОСТу, в соответствии с требованиями. Предоставляются исчерпывающие и содержательные консультации. Вам будет предложено несколько вариантов решения проблемы. Компания располагает огромным количеством готовых проектов.
Pingrutty
| #
darkmarket url darknet links
Toliknaf
| #
dark websites darkmarket 2025
Sazrhuf
| #
Приобрести диплом на заказ можно через официальный сайт компании. uat.editorlog.com/read-blog/4613_mozhno-li-kupit-diplom-v-reestre.html
JerryBak
| #
ai porn free nude ai art
Edwardveict
| #
Klavier noten noten auf klavier
BlueZ8
| #
https://chickennuggetsofwisdom.com/bilety-onlayn-v-isaakievskiy-sobor.html Как реализовать путешествие?
ScottRiz
| #
Piano sheet music sheet music and piano
Kevinwhast
| #
Полезная информация yanglish.ru свежие материалы и удобная навигация — всё, что нужно для комфортного пользования сайтом. Заходите, изучайте разделы и находите то, что действительно важно для вас.
Robertcek
| #
Сделаем официальную прописку в жилом фонде Москвы, с полным комплектом документов и подтверждением от собственника https://reg-v-msk.ru/
Willieavamy
| #
I don’t get why people hate on online casinos; it’s just entertainment! sugar rush
Rabyitabe
| #
darknet drug market dark web market
StormZ1
| #
Пошаговая инструкция по путешествию: http://roeliepost.com/2007/
wenizMiz
| #
Tent3302.ru предлагает необходимые автодетали. Мы продаем запчасти на Газель Некст. Под заказ двигатель Газель Камминз 2.8 поставляем. Стараемся поддерживать цены на самом низком уровне. Продажи выполняем со склада за безналичный и наличный расчет. Ищете карбюратор газель 406? Tent3302.ru – тут можете детальную о нас информацию отыскать. Мы качественную продукцию продаем. Шины на Газель особенным спросом у клиентов пользуются. Стараемся максимально сократить транспортные и накладные расходы. Вы можете быть уверены в вежливом и грамотном обслуживании.
Hijamnetly
| #
На сайте https://pilmi.ru вы найдете огромное количество фильмов самых разных жанров, включая комедии, мелодрамы, драмы, романтические. Для того чтобы подыскать наиболее подходящий вариант, нужно воспользоваться специальным каталогом. Вашему вниманию представлены и ожидаемые любопытные новинки этого года, которые произведут эффект. Все это можно просматривать в любое время и на различных устройствах. Прямо сейчас воспользуйтесь возможностью посмотреть азиатское кино. Используйте поиск для облегчения выбора.
SilverV6
| #
Путешествие для стартапов: https://brimfieldchamber.com/2013/11/the-bible-is-missionary-book.html
internetchelyabCah
| #
домашний интернет челябинск
domashij-internet-chelyabinsk003.ru
интернет тарифы челябинск
1win_qqpi
| #
1вин. http://www.1win7015.ru .
Prodvijenie kriptovaluta_pwEn
| #
appstore отзывы appstore отзывы .
Willieavamy
| #
Online casinos should cap losses—protect players! aviator casino
DonDonfaita
| #
darkmarket dark markets
Larryfut
| #
Врачи подбирают индивидуальное решение — от одиночного импланта до полного протезирования на имплантах https://www.storeboard.com/blogs/health/orthodontic-treatments-braces-and-beyond/6146414
AlphaReaper
| #
Памятка для работы с путешествием: https://uniqueblogdesigns.com/
JamesSex
| #
Возможна прописка как в пределах МКАД, так и в районах Новой Москвы https://reg-msk99.ru/
Willieavamy
| #
I love the live roulette—real deal! gransino
get cheap femara without prescription
| #
where buy cheap femara price
Vortex_Hacker
| #
Полезные сервисы для https://fraseseproverbios.com/videos/quid-des-blogs-face-aux-reseaux-sociaux/ работы с путешествием.
Pingrutty
| #
darknet market list darknet market
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наши товары:
Плоская мишень из иттрия, 3N, 4N
Donaldlaf
| #
darkmarket tor drug market
Thunder_Reaper
| #
Мне https://fundraising-newsletters.com/vyezdnye-ekskursii-iz-sankt-peterburga.html самому помог этот ресурс по путешествию.
Willieavamy
| #
Online poker pumps—bluff high! teen patti
Willieavamy
| #
Online poker is a blast—bluffing rocks! Gioco Del Pollo
protonix pantoprazole 40mg
| #
Medication information sheet. Drug Class.
protonix pantoprazole 40mg
All what you want to know about medicine. Get here.
casinos sin licencia en espana_jjpl
| #
casino sin licencia espa?a casino sin licencia espa?a .
Richardskash
| #
веб-сайт https://xn—-jtbjfcbdfr0afji4m.xn--p1ai/
Thunder186
| #
Конкретные шаги для работы с путешествием: https://ragdollresearch.org/vystavki-v-moskve.html
Toliknaf
| #
darknet market lists dark market list
can i buy generic synthroid tablets
| #
can i get cheap synthroid pill
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наша продукция:
Пружина из драгоценных металлов платиновая 650х7х0.9 мм Пл99.9 ТУ Выберите идеальные пружины из золота, серебра или платины, чтобы дополнить ваше украшение. Найдите пружины с изысканным дизайном и прочностью. Они предлагают широкий выбор для того, чтобы подчеркнуть уникальность вашего украшения.
StephanEteri
| #
ссылка на сайт https://falcoware.com/rus/match3_games.php
Jasonled
| #
выберите ресурсы https://xn—-jtbjfcbdfr0afji4m.xn--p1ai/
Silver258
| #
Как монетизировать https://abktechnologies.com/murmansk-zimoi.html путешествие в бизнесе?
Larryfeddy
| #
посмотреть в этом разделе https://falcoware.com/rus/match3_games.php
GeorgeTom
| #
Source:
snowlands.ru
Jamesker
| #
этот сайт https://xn—-jtbjfcbdfr0afji4m.xn--p1ai/
site
| #
You said that effectively!
Rabyitabe
| #
darknet site darknet sites
NightZ3
| #
Будущее путешествия: http://www.projectorlampsworld.com/vystavki-v-pitere
http://proekt.media/wp-content/uploads/2018/09/Listov_approved.jpg
| #
http://proekt.media/wp-content/uploads/2018/09/Listov_approved.jpg
Sazrwdw
| #
Приобретение документа о высшем образовании через надежную компанию дарит множество плюсов. Данное решение дает возможность сэкономить как личное время, так и значительные финансовые средства. Тем не менее, плюсов значительно больше.Мы готовы предложить дипломы психологов, юристов, экономистов и любых других профессий. Дипломы производят на подлинных бланках. Доступная стоимость сравнительно с крупными тратами на обучение и проживание. Покупка диплома о высшем образовании из российского ВУЗа будет выгодным шагом.
Приобрести диплом о высшем образовании: fastdiploms.com/kupit-diplom-s-registratsiej-bez-lishnix-problem/
Robertded
| #
авто напрокат сутки аренда авто напрокат
JamesStara
| #
машина в аренду владивосток по суточно авто прокат аренда владивосток
ArthurCeday
| #
аренда машины в махачкале https://arenda-avtomobilya-mahachkala.ru/
Storm_Code
| #
Изучите этот источник о путешествии: https://ragdollresearch.org/vystavki-v-spb.html
DannyHic
| #
аренда автомобиля для поездки в спб аренда авто на сутки в санкт петербурге
Willieavamy
| #
Are online casino jackpots real, or just bait to keep you playing? andar bahar
Willieavamy
| #
The live dealer feature online is a total game-changer! mines
Willieavamy
| #
The online thrill strikes—stay steady! book of ra
Willieavamy
| #
Online poker is wild—bluffing is a rush! aviator
DonDonfaita
| #
dark market 2025 dark web market list
JamesSex
| #
С нами можно оформить прописку, которая действительно работает и проходит проверки со стороны госорганов: сделать прописку в москве для граждан рф официально цена
Pingrutty
| #
darknet market lists dark websites
EagleZ2
| #
Основы https://fundraising-newsletters.com/vystavki-v-spb-segodnya.html путешествия для новичков.
Sazrtaj
| #
папка диплом купить спб
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Плоская металлическая мишень Церия
Willieavamy
| #
I’ve never tried an online casino—where should I start? plinko
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их соответствие стандартам. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы а также предоставлять решения под требования вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
поставляемая продукция:
Фольга кобальтовая РРџ59 Фольга кобальтовая РРџ59 – это высококачественный материал, предназначенный для различных промышленных нужд. РћРЅР° обладает отличной стойкостью Рє РєРѕСЂСЂРѕР·РёРё Рё высокой температуре, что делает ее идеальным выбором для применения РІ тяжелых условиях. Фольга используется РІ авиации, электронике Рё РґСЂСѓРіРёС… отраслях, РіРґРµ требуется надежность Рё долговечность. Если РІС‹ хотите купить Фольга кобальтовая РРџ59, РІС‹ получите РїСЂРѕРґСѓРєС‚, который соответствует самым высоким стандартам качества. Обеспечьте надежность ваших проектов СЃ этой фольгой!
Vortex29
| #
Какой https://tweetmeharder.com/vystavki-v-ermitazhe.html вариант предпочесть в путешествии?
casinos sin licencia en espana_repl
| #
casas apuestas sin licencia http://www.casinossinlicenciaespanola.com .
Jimmyreink
| #
Счетчики Газа с магнитом Счетчики с пультом: Экономия или обман? В современном мире, когда цены на энергоресурсы неуклонно растут, многие ищут способы снизить свои коммунальные платежи. Одним из таких способов стали счетчики с пультом дистанционного управления, а также различные методы “модификации” приборов учета. Но так ли это выгодно и законно, как кажется на первый взгляд?
Willieavamy
| #
Online poker is wild—bluffing is a rush! book of dead
Donaldlaf
| #
tor drug market darknet drugs
Willieavamy
| #
Online casinos should show odds clearer! plinko
Voyager2735
| #
Вебинар о путешествии: https://yourecumbentbike.com/vystavki-vermitazhe.html
inernetadresCah
| #
интернет по адресу красноярск
domashij-internet-krasnoyarsk001.ru
интернет провайдеры в красноярске по адресу дома
Cazroji
| #
Где приобрести диплом по необходимой специальности?
Мы изготавливаем дипломы любой профессии по доступным тарифам. Важно, чтобы дипломы были доступными для большого количества граждан. Быстро приобрести диплом о высшем образовании diplomv-v-ruki.ru/diplom-s-reestrom-pravda-ili-mif-tseni-i-riski/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние достижения.
Наши товары:
Мишень для распыления скандия
Willieavamy
| #
Online casino ads blast—quiet it! plinko
can you buy trileptal price
| #
Medication prescribing information. Generic Name.
can you buy trileptal price
Actual information about pills. Read information now.
Toliknaf
| #
darknet drug market dark web sites
Iron506
| #
Последние обновления по https://fundraising-newsletters.com/vystavki-v-ermitazhe-seychas.html путешествию.
order bactrim pill
| #
where to buy cheap bactrim for sale
Prodvijenie kriptovaluta_woer
| #
appstore отзывы appstore отзывы .
Omega479
| #
Глубокий анализ путешествия: https://stuttgart-isst.com/transfer/
mostbet_kvSt
| #
мостбет войти http://www.mostbet5002.ru .
1win_piSi
| #
1win вход на сайт https://1win7003.ru/ .
Rabyitabe
| #
darkmarket link darkmarket
Uazrore
| #
Всех приветствую!
Для некоторых людей, заказать диплом университета – это необходимость, уникальный шанс получить достойную работу. Впрочем для кого-то – это очевидное желание не терять время на учебу в институте. Что бы ни толкнуло вас на это решение, мы готовы помочь вам. Быстро, профессионально и по разумной стоимости изготовим документ любого ВУЗа и года выпуска на подлинных бланках с реальными подписями и печатями.
Главная причина, почему многие покупают документы, – получить хорошую работу. Допустим, способности и опыт дают возможность человеку устроиться на работу, а документального подтверждения квалификации не имеется. При условии, что для работодателя важно наличие “корочек”, риск потерять хорошее место довольно высокий.
Приобрести документ ВУЗа вы имеете возможность у нас в столице. Мы оказываем услуги по продаже документов об окончании любых университетов Российской Федерации. Вы сможете получить диплом по любой специальности, любого года выпуска, в том числе документы старого образца. Гарантируем, что при проверке документов работодателем, подозрений не возникнет.
Разных ситуаций, которые вынуждают приобрести диплом много. Кому-то очень срочно потребовалась работа, а значит, необходимо произвести впечатление на начальника при собеседовании. Некоторые желают попасть в большую компанию, чтобы повысить собственный статус в социуме и в последующем начать свой бизнес. Чтобы не терять впустую годы жизни, а сразу начать эффективную карьеру, применяя врожденные способности и полученные знания, можно купить диплом прямо в онлайне. Вы сможете стать полезным в обществе, обретете финансовую стабильность максимально быстро и легко- купить аттестат
Storm_Voyager
| #
http://www.projectorlampsworld.com/vystavki-v-moskve Заинтересовался путешествием. Ответ здесь.
Pingrutty
| #
darknet websites dark market list
Sazrhvl
| #
Студенческая жизнь прекрасна, пока не приходит время писать диплом, как это случилось со мной. Не стоит отчаиваться, ведь существуют компании, которые помогают с написанием и защитой диплома на высокие оценки!
Сначала я искал информацию по теме: купить диплом образование нижнем новгороде, сайт купить диплом, купить диплом в липецке, купить диплом в набережных челнах, купить диплом о среднем образовании в тольятти, а потом наткнулся на diplomybox.com/kupit-diplom-izhevsk
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Высокочистые гранулы титана
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наши товары:
Пружина из драгоценных металлов золотая 1000х5х0.4 мм ЗлМ98 ТУ Выберите идеальные пружины из золота, серебра или платины, чтобы дополнить ваше украшение. Найдите пружины с изысканным дизайном и прочностью. Они предлагают широкий выбор для того, чтобы подчеркнуть уникальность вашего украшения.
Willieavamy
| #
I love the low bets online—soft go! plinko
casinos sin licencia en espana_jipl
| #
casinos sin licencia en espa?a casinos sin licencia en espa?a .
Warden4964
| #
Сравнение лучших https://kittyspit.net/vystavki-v-pitere.html ресурсов по путешествию.
cost cheap atacand price
| #
where to get atacand no prescription
https://www.proekt.media/investigation/sosed-boris-listov/
| #
https://www.proekt.media/investigation/sosed-boris-listov/
DonDonfaita
| #
bitcoin dark web dark web market
1win_dfKi
| #
1win aplicația https://1win5024.ru .
mostbet_ygSl
| #
поддержка мостбет поддержка мостбет .
Eagle_Sentinel
| #
Где смотреть актуальные данные о путешествии? https://genesis-growers.com/interesnye-vystavki-v-sankt-peterburge.html
Sazrhfq
| #
Купить диплом университета!
Приобрести диплом института по выгодной цене возможно, обратившись к надежной специализированной фирме. Купить диплом: asxdiploman.com/ofitsialnij-diplom-s-reestrom-kupit-tsena-garantiya
Blue402
| #
Топовый ресурс по теме путешествия: https://comprehensivecommunitysolutions.org/master-klassy-v-moskve.html
RichardDoult
| #
проект перепланировки помещения
1win_pxKi
| #
1win metode de plată 1win5024.ru .
Donaldlaf
| #
darkmarket list darknet market lists
Iron226
| #
Чек-лист https://printgrowstrees.com/master-classi/ для работы с путешествием.
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица товара подтверждена соответствующими документами, подтверждающими их происхождение. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы по мере того как адаптировать решения под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
РўСЂСѓР±Р° кобальтовая HAYNES NS-163 РўСЂСѓР±Р° кобальтовая HAYNES NS-163 – это высококачественный РїСЂРѕРґСѓРєС‚, специально разработанный для работы РІ экстремальных условиях. РћРЅ отличается отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью Рё высокой прочностью, что делает его идеальным выбором для промышленных применений. Состав трубы обеспечивает надежность Рё долговечность эксплуатации. Рспользование этой трубы гарантирует долгий СЃСЂРѕРє службы даже РІ агрессивных средах. Если РІС‹ ищете надежное решение, РЅРµ упустите возможность купить РўСЂСѓР±Р° кобальтовая HAYNES NS-163 Рё обеспечьте себе качество РЅР° долгие РіРѕРґС‹.
mostbet_wgSl
| #
мостбет зеркало https://mostbet5003.ru .
mostbet_qfmt
| #
скачать мостбет официальный сайт скачать мостбет официальный сайт .
Dark_Byte
| #
Доступно объяснено о путешествии: https://bumpbrooklyn.com/interesnye-master-klassy-v-spb.html
Stephenindix
| #
https://opechatano.ru/kupit-plombirator-s-gravirovkoy2/kupit-plombirator-s-gravirovkoy/
Toliknaf
| #
darknet drug market dark market list
can i purchase generic valtrex without dr prescription
| #
Pills information for patients. Drug Class.
can i purchase generic valtrex without dr prescription
Actual news about pills. Read now.
site
| #
site
blog topic
Eagle_Overlord
| #
Нужны реальные кейсы по путешествию. https://kittyspit.net/razvlecheniya-v-moskve.html
Pingrutty
| #
onion dark website darknet markets 2025
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
поставляемая продукция:
Труба из драгоценных металлов серебряная 700х6х0.5 мм Ср99.99 ТУ Гарантированное качество и изысканный дизайн – купите драгоценные трубы для ювелирных украшений, машиностроения и архитектуры. Широкий выбор и высокое качество! Доставка по России.
Willieavamy
| #
Online gambling is too open—tighten up! pinup
Rabyitabe
| #
darknet markets 2025 dark web marketplaces
Vortex931
| #
Пример из практики: Путешествие – https://fundraising-newsletters.com/kakie-est-razvlecheniya-v-pitere.html
inernetadresCah
| #
интернет провайдеры по адресу дома
domashij-internet-krasnoyarsk002.ru
узнать провайдера по адресу красноярск
cost of generic tricor price
| #
can i buy tricor prices
Warden9754
| #
Видел прогнозы, что Путешествие будет главным трендом. Соглашусь? https://everytraycounts.org/ermitazh.html
Willieavamy
| #
Online gambling feels loose—risk high! aviator
Prodvijenie kriptovaluta_eber
| #
улучшение репутации криптовалюты улучшение репутации криптовалюты .
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
поставляемая продукция:
Медный двухраструбный отвод 45 градусов под пайку 35х29х1 мм 23х25 мм твердая пайка М1М ГОСТ 32590-2013 Приобретите высококачественные Медные отводы 45 градусов под пайку от компании Редметсплав.рф и улучшите вашу трубопроводную систему. Прочные, долговечные и устойчивые к коррозии отводы обеспечат надежное соединение под нужным углом, обеспечивая оптимальное распределение потока и высокую производительность вашей системы.
DonDonfaita
| #
darknet drug store darknet market
https://www.proekt.media/investigation/2018/10/03/sosed-boris-listov/
| #
https://www.proekt.media/investigation/2018/10/03/sosed-boris-listov/
Willieavamy
| #
I love the pay flow online—easy run! andar bahar
Shadow323
| #
Как сэкономить на путешествии: http://zaiprotiv.info/novosti/9569-toshnotvornye-trenirovki-amerikanskoj-armii
Mazryun
| #
Где заказать диплом по необходимой специальности?
Получаемый диплом с нужными печатями и подписями отвечает условиям и стандартам, никто не сможет отличить его от оригинала – даже со специальным оборудованием. Не откладывайте свои цели на несколько лет, реализуйте их с нашей помощью – отправляйте заявку на диплом сегодня! Диплом о среднем специальном образовании – быстро и просто! yourealtynews.ru/nastoyashhie-diplomyi-po-dostupnoy-tsene-konfidentsialnost-100
Phantom508
| #
Секретные методы путешествия: https://artisangourmetmarket.com/sobor-vspb.html.
GeorgeTom
| #
Source:
snowlands.ru
dahebDex
| #
Looking for gifts to Ukraine? Ukraineflora.com is the best choice for any event. We have a large assortment of flowers in Ukraine! Look at it now, we are sure you will like everything. Now it is not difficult to make flower delivery in Ukraine, just go to the portal and get acquainted with our offers in more detail, here are fresh flowers, bouquets, and baskets. In addition, you have the opportunity to send gifts to Ukraine. Gift delivery in Ukraine is an opportunity to please your relatives even more.
where to buy prednisone pill
| #
prednisone 1 mg kg day
Willieavamy
| #
I don’t trust online—too open! plinko
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их качество. Дружелюбная помощь – наш стандарт – мы на связи, чтобы ответить на ваши вопросы а также предоставлять решения под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Р’РёСЃРјСѓС‚ C99740 – UNS Р’РёСЃРјСѓС‚ C99740 – UNS – это высококачественный материал, идеально подходящий для различных промышленных приложений. Обладая уникальными физико-химическими свойствами, РѕРЅ демонстрирует отличную РєРѕСЂСЂРѕР·РёРѕРЅРЅСѓСЋ стойкость Рё стабильность РїСЂРё высоких температурах. Рто делает его незаменимым РІ производстве сплавов Рё РІ качестве легирующей добавки. Если РІС‹ хотите купить Р’РёСЃРјСѓС‚ C99740 – UNS, это отличный выбор для повышения эффективности ваших процессов. Выбирайте надежное сырье, Рё РІС‹ РЅРµ пожалеете!
Zivutrttar
| #
Перевозка негабаритных грузов и доставка по всей территории России и СНГ на сайте https://negabarit-24.ru/ – это профессиональные услуги от нашей компании. Перевозка негабаритных грузов по России и странам СНГ, ЖД перевозки, перевозки манипулятором и международные перевозки. Гибкие варианты оплаты. Соблюдение сроков. Страхование грузов. Подробнее на сайте.
Hacker9919
| #
Как применять путешествие в бизнесе? https://www.redcornpottery.com/kreiser-avrora/
brwin
| #
Registre-se no brwin – https://www.brwin-br.com e Comece com 100$ de Bônus para Apostar
воронин владимир александрович фск
| #
воронин владимир александрович фск
Donaldlaf
| #
darknet links bitcoin dark web
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
Наша продукция:
Лента никелевая 1.8С…50 РјРј РќРџРћРВРГОСТ 2170-73 Познакомьтесь СЃ высококачественной никелевой лентой РѕС‚ Редметсплав.СЂС„. РЁРёСЂРѕРєРёР№ выбор размеров Рё толщин. Прочность, устойчивость Рє высоким температурам. Применение РІ различных отраслях. Рдеально РїРѕРґС…РѕРґРёС‚ для электротехники, химической промышленности Рё медицинской техники.
mostbet_thSl
| #
скачать mostbet на телефон http://www.mostbet5003.ru .
Steel200
| #
Как устроен механизм путешествия? https://soupswap.com/bolshoj-teatr/
Willieavamy
| #
I love the poker range online—new always! plinko
Pingrutty
| #
dark market 2025 darkmarket url
where can i get paxil
| #
Medicament information. Short-Term Effects.
where can i get paxil
All trends of medication. Get here.
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их происхождение. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы и находить ответы под специфику вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
поставляемая продукция:
Рзделия РёР· висмута 42 3991 – CSN/STN 423991 Рзделия РёР· висмута 42 3991 – CSN/STN 423991 имеют уникальные физико-химические свойства, благодаря которым РѕРЅРё находят применение РІ различных отраслях. Рти изделия отличаются высокой устойчивостью Рє РєРѕСЂСЂРѕР·РёРё Рё механическим повреждениям. РћРЅРё идеально РїРѕРґС…РѕРґСЏС‚ для создания специализированных деталей Рё компонентов. Если РІС‹ ищете надежное решение, то вам стоит купить Рзделия РёР· висмута 42 3991 – CSN/STN 423991. Внешний РІРёРґ этих изделий также привлекает благодаря своей эстетике. Выбирайте качественное, выбирайте СЃ СѓРјРѕРј!
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Керамическая мишень для распыления РѕРєСЃРёРґР° олова – 4N, Плоская форма
GunefUsano
| #
На сайте http://www.lilibum.ru вы найдете много анкет реальных людей для того, чтобы завести знакомства, найти свою вторую половинку и просто интересно провести время, если пригласите кого-то на свидание. Для того чтобы составить мнение о человеке, можно обменяться фотографиями и пообщаться на определенные темы, которые вас интересуют. Самые привлекательные и интересные люди вашего города находятся на этом портале. Знакомьтесь, влюбляйтесь и создавайте семьи. Для получения доступа ко всем функциям, пройдите регистрацию.
EagleReaper
| #
Видел прогнозы, что Путешествие будет главным трендом. Возражения? https://builtbyitc.com/bilety-v-isakii.html
Toliknaf
| #
darknet markets darknet websites
Robertcek
| #
Прописка оформляется только в жилом фонде, в соответствии с требованиями закона https://reg-v-msk.ru/
mostbet_kvmt
| #
motbet http://www.mostbet6033.ru .
Sazrivg
| #
Заказать документ ВУЗа вы можете у нас. Мы оказываем услуги по изготовлению и продаже документов об окончании любых ВУЗов РФ. Вы получите необходимый диплом по любым специальностям, включая документы старого образца. Гарантируем, что в случае проверки документов работодателем, подозрений не появится. diplomidlarf.ru/vnesenie-diploma-v-reestr-bistro-i-nedorogo-2/
ShadowV3
| #
Секретные методы https://tncmusic.net/bilety-v-pushkinskij-muzej путешествия.
OmegaV2
| #
https://genesis-growers.com/bilety-onlayn-v-isaakievskiy-sobor.html Как сделать путешествие?
Rabyitabe
| #
dark web marketplaces onion dark website
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их происхождение. Превосходное обслуживание – наш стандарт – мы на связи, чтобы разрешать ваши вопросы и находить ответы под особенности вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
поставляемая продукция:
Полоса вольфрамовая ВМ (20-30) Полоса вольфрамовая ВМ (20-30) – это специализированный материал для различных промышленных и научных применений. Обладая высокой прочностью и термостойкостью, вольфрамовые полосы подходят для использования в условиях экстремальных температур и нагрузок. Они находят свое применение в электронике, авиастроении и других высокотехнологичных отраслях. Если вы ищете надежное решение для своих проектов, купить Полоса вольфрамовая ВМ (20-30) будет отличным выбором, так как она обеспечивает высокую производительность и долговечность.
Cogovnzotly
| #
Отказное письмо https://www.rospromtest.ru/sertifikati/otkaznoe-pismo/ — документ, который подтверждает, что товару не требуется сертификат или декларация о соответствии. Отказное письмо оформляется на основании данных от заявителя без подачи заявки на сертификацию, эксперт на основании области применения продукции и ее кода ТН ВЭД определяет необходимость обязательной сертификации, о чем предоставляется ответ в письменном виде. Отказное письмо называют информационным письмом.
Willieavamy
| #
Online casinos help practice—nice try! mines game
Light582
| #
Эксклюзивное https://www.jamiechadwickracing.com/bilety-v-pushkinskij-muzej/ интервью с экспертом по путешествию.
where can i buy generic zebeta without a prescription
| #
can you buy generic zebeta without prescription
Master6850
| #
Советую проверенный ресурс по https://www.coachingparajovenes.com/russkij-muzei.html путешествию.
inernetadresCah
| #
подключение интернета по адресу
domashij-internet-krasnoyarsk003.ru
провайдеры интернета в красноярске по адресу проверить
Trefloi
| #
Приобрести диплом любого института. Заказ документа о высшем образовании через надежную фирму дарит ряд преимуществ для покупателя. Данное решение помогает сберечь как личное время, так и значительные средства. israelafrica.mn.co/posts/82618917
DonDonfaita
| #
darknet market dark web market list
betafudew
| #
Доверие – центр, который психологические услуги предоставляет. Мы помогаем людям справляться с трудностями, которые у них возникают в отношениях с собой и окружающими. Записаться на личный прием к специалисту можно по телефону, указанному на сайте. Ищете психотерапия? Ack-group.ru/stati-po-psikhologii/psikhoterapiya – здесь узнаете, что такое психотерапия, какие цели перед ней стоят и кому она показана. Также тут представлены расценки на психологические консультации. Готовы вас на пути к эмоциональному равновесию и самопознанию поддержать.
kepirdrind
| #
Если вы хотите быть в курсе самой актуальной, ценной и нужной информации относительно трейдинга и инвестиций, то скорее посетите этот полезный портал, на котором вы найдете все, что нужно на данную тему. На портале просто рассказывается то, как торговать на внебиржевом пространстве. https://fxidea.com/ предлагает акции всем начинающим. Быстрее изучите индикаторы, реальные отзывы, а также ознакомьтесь с работающими стратегиями. Вы получите доступ к ценным материалам, касающимся криптовалюты, а также узнаете, как заработать на форексе.
Sazrbgy
| #
Приобретение диплома ВУЗа через надежную компанию дарит массу преимуществ. Данное решение помогает сберечь как продолжительное время, так и серьезные финансовые средства. Тем не менее, на этом выгоды не ограничиваются, достоинств намного больше.Мы изготавливаем дипломы любой профессии. Дипломы изготавливаются на настоящих бланках государственного образца. Доступная цена по сравнению с большими тратами на обучение и проживание. Заказ диплома института станет рациональным шагом.
Заказать диплом о высшем образовании: diplomers.com/kupit-diplom-s-registratsiej-v-reestre-bistro-i-bezopasno/
Willieavamy
| #
Online casinos should demo—test out! F7 Casino
Willieavamy
| #
I hit a bonus online and won $40—sweet! tome of madness
GhostSpark
| #
https://workshop-summit.com/russkii-musei.html Советую качественный ресурс по путешествию.
Dnrtwzw
| #
Мы изготавливаем дипломы любой профессии по разумным тарифам. Мы предлагаем документы ВУЗов, которые находятся на территории всей России. Документы выпускаются на “правильной” бумаге высшего качества. Это позволяет делать государственные дипломы, которые невозможно отличить от оригинала. newsbeautiful.ru/kupit-diplom-prosto
Jamesdusty
| #
Ваши флористы – маги! Букет звучал, как симфония!
заказать цветы с доставкой в томске
mostbet_lkSl
| #
казино онлайн kg http://mostbet5003.ru .
1win_fvEl
| #
portofele electronice casino https://1win5025.ru .
Pingrutty
| #
darknet drug links darknet markets
skladi_ouSi
| #
временный склад временный склад .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Керамическая мишень для распыления оксида олова – 4N, Плоская форма
ThunderZ1
| #
Где брать актуальные https://sylvanaisthebest.com/razvlecheniya/ данные о путешествии?
where can i buy dipyridamole pill
| #
can you buy dipyridamole without prescription
dragon money_pmSn
| #
драгон http://dragon-money30.com .
dragon money_llOa
| #
кэшбэк на драгон мани http://dragon-money33.com/ .
FofebRof
| #
Посетите сайт https://schetki.spb.ru/ и вы сможете подобрать и купить машинку для чистки обуви. Ознакомьтесь с нашим ассортиментом машинок для чистки обуви – для дома или офиса, для чистки подошвы обуви, бахилонадевателями, комплектующими и другим ассортиментом. Также обслуживаем и предоставляем в аренду машины для чистки обуви и аппараты для надевания бахил. Доставляем по всем регионам России.
Donaldlaf
| #
darknet market links dark market 2025
1win_ndMt
| #
1win online http://1win5030.ru .
can i purchase generic clomid without rx
| #
Medication prescribing information. Effects of Drug Abuse.
can i purchase generic clomid without rx
Best information about drug. Read information now.
dragon money_vgKl
| #
тревожная кнопка охрана цена http://www.trevros.ru .
TigigNoics
| #
Автозапчасти купить в Минске или с доставкой по всей Беларуси на сайте https://proparts.by/ – это оригинальные и лицензионные автозапчасти в огромном ассортименте для всех марок автомобилей, подбор по марке авто и VIN коду, система скидок. Мы предлагаем несколько каталогов на выбор: для иномарок, для китайских и отечественных автомобилей. Все в наличии, в том числе широкий ассортимент расходников для ТО.
1win_knpr
| #
1win,com https://1win7004.ru .
WilliamDix
| #
Мы изготавливаем дипломы любых профессий по приятным тарифам. Дипломы производятся на настоящих бланках государственного образца Приобрести диплом любого ВУЗа kupitediplom.ru
AlphaV3
| #
https://www.orlandosaving.com/employer-resources/master-klassy-v-moskve Примеры успеха в путешествии.
Toliknaf
| #
dark web market links dark markets
Lazrsoq
| #
Где приобрести диплом специалиста?
Приобрести диплом института по доступной цене возможно, обратившись к проверенной специализированной фирме.: magazin-diplomov.ru
SteelVoyager
| #
Практические советы для работы с https://innovationplatformcapital.com/master-klassy путешествием.
Rabyitabe
| #
best darknet markets darknet drug market
Wolf_Pioneer
| #
Где учиться путешествию? https://kittyspit.net/masterklass-v-spb.html
1win_jeOr
| #
1win ставки официальный сайт https://1win7016.ru .
Jariorhdb
| #
Где купить диплом специалиста?
Мы предлагаем выгодно заказать диплом, который выполнен на оригинальном бланке и заверен печатями, водяными знаками, подписями должностных лиц. Данный диплом пройдет любые проверки, даже с применением профессионального оборудования. Решайте свои задачи быстро и просто с нашим сервисом.
Купить диплом любого ВУЗа diplomus-spb.ru/kupit-diplom-prepodavatelya-3/
Sazrdld
| #
Заказать диплом возможно через сайт компании. mkbox.ru/communication/forum/user/76817
Willieavamy
| #
Online slots grab me—those looks! plinko
Sazrcyj
| #
Купить диплом института!
Заказать диплом университета по невысокой стоимости возможно, обратившись к надежной специализированной компании. Приобрести диплом: diplomk-vo-vladivostoke.ru/kupit-diplom-s-zaneseniem-v-reestr-stoimost-9
Lokoyasneupt
| #
Check out our blog https://nashvillerelocationservices.com/ – it’s a valuable resource for all things relocating in the US. Whether you’re moving down the block or across the country, we give you practical, simple advice that really works. All articles follow a quick-answer format so you’ll actually get the knowledge you need.
Silver141
| #
Лучшие инструменты для работы с путешествием: https://ragdollresearch.org/masterklass-v-spb.html
Willieavamy
| #
I hit a bonus online and won $120—sweet yes! mines
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наши товары:
Мельхиоровая труба 50х2х4000 мм МНЖМц10-1-1 ГОСТ 10092-2006 Широкий выбор мельхиоровых труб различных размеров и форм. Прочные и устойчивые конструкции для строительства и изготовления металлических конструкций. Ознакомьтесь с нашим ассортиментом прямо сейчас!
mostbet_kmSt
| #
mostbet скачать на телефон бесплатно андроид https://www.mostbet5002.ru .
avto-vezu
| #
сколько стоит эвакуатор спб https://avto-vezu.ru/
DonDonfaita
| #
dark web link dark web market urls
Pingrutty
| #
darkmarket darknet market
Iron_Walker
| #
Какие технологии использовать для путешествия? https://abktechnologies.com/entertainment-in-moscow.html
dragon money_ndSn
| #
dragon money зеркало сегодня http://www.dragon-money30.com .
1win_blOr
| #
1 вин вход http://1win7016.ru .
1win_nnMt
| #
1вин https://1win5030.ru/ .
dragon money_asOa
| #
драгон http://www.dragon-money33.com .
1win_ioKi
| #
1win.pro 1win.pro .
Ghost_Code
| #
Интерактивный https://soupswap.com/master-classi/ курс по путешествию.
cost of cheap proscar without insurance
| #
can i get generic proscar pills
Donaldlaf
| #
darknet markets url dark web market
dragon money_vmKl
| #
тревожная кнопка охрана росгвардия тревожная кнопка охрана росгвардия .
Willieavamy
| #
The online ease is tricky—heads up! mines
Williamreeda
| #
Наш сервисный центр Bosch в Москве предлагает квалифицированный ремонт техники Bosch от ведущего немецкого бренда. Наши опытные мастера оперативно и надежно устранят поломки любой сложности вашей техники Bosch. Используем только оригинальные запчасти и проверенные методы для эффективного восстановления работоспособности вашей техники. Предлагаем выезд мастера на дом или ремонт в условиях стационарного центра. Доверьте заботу о вашей технике опытным специалистам. Заказать перевес дверей холодильника bosch можно прямо сейчас.
Willieavamy
| #
I lost big online—reset time! aviator
1win_cnOr
| #
1win скачать kg 1win7016.ru .
SteelHacker
| #
Лучшие инструменты для работы с путешествием: https://artscenewarehouse.com/master-classi/
inernetkrdCah
| #
узнать провайдера по адресу краснодар
domashij-internet-krasnodar001.ru
интернет провайдеры краснодар по адресу
how to get olmesartan pills
| #
Medicine information sheet. Drug Class.
how to get olmesartan pills
All about meds. Read here.
Trefzdd
| #
Заказать диплом о высшем образовании. Покупка документа о высшем образовании через надежную компанию дарит немало плюсов. Это решение позволяет сэкономить как личное время, так и значительные деньги. d6united.mn.co/posts/82618654
Toliknaf
| #
dark market 2025 onion dark website
Steel224
| #
Путешествие для стартапов: https://georgiaempowerment.org/master-klassy-v-spb.html
can i get floxin pill
| #
can you buy floxin prices
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их соответствие стандартам. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы по мере того как адаптировать решения под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Порошок кобальтовый Wallex 55 Порошок кобальтовый Wallex 55 – высококачественный РїСЂРѕРґСѓРєС‚, предназначенный для различных промышленных Рё научных применений. Ртот порошок обладает отличными свойствами, такими как высокая термостойкость Рё стабильность РїСЂРё работе СЃ различными материалами. РћРЅ идеально РїРѕРґС…РѕРґРёС‚ для использования РІ metallurgy, электронике Рё специальных сплавах.Если вас интересует надежность Рё производительность, вам стоит купить Порошок кобальтовый Wallex 55. Р’С‹ получите РїСЂРѕРґСѓРєС‚, который соответствует самым строгим стандартам качества Рё обеспечивает отличные результаты РІ каждом проекте. РќРµ упустите возможность улучшить СЃРІРѕРё производственные процессы!
dragon money_oaSn
| #
драгон мани вывод денег https://www.dragon-money30.com .
Rabyitabe
| #
best darknet markets dark web market urls
Willieavamy
| #
I love the payment options at online casinos—handy! plinko
1win_uvpr
| #
1 вин войти 1 вин войти .
Willieavamy
| #
Online casinos feel risky—double watch! aviator
Willieavamy
| #
Online casinos can pull—set edge! plinko
Walker8148
| #
Ответы на частые вопросы о путешествии: https://tncmusic.net/master-classi
dragon money_xiOa
| #
драгон мани официальный http://dragon-money33.com/ .
Willieavamy
| #
Online casinos should care—shield us! plinko
cikavtob
| #
Time1c.ru предлагает купить 1С. В нашей команде исключительно проверенные временем профессионалы. Они готовы проконсультировать вас. Вы можете протестировать решение 1С в облаке. Заполните форму прямо сейчас на нашем сайте. Укажите ваше имя, телефон для связи и e-mail адрес. Ищете ут демо? Time1c.ru – здесь можете ознакомиться с ценами. Также тут есть удобный поиск и блог. Расскажем, как 1С ERP управлять помогает ресурсами и проектами. Вы узнаете о преимуществах использования 1С Бухгалтерия для строительной отрасли. Настало время бизнес развивать!
Shadow_Byte
| #
Детальный анализ путешествия: http://gritandglimmer.com/seven-reasons-to-love-tyler-farrar/
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
Наши товары:
Кольцо РёР· драгоценных металлов платиновое 200С…30С…3.5 РјРј ПлН95.5 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
Mazrmbb
| #
Где заказать диплом специалиста?
Готовый диплом со всеми печатями и подписями отвечает требованиям и стандартам Министерства образования и науки, никто не сможет отличить его от оригинала – даже со специально предназначенным оборудованием. Не следует откладывать собственные мечты и задачи на потом, реализуйте их с нами – отправляйте быструю заявку на изготовление диплома уже сегодня! Получить диплом о среднем специальном образовании – запросто! forum-ahangaran2007.flybb.ru/viewtopic.phpf=5&t=1208
Timothy_Rep
| #
Наша команда занимается профессиональным демонтажем загородных домов любого типа. Мы быстро и безопасно сносим строения из дерева, кирпича, панелей и каркаса, независимо от сложности работ. Все этапы выполняем в строгом соответствии с действующими нормами, организуем вывоз строительного мусора и готовим участок для последующего строительства. Работаем как с применением спецтехники, так и вручную: демонтаж домов цена
DonDonfaita
| #
tor drug market darknet markets links
MiguelJaw
| #
https://blog-cheeses.ru/
dragon money_fdKl
| #
установка тревожной кнопки росгвардия установка тревожной кнопки росгвардия .
Alfredomice
| #
Создай идеальный взгляд! Наращивание ресниц Ивантеевка с учётом формы глаз и пожеланий клиента. Работаем с проверенными материалами, соблюдаем стерильность и комфорт.
Dark704
| #
Конкретные шаги для работы с путешествием: http://roeliepost.com/2013/08/in-my-fight-against-the-trade-in-children-i-will-not-bow-to-intimidation/
ThunderX7
| #
http://www.life-renewal.org/product/hot-chocolate-and-handmade-cakes-gift-box/ Секретные методы путешествия.
Donaldlaf
| #
darknet markets url dark markets 2025
Sazrmkg
| #
Купить диплом под заказ в столице вы можете используя официальный портал компании. premium.listbb.ru/viewtopic.phpf=2&t=1259
Willieavamy
| #
Online casinos need better limit tools. fortune mouse
Walterwromi
| #
женские сайты
Iron_Master
| #
Тесты по путешествию: https://walnutridgefd.com/neobychnye-razvlecheniya-v-spb.html
Toliknaf
| #
dark market darknet drug store
skladi_nuSi
| #
индивидуальное хранение вещей в москве недорого индивидуальное хранение вещей в москве недорого .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наши товары:
Тигель высокочистого молибдена
Dark423
| #
Обнаружил, что по путешествию лучше всего https://crististefan.com/free-30-minute-consult/ читать здесь.
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их происхождение. Опытная поддержка – наш стандарт – мы на связи, чтобы разрешать ваши вопросы по мере того как находить ответы под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
поставляемая продукция:
Р’РёСЃРјСѓС‚ C 6804 – JIS H 3250:2015 Р’РёСЃРјСѓС‚ C 6804 – JIS H 3250:2015 представляет СЃРѕР±РѕР№ высококачественный материал, соответствующий современным стандартам. Ртот сплав обладает уникальными свойствами, такими как высокая коррозионная стойкость Рё отличная мажущая способность, что делает его идеальным выбором для различных промышленных применений.Приобретая Р’РёСЃРјСѓС‚ C 6804 – JIS H 3250:2015, РІС‹ получаете надежный РїСЂРѕРґСѓРєС‚, который прослужит долго Рё обеспечит высокую эффективность РІ работе. РњС‹ гарантируем высокое качество Рё доступные цены. РќРµ упустите возможность купить Р’РёСЃРјСѓС‚ C 6804 – JIS H 3250:2015 Рё улучшить производственные процессы.
Willieavamy
| #
The sounds online grab—can’t stop! aviator
Willieavamy
| #
The payout waits online lag—push it! fortune tiger
Trefmij
| #
Приобрести диплом о высшем образовании. Покупка документа о высшем образовании через качественную и надежную компанию дарит ряд достоинств для покупателя. Данное решение позволяет сберечь время и значительные деньги. pika.listbb.ru/posting.php?mode=post&f=9
Rabyitabe
| #
darkmarket url darknet markets url
can you get xenical pill
| #
Medication information leaflet. What side effects can this medication cause?
can you get xenical pill
Best about medicine. Get information here.
Code4736
| #
Профессионалы рекомендуют эти материалы по http://www.derbycityprints.com/category/relaxing-vacation-at-a-relaxed-price/ путешествию.
Garry_hiz
| #
Наша команда занимается профессиональным демонтажем загородных домов любого типа. Мы быстро и безопасно сносим строения из дерева, кирпича, панелей и каркаса, независимо от сложности работ. Все этапы выполняются в строгом соответствии с действующими нормами. Организуем вывоз мусора и полностью готовим участок для последующего строительства. Работаем как с применением спецтехники, так и вручную: снос дома
Willieavamy
| #
Online casinos should show—odds up! plinko
mostbet_cuml
| #
мостбет скачать андроид https://mostbet6043.ru .
JasonWrami
| #
Как только заходишь — ощущение, будто ты в кабине, к прыжку в гиперпространство. Всё по курсу: меню лаконичный.
Игры стартуют мгновенно. Игровые автоматы на деньги — это не просто игровая зона, а ядро миссии, где каждый ставка — телепорт.
Результаты — в один момент, ровно.
Willieavamy
| #
I love the online calm—no noise! plinko
1win_dfMt
| #
1winn http://1win5030.ru .
Eagle267
| #
Преодоление http://www.thefonestuff.com/gary-vasquez-bio-aka-gangsta/ трудностей при изучении путешествия.
luck
| #
Cadastre-se no luck e Ganhe 100$ de Bônus de Boas-Vindas!
buying starlix without insurance
| #
where to get generic starlix without prescription
DonDonfaita
| #
dark market link bitcoin dark web
Byte9075
| #
Может кто знает http://www.norsadfinance.com/rants/rant118.htm разбирается в путешествии? Куда зайти?
Dnrtoab
| #
Мы предлагаем дипломы любой профессии по выгодным тарифам. Мы предлагаем документы техникумов, расположенных в любом регионе Российской Федерации. Документы выпускаются на бумаге самого высокого качества. Это позволяет делать государственные дипломы, которые невозможно отличить от оригиналов. lider036.5nx.ru/viewtopic.phpf=26&t=2256
inernetkrdCah
| #
интернет провайдеры по адресу краснодар
domashij-internet-krasnodar002.ru
подключить интернет по адресу
mostbet_tpml
| #
mostbet kg скачать http://mostbet6043.ru .
Donaldlaf
| #
darkmarket link darknet marketplace
CyberSentinel
| #
Чек-лист для работы с https://tncmusic.net/na-korablike путешествием.
Sazrrnr
| #
Приобрести документ о получении высшего образования вы можете в нашей компании. Мы оказываем услуги по изготовлению и продаже документов об окончании любых университетов России. Вы сможете получить необходимый диплом по любым специальностям, любого года выпуска, в том числе документы Советского Союза. Даем гарантию, что в случае проверки документа работодателями, подозрений не появится. freediplom.com/kupit-diplom-texnikuma-s-zaneseniem-v-reestr-otzivi-7/
betsury
| #
O betsury oferece uma excelente oportunidade
para quem deseja começar sua experiência no cassino online com um bônus de
100$ para novos jogadores! Ao se registrar no site, você garante esse
bônus exclusivo que pode ser utilizado em diversos jogos de cassino, como slots, roleta e
poker. Esse é o momento perfeito para explorar o mundo das apostas com um saldo
extra, aproveitando ao máximo suas apostas sem precisar investir um grande valor logo de início.
Não perca essa oportunidade e cadastre-se já!
Diplomi_saKt
| #
Заказать диплом университета!
Мы готовы предложить документы любых учебных заведений, которые расположены на территории всей России.
good-diplom.ru/realno-li-kupit-diplom-s-reestrom-2/
PhantomGuardian
| #
Что нельзя делать https://www.godsavethecuisine.com/osobnyak-brusnitsynyh.htm с путешествием?
Toliknaf
| #
dark market 2025 darkmarket list
mostbet_wuml
| #
мостбет скачать андроид https://mostbet6043.ru/ .
Willieavamy
| #
I lost time online—fast gone! plinko
Sazrzwl
| #
Приобрести диплом возможно через сайт компании. westorebd.com/employer/frees-diplom
Alpha915
| #
https://www.6ixcycle.com/odnodnevnye-po-moskve.html Список инструментов по путешествию.
Rabyitabe
| #
dark web market links darknet links
StevenGaf
| #
список дел Планирование: Путь к Успеху, Гармонии и Свободе от Выгорания В бешеном ритме современной жизни, где каждый день бросает нам вызов, планирование становится не просто полезным навыком, а жизненной необходимостью. Это компас, указывающий верное направление среди бескрайнего моря возможностей и задач. Планирование – это искусство управления своим временем, своей энергией и, в конечном итоге, своей жизнью.
fixitcaree
| #
Сервисный центр ФикситЦентр – выполнит срочный ремонт радиосистем в Москве
Аренда авто Краснодар
| #
Аренда авто Краснодар
Willieavamy
| #
The visuals in online casino games are stunning! sugar rush
BlueX2
| #
Как http://www.wilwegman.com/1-General-Fishing.html сэкономить на путешествии.
JamesKen
| #
Jon Jones profile john jones ufc a fighter with a unique style, record-breaking achievements and UFC legend status. All titles, weight classes and the path to the top of MMA – on one page.
YuwiyEnuts
| #
На сайте https://xn—-7sbjhqn0bhjc0lk.xn--p1ai/ получите юридическую консультацию по различным вопросам. Компания Стратегия работает как с физическими, так и частными лицами. На все услуги установлены привлекательные расценки, чтобы воспользоваться ими смог каждый. Предприятие работает в этой сфере более 11 лет. В команде трудятся 11 специалистов, которые справятся с задачей независимо от сложности. К вашим услугам досудебный юрист, досудебное урегулирование, кредитный, семейный юрист, решение страховых споров.
Rockylet
| #
спарк ру Новости азартного мира: от Спарк-Ру до онлайн-казино Мир азартных игр находится в постоянном движении, и чтобы оставаться в курсе последних событий, необходимо следить за ключевыми источниками информации. Одним из таких источников является Спарк-Ру – база данных, содержащая информацию о юридических лицах, включая те, что связаны с игорным бизнесом. Анализ данных Спарк позволяет отслеживать финансовые показатели казино, изменения в структуре собственности и другие важные аспекты деятельности.
CegohsSit
| #
Центр ортодонтии и эстетической стоматологии в Москве https://ortodont-msk.com/ это квалифицированные услуги от врачей ортодонтов высшей категории, которые занимают первое место в рейтинге врачей ортодонтов в Москве. Посетите наш сайт, ознакомьтесь с нашими услугами, такими как исправление прикуса взрослым и детям, нашими брекет системами и другими услугами, галереей работ.
Willieavamy
| #
The mobile casino setup is rough—improve! plinko
KennethPob
| #
Онлайн-казино JoyCasino thejoycasino-ru яркий дизайн, популярные слоты, live-игры и щедрые бонусы. Простой вход, быстрые выплаты и надёжная поддержка. Играйте с комфортом и без лишних рисков.
leptozan
| #
leptozan is an innovative weight loss formula crafted to help you naturally sculpt the body you’ve always desired.
Willieavamy
| #
Online bingo flows—great crew! jackpotraider
can i buy cheap seroquel tablets
| #
Pills prescribing information. Cautions.
can i buy cheap seroquel tablets
Some what you want to know about pills. Get information here.
DonDonfaita
| #
dark web drug marketplace darknet market links
mitolyn
| #
mitolyn is a natural dietary supplement specifically outlined to enhance metabolism and support weight loss.
Willieavamy
| #
I wish online helped new—tough kick! craps
Darylkip
| #
?fsan?vi bokscu Mayk Tayson mike-tyson ag?r c?kid? s?ksiz cempion v? tarixin ?n tan?nm?s idmanc?lar?ndan biridir. Gucu, sur?ti v? xarizmas? onu boks simvoluna cevirdi.
RakijamPreap
| #
Посетите B2BOX https://b2box.ru/ – это уникальное комплексное решение для создания упаковки всех видов и направлений. Мы делаем упаковку, которая продает! Упаковочные решения для ритейла, создание индивидуальной и SRP упаковки, услуги дизайнерского бюро, производство транспортной упаковки, собственное плоттерное производство, все виды печати – это и многое другое вы найдете, посетив наш сайт.
Thomaspoext
| #
Подсказали, как продлить жизнь букету!
цветы томск
Davidsem
| #
Professional fighter Rafael Fiziev rafael-fiziev.com/ is a UFC star known for his explosive technique and spectacular fights. A lightweight fighter with a powerful punch and a strong Muay Thai base.
VortexX2
| #
http://zaiprotiv.info/novosti/3127-nazvany-prichiny-razliva-nefti-v-meksikanskom-zalive Эволюция развития путешествия.
mostbet_rnmt
| #
мостбет скачать http://mostbet6033.ru .
Donaldlaf
| #
darknet market lists darkmarket 2025
1win_bhMt
| #
баланс 1win https://1win5030.ru/ .
Phantom_Byte
| #
Оптимизация процессов через путешествие: https://printgrowstrees.com/na-teplohode-s-uzhinom/
where buy generic septra without a prescription
| #
can you get cheap septra without rx
jogodeouro
| #
Cadastre-se no jogodeouro e Ganhe 100$ para Apostar!
1win_gxOr
| #
1win партнерка вход http://1win7016.ru .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
Наша продукция:
Проволока вольфрамовая 1300 мм ВРН ГОСТ 19671-91 Купить вольфрамовую проволоку у производителя. Широкий выбор диаметров и нарезка по размеру. Высокая теплопроводность и стойкость к коррозии. Доставка по России.
Willieavamy
| #
I love the online promos—fresh take! Fortune Dragon
Toliknaf
| #
darkmarket darknet drug market
Hacker9534
| #
Предлагаю подискутировать о путешествии: https://www.teachersassistantpro.com/about-the-author.html.
TravisCauby
| #
https://dzen.ru/77volvo.ru Премиальное Обслуживание Lixiang, Volvo и Land Rover в Москве В динамичном ритме столичной жизни ваш автомобиль – это не просто средство передвижения, это важная часть вашего комфорта и имиджа. Осознавая это, наш сервисный центр предлагает полный спектр услуг для владельцев автомобилей премиум-класса: Lixiang, Volvo и Land Rover. Мы специализируемся на поддержании безупречного состояния этих марок, обеспечивая надежность и долговечность вашего авто.
Willieavamy
| #
The table games online spark—class act! sugar rush
1win_ykEl
| #
1win pariuri 1win pariuri .
Rabyitabe
| #
dark web drug marketplace onion dark website
Ghost_Hacker
| #
http://www.portrushseatours.com/tours/view/rathlin.html Как сэкономить на путешествии.
Willieavamy
| #
The payout waits online lag—push it! razor returns
Xazrakd
| #
Купить диплом института!
Мы можем предложить дипломы любых профессий по приятным ценам. Вы приобретаете диплом в надежной и проверенной компании. : kontorka.flybb.ru/viewtopic.php?f=9&t=2522
Williamemifs
| #
https://assa0.myqip.ru/?1-1-0-00002581-000-0-0-1742429880
mostbet_hnpl
| #
мостбет официальный сайт mostbet6038.ru .
fixitcaree
| #
Сервисный центр ФикситЦентр – выполнит срочный ремонт экшен-камер в Москве
imperador bet
| #
Comece no imperador bet com 100$ de Bônus de Boas-Vindas!
Willieavamy
| #
Online casinos should curb—care play! fortune tiger
Willieavamy
| #
I won spins online and got $80—sweet! plinko
inernetkrdCah
| #
интернет по адресу
domashij-internet-krasnodar003.ru
интернет по адресу
mostbet_rkmt
| #
мостюет http://www.mostbet6033.ru .
Shannontes
| #
elonbet
VortexZ1
| #
Мне самому помог этот ресурс по путешествию: http://www.thediv.org/
Willieavamy
| #
Online casinos help practice—good spot! dragon hatch
DonDonfaita
| #
darknet market links darknet market list
Iariorund
| #
Заказать диплом института по выгодной стоимости вы сможете, обратившись к надежной специализированной фирме. Купить документ ВУЗа вы можете в нашем сервисе. good-diplom.ru/kupit-diplom-s-vneseniem-v-reestr-bistro-i-udobno-3
Lazrgsj
| #
Быстро и просто заказать диплом ВУЗа!
Мы предлагаем дипломы любой профессии по приятным тарифам— diplom-zentr.com/kupite-diplom-o-meditsinskom-obrazovanii-legalno/
Willieavamy
| #
I won big online—wild joy! plinko
Shannontes
| #
elonbet
SilverSlayer
| #
http://civilhomelanddefense.us/tag/genesis/ Преодоление трудностей при изучении путешествия.
Lazrmri
| #
Диплом университета Российской Федерации!
Без присутствия диплома очень нелегко было продвинуться по карьерной лестнице. Поэтому решение о заказе диплома можно считать целесообразным. Приобрести диплом ВУЗа l90226mw.beget.tech/2025/03/24/zakazhite-diplom-s-vneseniem-v-reestr.html
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их качество. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы а также находить ответы под требования вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
Проволока магниевая M10501 – UNS Полоса магниевая M10501 – UNS – это высококачественный РїСЂРѕРґСѓРєС‚, изготовленный РёР· магния, который обладает отличными механическими свойствами Рё РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью. Ртот материал широко используется РІ aerospace, автомобилестроении Рё РґСЂСѓРіРёС… отраслях, РіРґРµ требуется высокая прочность Рё легкость. Если РІС‹ ищете надежный Рё прочный материал, то вам стоит купить Полоса магниевая M10501 – UNS. Благодаря СЃРІРѕРёРј уникальным характеристикам, эта полоса идеально РїРѕРґС…РѕРґРёС‚ для изготовления различных компонентов Рё конструкций. Заказывая Сѓ нас, РІС‹ получаете качественный РїСЂРѕРґСѓРєС‚ СЃ быстрой доставкой.
exploring the dual faces of diclofenac unveiling the diverse roles of voltaren cataflam
| #
Medicines information sheet. Long-Term Effects.
exploring the dual faces of diclofenac unveiling the diverse roles of voltaren cataflam
Some what you want to know about drugs. Read information now.
1win_hopr
| #
1 вин вход в личный кабинет 1 вин вход в личный кабинет .
realbet
| #
Aposte no realbet – https://realbet-br.com: Cadastre-se e Receba
100$ de Bônus Imediato!
ShadowZ1
| #
Базовые принципы https://brimfieldchamber.com/2014/05/the-problem-of-cheap-law.html путешествия для новичков.
1win_ttpr
| #
1вин кыргызстан 1вин кыргызстан .
Donaldlaf
| #
onion dark website darknet sites
CyberMaster
| #
Инфографика по путешествию: https://www.scotchcinema.com/a-culture-of-no-sleep.html
Mazrwgj
| #
Купить диплом университета!
Мы предлагаем документы об окончании любых университетов Российской Федерации. Документы изготавливаются на фирменных бланках государственного образца. usa.life/read-blog/133381_kupit-diplom-ob-obrazovanii-zanesennyj-v-reestr.html
Toliknaf
| #
darknet market lists darkmarket url
mostbet_lgmt
| #
мостбет казино войти https://mostbet6033.ru/ .
mostbet_czsa
| #
mostbet casino https://mostbet5004.ru/ .
Jariorjcb
| #
Где приобрести диплом специалиста?
Наши специалисты предлагают быстро и выгодно заказать диплом, который выполнен на оригинальной бумаге и заверен печатями, водяными знаками, подписями должностных лиц. Наш документ способен пройти любые проверки, даже при использовании профессионального оборудования. Решайте свои задачи быстро с нашей компанией.
Заказать диплом любого ВУЗа diplomskiy.com/kupit-diplom-v-voronezhe-11/
Rabyitabe
| #
darknet marketplace darkmarket link
Willieavamy
| #
The live dealers online rock—so real! aviator
1win_mcEl
| #
descărca 1win 1win5025.ru .
Аренда авто Краснодар
| #
Аренда авто Краснодар
can you get cheap glucotrol prices
| #
how can i get generic glucotrol without a prescription
DonDonfaita
| #
darkmarket link darknet market list
Braceface
| #
Braceface
GeorgeCow
| #
King of the ring https://roy-jones.com Roy Jones is a fighter with a unique style and lightning-fast reactions. His career includes dozens of titles, spectacular knockouts and a cult status in the boxing world.
KennethUnild
| #
Football genius https://luka-modric.com Luka Modric – from humble beginnings in Zagreb to a world-class star. His path inspires, his play amazes. The story of a great master in detail.
Shannontes
| #
elonbet
Walterfobby
| #
Свежие новости https://actualnews.kyiv.ua Украины и мира онлайн: события, аналитика, интервью и факты. Будьте в курсе главного — обновления 24/7, объективная подача и лента новостей в реальном времени.
Jeraldler
| #
Автопортал для водителей https://addinfo.com.ua и автолюбителей: свежие авто новости, сравнения моделей, рейтинги, советы по выбору и обслуживанию автомобилей. Полезная информация каждый день.
1win_kamn
| #
1win betting https://1win17.com.ng .
fixitcaree
| #
Сервисный центр ФикситЦентр – выполнит срочный ремонт ручных массажеров в Москве
Cazrxji
| #
Заказать диплом о высшем образовании!
Мы готовы предложить документы учебных заведений, расположенных в любом регионе РФ. Документы выпускаются на “правильной” бумаге высшего качества: vagyonor.hu/employer/aurus-diploms
trevojnaya knopka rosgvardii_hppn
| #
установить тревожную кнопку росгвардии установить тревожную кнопку росгвардии .
Create a free account
| #
Your point of view caught my eye and was very interesting. Thanks. I have a question for you.
1win_zumn
| #
1win casino http://www.1win17.com.ng .
mostbet_wpml
| #
мостбет промокод http://mostbet6043.ru .
seroquel and general anaesthetic
| #
Medicines information for patients. What side effects can this medication cause?
seroquel and general anaesthetic
Some about medicament. Get information now.
Sazrktt
| #
Купить диплом института !
Покупка диплома ВУЗа России в нашей компании – надежный процесс, поскольку документ будет заноситься в государственный реестр. Приобрести диплом об образовании diplom-club.com/kupit-diplom-s-reestrom-tsena-7
Cazrzfo
| #
Привет!
Мы готовы предложить дипломы любой профессии по приятным тарифам. Стоимость будет зависеть от той или иной специальности, года получения и ВУЗа: rdiplomans.com/
premier bet
| #
premier bet Oferece 100$ de Bônus para Novos Jogadores – Registre-se Já!
HebohnWerse
| #
Go to the site https://betwave.ro/en where you will find a list of all the best online casinos, as well as useful and complete information about each of them – how to play, what bonuses there are, how to withdraw money, which casinos have a minimum deposit, registration steps and advantages of each of them, as well as what are the biggest bonuses in online casinos. More details on the site.
Willieavamy
| #
I wish online casinos had better tutorials for beginners! legacy of dead
inernetsamaraCah
| #
подключение интернета по адресу
domashij-internet-samara001.ru
узнать провайдера по адресу самара
Willieavamy
| #
I hit a progressive jackpot online once—life-changing moment! Gioco Del Pollo
Willieavamy
| #
The slot themes online glow—play on! sugar rush
Lazrlot
| #
Заказать диплом ВУЗа!
Мы предлагаем дипломы любых профессий по разумным ценам— kupitediplom0027.ru/diplom-9-klassov-kupit/
Donaldlaf
| #
darkmarket darknet sites
Jariorlij
| #
Где приобрести диплом специалиста?
Мы предлагаембыстро купить диплом, который выполнен на оригинальной бумаге и заверен печатями, водяными знаками, подписями должностных лиц. Наш документ способен пройти лубую проверку, даже при использовании специально предназначенного оборудования. Достигайте свои цели быстро и просто с нашими дипломами. Купить диплом любого ВУЗа! telegra.ph/Kupit-diplom–prosto-i-bystro-03-13
Shannontes
| #
elonbet
Pingrutty
| #
dark market url darkmarket url
Willieavamy
| #
I won a tournament online—bragging rights for days! betonred
Keithclade
| #
купить баню из бруса под ключ недорого Строительство бани под ключ в Ленинградской области: цены, проекты от производителя. Мечтаете о собственной бане на даче или загородном участке в Ленинградской области? Строительная компания бани-бани предлагает профессиональное строительство бань в СПб по выгодным ценам. Купить баню без предоплаты!
casinos sin licencia en espana_ddpl
| #
casino online sin licencia casino online sin licencia .
Toliknaf
| #
dark web market urls onion dark website
Stevenhaxia
| #
Женский журнал https://asprofrutsc.org о стиле, красоте, психологии, отношениях и саморазвитии. Актуальные статьи, советы экспертов, тренды и вдохновение — всё для современной женщины.
WilliamFlali
| #
Онлайн-журнал для женщин https://chernogolovka.net всё о жизни, любви, красоте, детях, финансах и личностном развитии. Простым языком о важном — полезно, интересно и по делу.
Frankmib
| #
Автомобильный портал https://avto-limo.zt.ua для тех, кто за рулём: автообзоры, полезные советы, новости индустрии и подбор авто. Удобный поиск, свежая информация и всё, что нужно автолюбителю.
Michaelgow
| #
Авто журнал онлайн https://clothes-outletstore.com всё о мире автомобилей: новости, тест-драйвы, обзоры, советы, новинки автопрома и технологии. Читайте с любого устройства — всегда в курсе автоиндустрии.
trevojnaya knopka rosgvardii_ivpn
| #
тревожная кнопка мвд тревожная кнопка мвд .
can you buy cymbalta without rx
| #
can i purchase cymbalta online
batbet
| #
O batbet oferece uma excelente oportunidade para quem deseja começar sua experiência no cassino
online com um bônus de 100$ para novos jogadores! Ao se registrar no site, você garante esse bônus exclusivo que pode ser utilizado em diversos jogos de cassino, como slots, roleta e poker.
Esse é o momento perfeito para explorar o mundo das apostas com
um saldo extra, aproveitando ao máximo suas apostas sem precisar investir um grande valor logo de início.
Não perca essa oportunidade e cadastre-se já!
Rabyitabe
| #
darknet market lists darkmarket
solicitor
| #
Fine material, Thank you.
Davidguicy
| #
Hi, what is your hobby? what do you do in spare time? personally love to play pokies
Shannontes
| #
elonbet
Edwardmoope
| #
https://hm-notes.ru/kak-sohranit-zhenskoe-zdorove-sidya-za-rukodeliem/
cost of cheap elavil without prescription
| #
Meds information sheet. Cautions.
cost of cheap elavil without prescription
Everything information about medicine. Read information now.
Willieavamy
| #
Online casinos are perfect for strategy practice! plinko
Kelvinwooff
| #
проект перепланировки цена
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние достижения.
Наша продукция:
Керамическая мишень для распыления оксида олова
DonDonfaita
| #
darknet market dark markets 2025
Shannontes
| #
elonbet
apostebet
| #
apostebet Oferece 100$ de
Bônus para Novos Jogadores – Registre-se Já!
Willieavamy
| #
I lost $100 in an online casino—lesson learned, set a budget! fruit cocktail
razrabotka saita na bitriks_ajKt
| #
разработка битрикс стоимость razrabotka-saita-bx.ru .
Netawrfum
| #
Ищете сельхозтехнику и оборудование по самым выгодным ценам от производителя? Посетите сайт Производителя ООО МИТЕХ https://mi-teh.ru/ где вы также найдете запасные части и комплектующие к сельхоз машинам и тракторам. Ознакомьтесь с нашим ассортиментом продукции на сайте.
https://russiancouncil.ru/petr-aven/?str=Е
| #
https://russiancouncil.ru/petr-aven/?str=Е
Edwardmoope
| #
https://www.consmed.ru/news/view/2658/
trevojnaya knopka rosgvardii_wtpn
| #
тревожная кнопка цена http://trevros.ru/ .
Shannontes
| #
elonbet
Pingrutty
| #
dark web sites darknet markets onion
Аренда авто Краснодар
| #
Аренда авто Краснодар
Diplomi_vzKt
| #
Купить диплом о высшем образовании!
Мы можем предложить документы учебных заведений, которые находятся в любом регионе РФ.
freediplom.com/kupit-diplom-instituta-s-reestrom-2/
Sazrfge
| #
Всегда думал что купить диплом о высшем образовании это миф и нереально, но все оказалось не так, изначально искал информацию про: купить диплом менеджера по продажам, купить диплом молдова, купить диплом невролога, купить диплом нотариуса, купить диплом о высшем образовании московский государственный, потом про дипломы вузов, подробнее здесь diplomybox.com/attestat-9-klassov
Donaldlaf
| #
darknet markets onion address darkmarket list
sex địt máy bay VN
| #
sex địt máy bay VN
A Brief History of Elemental Analysis » Artemis Analytical
Lazrsmz
| #
Приобрести диплом любого ВУЗа!
Мы изготавливаем дипломы психологов, юристов, экономистов и прочих профессий по приятным ценам— diplom-ryssia.com/kupit-attestat-11-klassa-bistro-i-nadezhno-2/
Toliknaf
| #
darknet markets bitcoin dark web
Iariorpkt
| #
Купить диплом ВУЗа по выгодной цене вы можете, обращаясь к проверенной специализированной фирме. Купить документ о получении высшего образования вы имеете возможность в нашей компании. diplomus-spb.ru/kupit-diplom-provedennij-cherez-reestr-5
Shannontes
| #
elonbet
inernetsamaraCah
| #
провайдер интернета по адресу самара
domashij-internet-samara002.ru
провайдер по адресу
mua bán nội tạng trẻ em
| #
mua bán nội tạng trẻ em
A Brief History of Elemental Analysis » Artemis Analytical
Rabyitabe
| #
dark web link darkmarket 2025
razrabotka saita na bitriks_mcKt
| #
разработка сайта на 1с битрикс цена https://www.razrabotka-saita-bx.ru .
where buy generic mobic for sale
| #
how can i get generic mobic without dr prescription
Willieavamy
| #
I love the live roulette—real deal! gransino
site
| #
You actually said that superbly.
CadecenPaund
| #
На сайте https://arenda24.by/ каждый желающий получает возможность взять в прокат спецтехнику, садовый инвентарь для выполнения всех необходимых хозяйственных работ. Если нашли подходящее объявление, то свяжитесь и договоритесь с продавцом для того, чтобы он на выгодных условиях передал технику. Напротив каждого варианта имеются его характеристики, особенности, детальное описание, чтобы потенциальный клиент сразу же сориентировался. Постоянно появляются новые объявления, что позволит найти все, что нужно.
azithromycin hpv
| #
Drug information for patients. Long-Term Effects.
azithromycin hpv
Some trends of drugs. Read now.
Pingrutty
| #
darknet links darknet sites
Willieavamy
| #
I lost big online—reboot now! book of dead
Willieavamy
| #
The live aid online saves—big aid! slot gallina
legal practice
| #
Wonderful postings, Thanks.
casinos sin licencia en espana_gipl
| #
casinos sin licencia espa?a casinos sin licencia espa?a .
Shannontes
| #
elonbet
Donaldlaf
| #
dark market url bitcoin dark web
Normandeary
| #
заборы В современном строительстве выбор материалов играет ключевую роль в долговечности и эстетическом виде здания. Кровельные материалы и фасадные материалы формируют не только внешний облик, но и обеспечивают защиту от атмосферных воздействий.
mostbet_tqmt
| #
mostbet.kg https://mostbet6033.ru .
Toliknaf
| #
dark market link darkmarket link
codbet
| #
Bônus de 100$ para Novos Jogadores no codbet – Cadastre-se Agora!
site
| #
Position nicely used..
Cazrktt
| #
Заказать диплом университета можем помочь. Купить диплом техникума, колледжа в Кургане – diplomybox.com/kupit-diplom-tekhnikuma-kolledzha-v-kurgane
mostbet_fxsa
| #
мостбет скачать казино http://mostbet5004.ru/ .
site
| #
You said it very well.!
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наши товары:
Лист судостроительный 12x1500x4000 D EN10025:3 Приобретите высококачественные листы судостроительные от компании Редметсплав.рф для надежного и долговечного использования в вашем судостроительном проекте. Различные размеры и толщины, прочность и устойчивость к коррозии делают эти листы идеальным выбором для любого судостроительного проекта.
mostbet_ozpl
| #
mostbet casino https://mostbet6038.ru/ .
Cazrvht
| #
Мы можем предложить дипломы любой профессии по выгодным ценам. Купить диплом в городе Ковров — kyc-diplom.com/geography/kovrov.html
dragon money_rxOa
| #
драгон мани официальный сайт регистрация http://dragon-money33.com/ .
Rabyitabe
| #
dark market onion dark web link
Машины страшилки
| #
Машины страшилки
1win_myEl
| #
1 win md https://1win5025.ru/ .
Pingrutty
| #
dark web marketplaces darknet market lists
Shannontes
| #
elonbet
Pixunlipglice
| #
Система менеджмента управления качеством содержит критерии менеджмента и стандарты, по которым проводится последующая сертификация предприятия. Чтобы получить сертификат СМК, нужно обратиться в аккредитованный сертификационный центр. Сертификация систем менеджмента качества является документальным подтверждением того, что система менеджмента сертифицированной компании соответствует требованиям того или иного стандарта. Подробнее: https://ok.ru/group/70000034956977
Willieavamy
| #
The poker online shines—real kick! aviator
mostbet_dbsa
| #
поддержка мостбет http://www.mostbet5004.ru .
Willieavamy
| #
Online gambling can fly—hold back! dragon tiger
Willieavamy
| #
The online bonuses shine—jump in! mines
Shannontes
| #
elonbet
Willieavamy
| #
Online casinos should give more demos. betonred
DonDonfaita
| #
darkmarket darknet drug market
can i get cheap isordil without insurance
| #
buying generic isordil online
dragon money_rqSn
| #
драган мани http://www.dragon-money30.com .
inernetsamaraCah
| #
провайдер интернета по адресу самара
domashij-internet-samara003.ru
проверить провайдера по адресу
winbra
| #
Aposte com 100$ de Bônus no winbra – Cadastre-se e Aproveite!
GisucphReofs
| #
Use our online currency calculator to find https://how-much-is.com/ out the exchange rate for dollars, euros, British Pounds, Russian rubles and other currencies.
neurontin is the brand name for
| #
Medicine prescribing information. What side effects can this medication cause?
neurontin is the brand name for
Best about medicine. Read now.
Аренда авто Краснодар
| #
Аренда авто Краснодар
Shannontes
| #
elonbet
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их происхождение. Превосходное обслуживание – наш стандарт – мы на связи, чтобы разрешать ваши вопросы по мере того как адаптировать решения под требования вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
Лист магниевый M12300 – UNS Лента магниевая M12300 – UNS представляет СЃРѕР±РѕР№ высококачественный РїСЂРѕРґСѓРєС‚, разработанный для удовлетворения потребностей современных технологий. Рта магниевая лента идеально РїРѕРґС…РѕРґРёС‚ для применения РІ различных отраслях, включая авиацию, автомобильный сектор Рё электронику. Благодаря своей легкости Рё прочности, лента обеспечивает надежную защиту Рё эффективность. Купить Лента магниевая M12300 – UNS можно для улучшения производственных процессов Рё повышения общей надежности изделий. Заказывайте сейчас, чтобы РЅРµ упустить возможность использовать инновационный материал для СЃРІРѕРёС… проектов.
Mazrgyw
| #
Приобрести диплом любого ВУЗа!
Мы предлагаем документы об окончании любых университетов России. Документы изготавливаются на оригинальных бланках государственного образца. twixxor.com/read-blog/18168_vysshee-obrazovanie-kupit-diplom-s-zaneseniem.html
Donaldlaf
| #
darknet drug store dark web drug marketplace
Toliknaf
| #
darkmarkets darknet drug market
Willieavamy
| #
I lost big online—reboot now! vincispin
Willieavamy
| #
I wish online casinos had lower stakes! mines
Pingrutty
| #
dark web market links best darknet markets
Shannontes
| #
elonbet
mostbet_mgmt
| #
motsbet https://mostbet6033.ru/ .
Rabyitabe
| #
darknet markets url dark market 2025
Willieavamy
| #
Online gambling is too accessible—needs control! mines
Willieavamy
| #
The poker graphics online are next-level—wow! tom of madness
hojisCop
| #
Одно из существенных преимуществ Апостолиди – возможность предоставлять необходимую информацию посетителям. Мы понятным языком сложные финансовые вопросы разъясняем. Благодаря регулярным обновлениям вы всегда будете в курсе важных событий. https://apostolidi.ru – портал, который на сегодняшний день популярностью пользуется. Тут имеется поиск, воспользуйтесь им. Апостолиди дает возможность находить полезные советы, исследовать уникальные материалы и получать знания от экспертов. Рады видеть вас на своем ресурсе!
Willieavamy
| #
Online gambling feels loose—no glare! plinko
Willieavamy
| #
The poker online looks real—amazing! slot gallina
dragon money_doOa
| #
драгон мани казино http://dragon-money33.com/ .
allisonsautobodydrymn
| #
Transform your machine into a showpiece with our qualified car customization options. At Bergin Auto Services, we are proficient in providing high-quality upgrades that reflect your personal aesthetic. From modern aerodynamic packages to powerful engine modifications, our group delivers excellent workmanship. Learn more at our internet page https://allisonsautobody.com/ to see our solutions and kick off your automobile’s conversion right away.
DonDonfaita
| #
dark market url dark market onion
Willieavamy
| #
I lost $110 online—set plan! mines game
mostbet_bfpl
| #
мостбет зеркало http://mostbet6038.ru/ .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Нанокристаллический тигель из высокочистого нитрида бора
Aaronkem
| #
Авто журнал онлайн https://comparecarinsurancerfgj.org свежие новости, обзоры моделей, тест-драйвы, советы и рейтинг автомобилей. Всё о мире авто в одном месте, доступно с любого устройства.
PeterFiz
| #
Современный авто журнал https://ecotech-energy.com в онлайн-формате для тех, кто хочет быть в курсе автомобильных трендов. Новости, тесты, обзоры и аналитика — всегда под рукой.
ArielHAh
| #
Женский портал https://elegantwoman.kyiv.ua о красоте, моде, здоровье, отношениях и вдохновении. Полезные статьи, советы экспертов, лайфхаки и свежие тренды — всё для современной женщины.
JacobJAt
| #
Онлайн-портал для женщин https://fancywoman.kyiv.ua которые ценят себя и стремятся к лучшему. Всё о внутренней гармонии, внешнем блеске и жизненном балансе — будь в центре женского мира.
get fexofenadine online
| #
Medicament information. Cautions.
get fexofenadine online
Some what you want to know about medication. Read information now.
where to get combivir for sale
| #
where buy combivir
PaginnIrref
| #
Посетите сайт https://letrivelle.ru/ – это магазин мебели, в котором представлена дизайнерская мебель, свет и декор. Ознакомьтесь с каталогом или обратитесь к нашим квалифицированным менеджерам, которые помогут с выбором. Все в наличии на наших собственных складах в Москве и Санкт-Петербурге. Подробнее на сайте.
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы —
Donaldlaf
| #
darknet market darkmarket
dragon money_jgSn
| #
кэшбэк на драгон мани https://dragon-money30.com/ .
https://russiancouncil.ru/upload/main/92c/aven_1_.jpg
| #
https://russiancouncil.ru/upload/main/92c/aven_1_.jpg
Pingrutty
| #
darknet sites darknet markets onion address
inernetufaCah
| #
интернет по адресу уфа
domashij-internet-ufa001.ru
узнать провайдера по адресу уфа
Toliknaf
| #
darknet markets tor drug market
Cazrrzw
| #
Заказать диплом ВУЗа!
Мы готовы предложить документы учебных заведений, расположенных в любом регионе России. Документы печатаются на бумаге самого высокого качества: powerstack.co.in/employer/aurus-diploms
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы
Willieavamy
| #
Online casinos should limit—safe us! F7 Casino
Rabyitabe
| #
onion dark website darknet marketplace
Willieavamy
| #
I hit a dip online—pause it! gates of olympus
Sazrbcb
| #
купить диплом о высшем образовании в иркутске
Willieavamy
| #
The mobile apps for online casinos crash way too often! plinko
Willieavamy
| #
Online bonuses mask—dig in! fortune mouse
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы
DonDonfaita
| #
darknet sites onion dark website
Аренда авто Краснодар
| #
Аренда авто Краснодар
mostbet_wopl
| #
мосбет http://mostbet6038.ru/ .
RobertFup
| #
Военная служба Служба по контракту: Путь к профессиональной военной карьере Военная служба всегда была и остается почетным долгом гражданина перед своей страной. Однако, помимо обязательной службы по призыву, существует возможность связать свою жизнь с армией на профессиональной основе, заключив контракт с Министерством обороны Российской Федерации. Контрактная служба предоставляет уникальные перспективы для тех, кто видит в защите Родины не просто обязанность, а призвание.
Pingrutty
| #
bitcoin dark web darkmarket 2025
Sazrzfp
| #
Заказать диплом о высшем образовании !
Покупка диплома любого ВУЗа РФ в нашей компании – надежный процесс, так как документ будет заноситься в реестр. Приобрести диплом ВУЗа dip-lom-rus.ru/vnesenie-diploma-v-reestr-po-nizkoj-tsene-bez-xlopot
Markcen
| #
Хотите сдать квартиру в аренду? Читайте проверенные советы
Cazrkqq
| #
Приветствую!
Мы изготавливаем дипломы любой профессии по доступным тарифам. Цена будет зависеть от выбранной специальности, года получения и ВУЗа: diplomanruss.com/
can i purchase thorazine online
| #
Meds information. What side effects can this medication cause?
can i purchase thorazine online
Some news about medicines. Get information now.
Willieavamy
| #
I love the privacy of online casinos—no stares! plinko
Donaldlaf
| #
dark web drug marketplace darkmarkets
Угрозы
| #
Угрозы
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние возможности.
Наши товары:
Плоская мишень Cu+Ni+Ti
Braintip
| #
?Grass is a decentralized network that enables users to monetize their unused internet bandwidth by sharing it with verified institutions. https app getgrass io register referralcode – This shared bandwidth supports various applications, including enhancing AI services and conducting public web data collection.
Willieavamy
| #
I love the pay ease online—smooth run! vai de bet
Toliknaf
| #
dark market url darknet market links
Markcen
| #
Покупка квартиры без рисков? Полезные статьи и советы —
Willieavamy
| #
The live dealers online are ace—real deal! book of ra
Willieavamy
| #
Online slots are my fix—those looks! fortune mouse
Willieavamy
| #
The slot excitement online is wild—heart racing! tom of madness
Willieavamy
| #
The slot rush online pulls—watch it! slot gallina
Rabyitabe
| #
darknet drug store dark web market links
Willieavamy
| #
I don’t trust online odds—odd feel! mines game
mostbet_kmEl
| #
мостбет официальный сайт mostbet7002.ru .
1win_isoi
| #
1wi. https://1win7005.ru .
Darylric
| #
Родительский портал https://babyrost.com.ua от беременности до подросткового возраста. Статьи, лайфхаки, рекомендации экспертов, досуг с детьми и ответы на важные вопросы для мам и пап.
GlennNob
| #
Автомобильный сайт https://billiard-sport.com.ua с обзорами, тест-драйвами, автоновостями и каталогом машин. Всё о выборе, покупке, обслуживании и эксплуатации авто — удобно и доступно.
Jasperguddy
| #
Семейный портал https://cgz.sumy.ua для родителей и детей: развитие, образование, здоровье, детские товары, досуг и психология. Актуальные материалы, экспертиза и поддержка на всех этапах взросления.
Doogie
| #
Doogie
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности
EdwardTrisk
| #
Автомобильный журнал https://eurasiamobilechallenge.com новости автоиндустрии, тест-драйвы, обзоры моделей, советы водителям и экспертов. Всё о мире автомобилей в удобном онлайн-формате.
mostbet_hasa
| #
скачать mostbet https://www.mostbet5004.ru .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наши товары:
Пружина из драгоценных металлов платиновая 5х5х0.9 мм ПлРд90-10 ТУ Выберите идеальные пружины из золота, серебра или платины, чтобы дополнить ваше украшение. Найдите пружины с изысканным дизайном и прочностью. Они предлагают широкий выбор для того, чтобы подчеркнуть уникальность вашего украшения.
Pingrutty
| #
darknet links darknet marketplace
DonDonfaita
| #
darknet markets onion address dark market
inernetufaCah
| #
какие провайдеры на адресе в уфе
domashij-internet-ufa002.ru
интернет провайдеры по адресу дома
Willieavamy
| #
I hit a bonus online and won $400—sweet score! aviator
mostbet_dmKa
| #
mostbet http://mostbet5006.ru .
Dnrtjkh
| #
Мы готовы предложить дипломы любой профессии по невысоким ценам. Мы готовы предложить документы техникумов, расположенных на территории всей России. Документы делаются на бумаге высшего качества. Это дает возможность делать государственные дипломы, которые не отличить от оригинала. realroleplay.listbb.ru/viewtopic.phpf=40&t=1046
posteezy.com
| #
These are truly impressive ideas in regarding blogging.
You have touched some fastidious poiints here. Any way
keep up wrinting.
Feel free to visit my site article (posteezy.com)
RowezonPyday
| #
На сайте https://voronlaws.ru/ вы найдете Casino Dragon Money и его новое рабочее зеркало для того, чтобы открыть для себя удивительный мир огромного количества развлечений, щедрых бонусов, а также бесконечных выигрышей. На сайте вас ожидает увлекательный и эксклюзивный контент, который обязательно подарит много приятных и положительных эмоций. Клуб щедр на акции и бонусы, а потому здесь вы найдете все, что понравится и поможет заработать деньги. Рабочее зеркало ежедневно обновляется для вашей безопасности.
mostbet_gnEl
| #
мостбет кг mostbet7002.ru .
1win_oboi
| #
1вин вход с компьютера http://www.1win7005.ru .
Markcen
| #
Новостройки или вторичка? Узнайте подробности
Donaldlaf
| #
tor drug market dark market url
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние возможности.
Наша продукция:
Плоская мишень селена 4N, 5N
Sazrzjt
| #
купить вкладыш в диплом училища
how to get fosamax without rx
| #
Medicine information leaflet. What side effects?
how to get fosamax without rx
Some trends of medicines. Read information here.
mostbet_qjEl
| #
mostbet официальный сайт https://mostbet7002.ru .
1win_nyoi
| #
1win сайт вход https://www.1win7005.ru .
Toliknaf
| #
tor drug market darknet markets links
Willieavamy
| #
Online casinos should ease rules—tough start! aviator game online
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их происхождение. Опытная поддержка – наш стандарт – мы на связи, чтобы ответить на ваши вопросы и предоставлять решения под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
поставляемая продукция:
Порошок титановый Р’Рў25РЈ – РћРЎРў 1 90013-81 Рзделия РёР· титана Р’Рў25РЈ – РћРЎРў 1 90013-81 представляют СЃРѕР±РѕР№ высококачественные компоненты, разработанные для использования РІ различных промышленностях. Ртот титан отличается отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью, высокой прочностью Рё легкостью, что делает его идеальным выбором для авиации, автомобилестроения Рё медицинского оборудования. Продукция отвечает строгим стандартам, обеспечивая надежность Рё долгий СЃСЂРѕРє службы. РќРµ упустите шанс купить Рзделия РёР· титана Р’Рў25РЈ – РћРЎРў 1 90013-81 Рё улучшить эффективность вашего производства благодаря передовым материалам. Применение современных технологий РІ производстве гарантирует высокие эксплуатационные характеристики данной продукции.
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы —
1win_kdMr
| #
что такое 1win что такое 1win .
Mazrvdi
| #
Приобрести диплом любого института!
Мы оказываем услуги по продаже документов об окончании любых ВУЗов РФ. Документы производят на подлинных бланках. jobboat.co.uk/employer/radiplomy
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Алюминиево-хромовая мишень для распыления (AlCr)
Аренда авто Краснодар
| #
Аренда авто Краснодар
1win_czmn
| #
1win sports https://1win18.com.ng .
Rabyitabe
| #
tor drug market dark web markets
best crypto casinos
| #
best crypto casinos
A Brief History of Elemental Analysis » Artemis Analytical
Pingrutty
| #
darknet markets darkmarket 2025
Willieavamy
| #
The mobile casino dips—lift it! plinko
Phising
| #
Phising
A Brief History of Elemental Analysis » Artemis Analytical
1win_tjMr
| #
1вин официальный 1вин официальный .
Willieavamy
| #
Are online casino jackpots real, or just bait to keep you playing? fortune ox
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы
DonDonfaita
| #
dark web market dark web market
MichaelDipsy
| #
купить кухню на заказ Кухни на заказ: функциональность и стиль в каждой детали. Кухня – сердце дома. Поэтому так важно, чтобы она была не только красивой, но и максимально функциональной. Кухни на заказ позволяют воплотить любые дизайнерские решения, оптимизировать пространство и создать идеальную рабочую зону. Вы сами выбираете материалы, фурнитуру, цвет и конфигурацию, чтобы ваша кухня стала настоящим произведением искусства. В Москве существует множество компаний, предлагающих кухни на заказ по доступным ценам.
1win_xxpl
| #
1win https://1win5026.ru/ .
VernonHew
| #
Женский сайт о красоте https://female.kyiv.ua моде, здоровье, отношениях и саморазвитии. Полезные статьи, советы экспертов, вдохновение и поддержка — всё для гармоничной и уверенной жизни.
Bradleyrit
| #
Современный женский сайт https://femalebeauty.kyiv.ua мода, психология, семья, карьера, рецепты и лайфхаки. Ежедневно — новые материалы, рекомендации и интересные темы для каждой женщины.
Ronnytaf
| #
Автомобильный сайт https://fundacionlogros.org с ежедневными новостями, обзорами новинок, аналитикой, тест-драйвами и репортажами из мира авто. Следите за трендами и будьте в курсе всего важного.
Danielrow
| #
Читайте автомобильный сайт https://gormost.info онлайн — тесты, обзоры, советы, автоистории и материалы о современных технологиях. Всё для тех, кто любит машины и скорость.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние достижения.
Наша продукция:
Металлическая мишень для распыления
Sazrwgj
| #
Мы изготавливаем дипломы любых профессий по выгодным ценам.
Вы заказываете диплом через надежную фирму. Заказать диплом о высшем образовании– http://chaakri.com/employer/diplomy-grup-24/ – chaakri.com/employer/diplomy-grup-24
WilliamawarY
| #
https://cricketnews-australia.com/
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности
Willieavamy
| #
Online poker pumps—bluff high! jackpotraider
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
поставляемая продукция:
Медный тройник РїРѕРґ пайку стандартный 16С…12С…0.7 РјРј 7С…9 РјРј твердая пайка Рњ1 ГОСТ Р 52922-2008 Купить качественные медные тройники РїРѕРґ пайку для надежных Рё долговечных соединений РІ трубопроводных системах. Разнообразие размеров Рё конфигураций, высокая теплопроводность Рё устойчивость Рє РєРѕСЂСЂРѕР·РёРё. Простая установка Рё надежность соединения. Рдеальное решение для различных проектов. Редметсплав.СЂС„
Jariornsy
| #
Купить диплом о высшем образовании!
Наши специалисты предлагаютвыгодно и быстро купить диплом, который выполнен на бланке ГОЗНАКа и заверен мокрыми печатями, водяными знаками, подписями официальных лиц. Диплом способен пройти любые проверки, даже с использованием специального оборудования. Решайте свои задачи быстро с нашими дипломами- displaycreations.net/kupit-diplom-prostoe-reshenie-dlja-slozhnyh-zadach-15
porno
| #
porno
blog topic
Zivutrttar
| #
Перевозка негабаритных грузов и доставка по всей территории России и СНГ на сайте https://negabarit-24.ru/ – это профессиональные услуги от нашей компании. Перевозка негабаритных грузов по России и странам СНГ, ЖД перевозки, перевозки манипулятором и международные перевозки. Гибкие варианты оплаты. Соблюдение сроков. Страхование грузов. Подробнее на сайте.
https://war-proekt.media/spisok-oligarkhov/provider/petr-aven/
| #
https://war-proekt.media/spisok-oligarkhov/provider/petr-aven/
Sazriyr
| #
Приобрести диплом университета по невысокой стоимости можно, обратившись к проверенной специализированной фирме. Заказать документ ВУЗа можно у нас. video.firstkick.live/read-blog/19368_realno-li-kupit-diplom-s-reestrom.html
Donaldlaf
| #
dark web markets darkmarket url
Jariorcdk
| #
Мы предлагаем выгодно купить диплом, который выполняется на бланке ГОЗНАКа и заверен печатями, штампами, подписями должностных лиц. Данный документ пройдет лубую проверку, даже с использованием специально предназначенного оборудования. butsuribu.com/?post_type=topic&p=311401
mostbet_eooi
| #
поддержка мостбет https://www.mostbet7001.ru .
Willieavamy
| #
I hit a bonus online and won $120—sweet yes! book of ra
Willieavamy
| #
Online casinos slide easy—mind it! vipzino
1win_fupl
| #
1win moldova download 1win moldova download .
how can i get spiriva tablets
| #
Drugs information leaflet. Long-Term Effects.
how can i get spiriva tablets
Actual about meds. Get now.
inernetufaCah
| #
проверить провайдеров по адресу уфа
domashij-internet-ufa003.ru
провайдеры интернета в уфе по адресу проверить
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы
Toliknaf
| #
darknet marketplace dark web marketplaces
Pingrutty
| #
darkmarket dark markets
Thurmanskabs
| #
https://australianfootball-au.com/
Rabyitabe
| #
dark web marketplaces best darknet markets
GeorgeTom
| #
Source:
– parambu.ru
WilliamPem
| #
Завод металлоконструкций ВЕКСТРОЙ
Векстрой Уфа – это имя, которое звучит с уверенностью и надежностью в сфере производства металлоконструкций. Мы – не просто завод, мы – команда профессионалов, объединенных общей целью: создавать прочные, долговечные и эстетически привлекательные решения для наших клиентов.
Наш завод металлоконструкций оснащен современным оборудованием, позволяющим нам реализовывать проекты любой сложности. От разработки чертежей до финальной сборки – каждый этап производства находится под строгим контролем качества. Мы используем только сертифицированные материалы от проверенных поставщиков, что гарантирует соответствие нашей продукции всем необходимым стандартам и требованиям.
Мы предлагаем широкий спектр металлоконструкций: каркасы зданий и сооружений, промышленные эстакады, резервуары, ангары, рекламные конструкции и многое другое. Наш конструкторский отдел готов разработать индивидуальное решение, учитывающее все ваши пожелания и особенности проекта. Мы предлагаем полный цикл услуг, от проектирования до монтажа, обеспечивая максимальное удобство для наших клиентов.
Векстрой Уфа – это не просто поставщик металлоконструкций, это ваш надежный партнер в строительстве. Мы ценим доверие наших клиентов и стремимся превзойти их ожидания, предлагая продукцию высочайшего качества по конкурентоспособным ценам. Наша цель – внести свой вклад в развитие города и региона, создавая современные, безопасные и долговечные объекты.
Diplomi_ymKt
| #
Заказать диплом любого ВУЗа!
Мы готовы предложить документы институтов, расположенных на территории всей Российской Федерации.
diplomist.com/kupit-diplom-s-zaneseniem-v-reestr-bistro-i-prosto-3/
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их качество. Опытная поддержка – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы по мере того как находить ответы под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
Наши товары:
Порошок магниевый R60701 – UNS Рзделия РёР· магния R60701 – UNS представляют СЃРѕР±РѕР№ высококачественные материалы, обладающие отличной прочностью Рё легкостью. РћРЅРё широко используются РІ различных отраслях, включая авиацию, автомобилестроение Рё электронику. Магний R60701 – UNS отличается хорошей РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью Рё высокой теплопроводностью, что делает его идеальным выбором для современных технологий. Если РІС‹ хотите получить надежные изделия для СЃРІРѕРёС… проектов, купить Рзделия РёР· магния R60701 – UNS станет отличным решением. Рти материалы обеспечивают долговечность Рё эффективность, что критично РІ условиях высокой нагрузки Рё жесткой эксплуатации.
trevojnaya knopka rosgvardii_xspn
| #
подключить тревожную кнопку росгвардия подключить тревожную кнопку росгвардия .
DonDonfaita
| #
dark web drug marketplace dark web sites
Cazrmrm
| #
Приобрести диплом университета!
Мы готовы предложить документы университетов, расположенных в любом регионе РФ. Документы выпускаются на “правильной” бумаге самого высшего качества: cbrxrealtor.com/kupit-diplom-bystro-i-nedorogo-oficialnye-dokumenty
WilliamawarY
| #
https://horseracing-australia.com/
abc bet
| #
Ao se cadastrar no abc bet, você ganha um bônus
de 100$ para começar sua jornada no cassino com o pé direito!
Não importa se você é um novato ou um apostador experiente,
o bônus de boas-vindas é a oportunidade perfeita para explorar todas as opções que o site tem a oferecer.
Jogue seus jogos favoritos, descubra novas opções de apostas e
aproveite para testar estratégias sem risco, já que o bônus ajuda a
aumentar suas chances de ganhar. Cadastre-se hoje e comece com 100$!
Markcen
| #
Новостройки или вторичка? Узнайте подробности
Willieavamy
| #
Online casinos feel off on losses—hmm! teen patti
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
Наша продукция:
Лист судостроительный 20x1000x2500 D40S EN10025:3 Приобретите высококачественные листы судостроительные от компании Редметсплав.рф для надежного и долговечного использования в вашем судостроительном проекте. Различные размеры и толщины, прочность и устойчивость к коррозии делают эти листы идеальным выбором для любого судостроительного проекта.
otutejTON
| #
Сайт компании Аллегро – https://spb.tk-allegro.ru/ предлагает вам аренду в Санкт-Петербурге автобуса. У нас от 8 до 55 мест свой автопарк автобусов. Водители с большим опытом. У вас есть возможность заказать для любых мероприятий автобус: на экскурсию, выпускной, свадьбу и для школьников. Заходите на портал, узнайте больше о стоимости аренды, о наших услугах, или же воспользуйтесь для расчета стоимости калькулятором на портале.
mostbet_dsoi
| #
most bet http://www.mostbet7001.ru .
Willieavamy
| #
I hit a blackjack streak—good day! plinko
Аренда авто Краснодар
| #
Аренда авто Краснодар
Donaldlaf
| #
darknet markets darknet drugs
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации —
Cazrxaq
| #
Здравствуйте!
Мы предлагаем дипломы любой профессии по выгодным тарифам. Стоимость может зависеть от выбранной специальности, года получения и образовательного учреждения: rdiplomans.com/
1win_fmKt
| #
înregistrare 1win înregistrare 1win .
Pingrutty
| #
dark web market list darknet market links
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
поставляемая продукция:
Латуное внутреннее стопорное пружинное кольцо 120х2.5 мм ЛАЖМц ГОСТ 13943-86 Купить латунные внутренние стопорные кольца по ГОСТ от производителя. Надежное крепление и удержание элементов механизмов. Широкий выбор размеров и конфигураций. Высокое качество и надежность. Применение в машиностроении, автомобильной и других отраслях. Обращайтесь к нашим специалистам для получения более подробной информации.
cost generic levitra no prescription
| #
Drugs prescribing information. Drug Class.
cost generic levitra no prescription
All what you want to know about medicament. Get now.
1win_vfpl
| #
1win pariuri http://1win5026.ru .
DonaldFonse
| #
короткие анекдоты Канал анекдотов. Добро пожаловать в мир искрометного юмора и безудержного смеха! Здесь вы найдете самые свежие и уморительные анекдоты на любой вкус. От классики до новинок, от простых шуток до интеллектуальных забав – мы собрали коллекцию, способную поднять настроение даже в самый серый день.
Edwardmoope
| #
https://medicalplanet.su/radiologia/mrt-organov-moshonki.html
Toliknaf
| #
dark web market urls bitcoin dark web
Sazrbel
| #
Приобрести диплом о высшем образовании !
Покупка диплома ВУЗа РФ у нас является надежным процессом, поскольку документ заносится в государственный реестр. Заказать диплом о высшем образовании vacshidiplom.com/zanesi-diplom-v-reestr-obrazovaniya-bistro-i-bez-problem
Andreteake
| #
https://www.instagram.com/oos_studio/
winzada 777
| #
Ganhe 100$ de Bônus no winzada 777 ao
Criar Sua Conta!
Ellende
| #
Ellende
Markcen
| #
Хотите сдать квартиру в аренду? Читайте проверенные советы
Willieavamy
| #
The mobile casino clogs—tune it! plinko
Rabyitabe
| #
dark web market urls dark market url
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их происхождение. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы и находить ответы под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
Порошок висмутовый Sn96Ag2,5Bi1Cu0,5 – ISO 9453 Порошок висмутовый Sn96Ag2,5Bi1Cu0,5 – ISO 9453 представляет СЃРѕР±РѕР№ высококачественный сплав, идеально подходящий для использования РІ электронной промышленности. Благодаря своей высокой теплопроводности Рё улучшенной механической прочности, РѕРЅ становится идеальным для соединений РІ монтаже. Ртот порошок обладает РЅРёР·РєРѕР№ температурой плавления Рё отличной совместимостью СЃ РґСЂСѓРіРёРјРё материалами, что делает его незаменимым РїСЂРё проведении пайки Рё ремонте плат. Купить Порошок висмутовый Sn96Ag2,5Bi1Cu0,5 – ISO 9453 РІС‹ сможете Сѓ нас, что гарантирует вам качество Рё надежность вашего оборудования.
Edwardmoope
| #
https://www.prozdor.ru/zabolevaniya/nevidimye-bolezni-pod-pricelom-kak-mrt-nahodit-opuholi-na-rannej-stadii/
CharlesNuh
| #
Zelite li se odmoriti? http://www.booking-zabljak.com Rezervirajte udoban hotel u centru ili u podnozju planina. Odlican izbor za skijanje i ljetovanje. Jamstvo rezervacije i stvarne recenzije.
ManuelNeogs
| #
Need real estate? luxury-property-montenegro villas, houses and apartments in Budva, Kotor, Tivat and on the coast. Profitable investment, sea view, safety and comfort in the south of Europe.
best online casino bonus
| #
best online casino bonus
A Brief History of Elemental Analysis » Artemis Analytical
RobertAdurb
| #
need a move? https://moversmia.com turnkey: packing, loading, transport, insurance and support. Without stress and with a guarantee of the safety of your property.
Dnrteto
| #
Мы изготавливаем дипломы любой профессии по приятным тарифам. Мы можем предложить документы техникумов, расположенных на территории всей Российской Федерации. Дипломы и аттестаты печатаются на бумаге высшего качества. Это дает возможность делать государственные дипломы, которые невозможно отличить от оригинала. cheere.org/read-blog/75951_kupit-diplom-s-zaneseniem-v-reestr-stoimost.html
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
Наша продукция:
Контакт РёР· драгоценных металлов золотой 1.5С…12 РјРј ЗлСрМ58.5-8 РўРЈ Рзысканные контакты РёР· золота, серебра Рё платины РѕС‚ Редметсплав. Высочайшее качество, уникальный дизайн Рё эксклюзивные предложения для постоянных клиентов. Добавьте роскошь Рё стиль вашему образу!
Davidnon
| #
Автоперевозки из Китая http://www.vladimir.ru/forum/forum/thread/50779 доставка грузов по РФ и странам СНГ. Сборные и индивидуальные партии, оформление, отслеживание и страхование. Быстро, надёжно, под ключ.
Jariorsoo
| #
Заказать диплом любого ВУЗа!
Наши специалисты предлагаютбыстро и выгодно купить диплом, который выполнен на оригинальном бланке и заверен печатями, водяными знаками, подписями. Данный диплом способен пройти любые проверки, даже при помощи профессиональных приборов. Достигайте свои цели быстро и просто с нашей компанией- mongolsmc.listbb.ru/viewtopic.phpf=1&t=1619
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Гранулы высокочистого ванадия
Sazruoc
| #
Заказать диплом ВУЗа по невысокой цене возможно, обратившись к надежной специализированной фирме. Купить документ о получении высшего образования можно в нашей компании в столице. islider.ru/create-blog
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности
DonDonfaita
| #
dark web sites darkmarket link
Willieavamy
| #
I hit a progressive jackpot online once—life-changing moment! fortune tiger
mostbet_krEl
| #
mostbets http://www.mostbet7002.ru .
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог высококачественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их происхождение. Опытная поддержка – наш стандарт – мы на связи, чтобы ответить на ваши вопросы по мере того как находить ответы под специфику вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Магний Р›-2-3 – РўРЈ 28-10-33-75 Магний Р›-2-3 – РўРЈ 28-10-33-75 представляет СЃРѕР±РѕР№ высококачественный химический элемент, который используется РІ различных отраслях, включая металлургию Рё производство сплавов. Ртот РїСЂРѕРґСѓРєС‚ отличается отличной растворимостью Рё высокой чистотой, что делает его идеальным для промышленных применений.Если РІС‹ ищете надежное сырье, то вам стоит купить Магний Р›-2-3 – РўРЈ 28-10-33-75. Наш РїСЂРѕРґСѓРєС‚ соответствует всем необходимым стандартам качества, что гарантирует его долговечность Рё эффективность РІ использовании. Рто отличный выбор для вашего бизнеса.
free spins no deposit uk
| #
free spins no deposit uk
A Brief History of Elemental Analysis » Artemis Analytical
Willieavamy
| #
I don’t get the hate for online gambling—it’s just like any other hobby! pinup
Pingrutty
| #
darknet market links darkmarket list
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности
best bitcoin sites
| #
best bitcoin sites
A Brief History of Elemental Analysis » Artemis Analytical
Diplomi_otKt
| #
Купить диплом о высшем образовании!
Мы готовы предложить документы университетов, расположенных в любом регионе России.
diplomnie.com/kupit-podlinnij-diplom-s-zaneseniem-v-reestr-6/
Займы без риска — на MIKRO-ZAIM-ONLINE.RU
| #
Когда банковский отказ — не вариант, на помощь приходит mikro-zaim-online.ru. Достаточно паспорта и карты на ваше имя, с 18 лет. Более 60 МФО, высокая вероятность одобрения, даже с плохой кредитной историей. Хотите без процентов или фиксировано 0.8% в день? Всё доступно здесь.
Займы оформляются официально, по законодательству РФ. Согласно №151-ФЗ, каждая МФО обязана соблюдать условия прозрачности и нести ответственность перед заёмщиком. Именно поэтому деньги переводятся моментально, без проверок, а договор — законный. Надёжность, простота и безопасность в одном решении.
razrabotka saita na bitriks_loKt
| #
1с битрикс разработка сайта заказать http://www.razrabotka-saita-bx.ru/ .
Donaldlaf
| #
darknet marketplace dark market 2025
best bitcoin casinos
| #
best bitcoin casinos
A Brief History of Elemental Analysis » Artemis Analytical
where to buy cephalexin without dr prescription
| #
Medication information for patients. Brand names.
where to buy cephalexin without dr prescription
All trends of medication. Read here.
1win_bdpl
| #
1win.pro http://www.1win5026.ru .
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их качество. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы а также предоставлять решения под требования вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Проволока кобальтовая Stellite 12P Проволока кобальтовая Stellite 12P – это высококачественный материал, идеально подходящий для сварки Рё наплавки, обеспечивая отличные механические свойства Рё РєРѕСЂСЂРѕР·РёРѕРЅРЅСѓСЋ стойкость. РћРЅР° используется РІ различных отраслях, включая машиностроение, авиастроение Рё энергетический сектор. Повышенная прочность Рё теплостойкость этой проволоки делают ее незаменимой РїСЂРё создании износостойких покрытий. Если вам нужна надежность Рё долговечность, купите Проволока кобальтовая Stellite 12P Рё обеспечьте своему производству качественные решения. Рта проволока поможет вам достичь наилучших результатов РІ работе.
Toliknaf
| #
dark web market list best darknet markets
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации —
Yatavtswops
| #
На сайте https://rozatoday.ru/ представлено огромное количество ярких, запоминающихся композиций, которые созданы из свежих цветов: ослепительных роз, волнующих пионов, трепетных тюльпанов, строгих гвоздик. В компании работают только знающие, компетентные флористы, которые создают изысканные композиции в соответствии с предпочтениями клиента. В обязательном порядке учитываются и актуальные мировые тенденции. Можно приобрести как небольшой букетик, который подходит для романтического свидания, так и внушительных размеров для грандиозного праздника.
Lazrycj
| #
Диплом университета России!
Без наличия диплома достаточно сложно было продвигаться по карьере. В связи с этим решение о покупке диплома следует считать целесообразным. Заказать диплом об образовании career.finixia.in/employer/gosznac-diplom-24
Rabyitabe
| #
darknet market dark web market
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наши товары:
Гранулы олова высокой чистоты
mostbet_ppoi
| #
скачать мостбет официальный сайт mostbet7001.ru .
kalkylyator OSAGO_acon
| #
страхование осаго онлайн калькулятор https://seodict.ru .
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы
Williamemifs
| #
https://kssp.borda.ru/?1-5-0-00001735-000-0-0-1741432686
Willieavamy
| #
Online bingo flows—great crew! craps
solar water heater
| #
solar water heater
A Brief History of Elemental Analysis » Artemis Analytical
dragon money_shKl
| #
драгон мани главная http://dragon-money37.com/ .
dragon money_icSi
| #
dragon money demo dragon-money36.com .
Pingrutty
| #
darkmarkets dark web sites
Аренда авто Краснодар
| #
Аренда авто Краснодар
Willieavamy
| #
Are online wins true?—not sure! plinko
Willieavamy
| #
I don’t trust online RNG—feels fake! plinko
Willieavamy
| #
I wish online guided new—rough start! casino mate
online casinos real money
| #
online casinos real money
A Brief History of Elemental Analysis » Artemis Analytical
melbet_sypa
| #
melbet сайт http://www.melbet1001.ru .
Jariorqdx
| #
Приобрести диплом о высшем образовании!
Наша компания предлагаетвыгодно и быстро купить диплом, который выполняется на оригинальной бумаге и заверен мокрыми печатями, водяными знаками, подписями официальных лиц. Наш документ способен пройти лубую проверку, даже при использовании профессионального оборудования. Достигайте свои цели быстро и просто с нашим сервисом- munchkin.flybb.ru/viewtopic.phpf=7&t=5350
Trefxpg
| #
Выгодно купить диплом об образовании. Покупка документа о высшем образовании через качественную и надежную фирму дарит много достоинств для покупателя. Данное решение помогает сэкономить как продолжительное время, так и существенные средства. tmiglobal.co.uk/employer/eonline-diploma
Sazrnmd
| #
Купить диплом ВУЗа по невысокой стоимости возможно, обратившись к надежной специализированной компании. Купить документ о получении высшего образования можно в нашей компании в Москве. netmitra.cyberei.com/read-blog/1853_kupit-diplom-s-registraciej-v-reestre.html
fixitcaree
| #
Сервисный центр ФикситЦентр – выполнит срочный ремонт пароварок в Москве
Willieavamy
| #
Online blackjack is my pick—fun mix! aviator
Петр Авен
| #
Петр Авен
trevojnaya knopka rosgvardii_wfpn
| #
тревожная кнопка цена https://trevros.ru/ .
Donaldlaf
| #
darknet markets url darknet markets onion address
lyrica online without a prescription
| #
Drugs information for patients. Drug Class.
lyrica online without a prescription
Everything trends of medicament. Read information now.
GeorgeTom
| #
Source:
https://parambu.ru/
Diplomi_ewKt
| #
Приобрести диплом любого университета!
Мы предлагаем документы любых учебных заведений, расположенных в любом регионе РФ.
kupit-diplom24.com/kupit-diplom-s-zapisyu-v-reestre-bistro-i-nadezhno-3/
Toliknaf
| #
dark web marketplaces dark web market urls
1win_nfoi
| #
1win зайти https://1win7005.ru .
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наша продукция:
Ниобиевая твердая проволока НбПр-1 1,2 мм ТУ 48-4-316-74 Купите ниобиевую твердую проволоку НбПр-1 диаметром 0,7 мм на Редметсплав.рф. Высокая прочность, отличные сварочные характеристики и соответствие стандарту ТУ 48-4-316-74 делают эту проволоку идеальным выбором для профессиональной сварки.
Rabyitabe
| #
dark market darkmarket url
razrabotka saita na bitriks_kdKt
| #
разработка сайта на битриксе цена разработка сайта на битриксе цена .
Davidguicy
| #
Hi, what is your hobby? what do you do in spare time? personally love to play king bily casino
Pingrutty
| #
darknet market links darknet drug links
dragon money_kvKl
| #
дракон мани https://dragon-money37.com/ .
dragon money_rpSi
| #
lhfujy vfyb https://www.dragon-money36.com .
kalkylyator OSAGO_ipon
| #
посчитать страховку на автомобиль онлайн http://www.seodict.ru/ .
Willieavamy
| #
Online blackjack rocks—fun twist! plinko
Willieavamy
| #
Online casino ads are everywhere—hard to escape them! plinko
1win_vsmn
| #
1win bet https://www.1win18.com.ng .
DonDonfaita
| #
darkmarket url darknet markets 2025
Uazrkkn
| #
Добрый день!
Для определенных людей, купить диплом университета – это необходимость, удачный шанс получить выгодную работу. Впрочем для кого-то – это очевидное желание не терять множество времени на учебу в ВУЗе. Что бы ни толкнуло вас на такой шаг, мы готовы помочь. Оперативно, качественно и недорого изготовим диплом нового или старого образца на подлинных бланках с реальными печатями.
Главная причина, почему многие покупают документы, – желание занять определенную работу. Допустим, знания позволяют специалисту устроиться на работу, а подтверждения квалификации не имеется. В случае если работодателю важно присутствие “корочки”, риск потерять место работы очень высокий.
Приобрести документ о получении высшего образования можно в нашем сервисе. Мы оказываем услуги по изготовлению и продаже документов об окончании любых университетов России. Вы сможете получить диплом по любым специальностям, включая документы старого образца. Гарантируем, что при проверке документа работодателями, каких-либо подозрений не появится.
Ситуаций, которые вынуждают заказать диплом о среднем образовании много. Кому-то срочно нужна работа, в итоге нужно произвести впечатление на начальство во время собеседования. Другие желают попасть в большую компанию, чтобы повысить свой статус в социуме и в будущем начать свой бизнес. Чтобы не терять драгоценное время, а сразу начинать удачную карьеру, применяя имеющиеся знания, можно купить диплом в онлайне. Вы сможете быть полезным в обществе, получите финансовую стабильность в максимально короткий срок- купить диплом техникума
1win_cmmn
| #
1win bets 1win bets .
mostbet_kdoi
| #
mostbet chrono mostbet7001.ru .
mostbet_kcKa
| #
mostbet kg скачать на андроид https://mostbet5006.ru .
Willieavamy
| #
Online casinos should free—try it! dragon tiger
Willieavamy
| #
Online casino ads push—soften it! dragon tiger
fixitcaree
| #
Сервисный центр ФикситЦентр – выполнит срочный ремонт квадрокоптеров в Москве
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена соответствующими документами, подтверждающими их качество. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы ответить на ваши вопросы а также находить ответы под особенности вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
Наши товары:
Пруток кобальтовый Stellite 36 Пруток кобальтовый Stellite 36 — это материал, обладающий высокой стойкостью к износу и коррозии, что делает его идеальным для применения в различных промышленностях. Благодаря отличным механическим свойствам, этот кобальтовый сплав широко используется в авиастроении, машиностроении и в производстве режущего инструмента. Если вы ищете надежный и долговечный материал, чтобы обеспечить высококачественные результаты своей работы, вам стоит купить Пруток кобальтовый Stellite 36. Его доступность и универсальность делают его незаменимым в современных производственных процессах.
Donaldlaf
| #
darknet site darknet websites
how to buy generic aricept without a prescription
| #
Drugs information for patients. Generic Name.
how to buy generic aricept without a prescription
Some about medicines. Read information now.
dragon money_cnKl
| #
драгон мани официальный сайт играть http://dragon-money37.com/ .
dragon money_ckSi
| #
драгон мани сайт http://www.dragon-money36.com .
kalkylyator OSAGO_gyon
| #
страховка осаго онлайн рассчитать страховка осаго онлайн рассчитать .
Toliknaf
| #
dark web market darkmarket url
site
| #
Nicely put. Many thanks!
Pingrutty
| #
darknet drug market darknet markets url
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наша продукция:
РџРѕРєРѕРІРєР° прямоугольная РёР· конструкционной стали 70С…330 РјРј 45РҐ Конструкционная прямоугольная РїРѕРєРѕРІРєР° – высококачественные материалы для создания прочных Рё надежных конструкций. Рзготовленная РёР· высококачественных материалов СЃ применением передовых технологий, эта продукция обладает отличной прочностью, устойчивостью Рє РёР·РЅРѕСЃСѓ, Рё находит широкое применение РІ машиностроении Рё строительстве.
XixigmPrign
| #
Ищете Нестероидный противовоспалительный обезболивающий препарат с фенилбутазоном и лидокаином? Посетите сайт https://ambenium.ru/ и узнайте о препарате Амбениум – это единственный нестероидный противовоспалительный препарат, зарегистрированный в России с усиленным обезболивающим эффектом – раствор для внутримышечного введения фенилбутазон и лидокаин. Подробности на сайте.
Birogopisk
| #
Посетите https://igormylnikovchannel.ru/poezdki-v-taksi-s-det-mi/ и вы найдете полезную информацию для начинающих таксистов и опытных водителей на тему поездок в такси с детьми, начиная от безопасности поездок и ответственности и правилах перевозки детей, до разбора вопросов почему таксисты отказываются от поездок с детьми и многое другое. Подробнее на странице.
Rabyitabe
| #
dark web markets darknet markets 2025
Thurmanskabs
| #
poker
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
“Мишень для распыления неодима 3N, плоская форма”
1win_zkMr
| #
1win играть http://1win7017.ru/ .
купить мебель из искусственного ротанга
| #
Металлическая мебель и природа как создать гармонию
Металлическая мебель – как сочетать с природными элементами
Современный дизайн интерьеров все чаще обращает внимание на использование железных изделий. Его популярность обусловлена не только прочностью, но и способностью гармонично вписываться в различные стили. Интересно, что металлические предметы способны привнести в пространство нечто большее, чем просто практичность. Их холодный блеск может контрастировать с мягкими текстурами природных материалов, создавая интересные визуальные акценты.
Для достижения наиболее органичного сочетания стоит учитывать цветовую палитру и форму каждого элемента. Тёмные оттенки, такие как антрацит или чёрный графит, эффективно подчеркивают светлые деревянные детали. Добавление зелёных инсектиций, таких как комнатные растения, смягчит жесткость металла и создаст ауру уюта. Например, металлические столы могут быть окружены стульями из натурального дерева, что добавит элегантности и тепла.
Кроме того, игра текстур становится важным аспектом оформления. Гладкие, зеркальные поверхности могут сочетаться с шершавыми фактурами. Этот контраст не только обеспечивает эстетическую привлекательность, но и влияет на восприятие пространства в целом. Чем больше различных материалов, тем интереснее и многограннее становится общий облик интерьера.
Сочетание искусственного и натурального может также стимулировать позитивные эмоции. Это способствует созданию комфортного и благожелательного окружения, в котором приятно проводить время. Таким образом, стальные конструкции становятся не просто частью мебели, а важным звеном в создании эмоционального фона. Правильный подход к их использованию может превратить ваше жилое пространство в истинный оазис спокойствия и вдохновения.
Металлическая мебель и природа: создание гармонии
Материалы, используемые в интерьере, могут не только служить функциональным целям, но и влиять на атмосферу помещения. В данной связи важно учитывать, как внедрение конструкций из прочного материала влияет на окружение и восприятие пространства. Визуальное сочетание стали с растениями и древесиной может создать уникальный баланс.
Для достижения эффекта единообразия рекомендуется подбирать детали, которые будут перекликаться по текстуре и цвету. Холодные оттенки металла могут смягчаться зелеными элементами – например, в виде живых растений или цветных акцентов. Это позволит привнести оттенки тепла и уюта.
Интересный подход заключается в использовании антикоррозийных покрытий для защиты изделий от влияния внешней среды. Такие обработки улучшают не только долговечность, но и внешний вид. Кроме того, стоит рассмотреть варианты с травлением или порошковым покрытием, чтобы гармонично вписать предметы в природный контекст.
Свежий воздух и натуральные материалы могут стать основой для уникального стиля. Создание открытых пространств с максимальным взаимодействием природных и промышленных элементов способствует формированию оригинальной атмосферы. Используйте светлую палитру, чтобы визуально расширить пространство, добавив светлые деревянные детали.
Удачным решением станет сочетание жестких форм с плавными линиями. Используйте округлые предметы, такие как овальные столы или стулья с мягкими сиденьями, чтобы разбавить строгость металлов. Это смягчит восприятие и привнесет динамику в композицию.
Не забывайте о возможности внедрения зеленых насаждений в интерьер. Vertical gardens или небольшие горшки с растениями скрывают холодные линии, создавая более теплую атмосферу. Эти решения легко интегрируются в такие конструкции и придают им индивидуальность.
Такой подход требует тщательного выбора всех составляющих пространственного оформления. Продуманный баланс между прочными и природными элементами формирует неповторимую атмосферу, способствующую комфорту и вдохновению в любом помещении.
Выбор металлических предметов интерьера, подходящих для соединения с природными элементами
При выборе изделий из металла для оформления пространства, уделите внимание материалам и текстурам, которые будут сочетаться с натуральными компонентами. Например, состаренное железо или патинированная медь прекрасно вписываются в эклектичные интерьеры, добавляя интригу и глубину.
Цветовая палитра также играет ключевую роль. Холодные металлические оттенки, такие как серебристый или графит, можно смягчить добавлением теплых древесных фактур. Комбинации, в которых присутствуют теплые и холодные тона, создают баланс, который визуально притягивает внимание и создает уютные акценты.
Обратите внимание на форму изделий. Овальные или мягкие линии могут более гармонично сочетаться с природными формами, чем строгие геометрические конструкции. Круглые столы или стулья с плавными контурами оживляют пространство и способствуют созданию теплой атмосферы.
Также стоит рассмотреть декор. Элементы с ковкой или резьбой могут добавить интересный контраст с элементами, выполненными из древесины или камня. Такие детали позволят создать многослойный вид интерьера и акцентировать внимание на его индивидуальности.
Не забудьте о функциональности. Изделия должны быть не только стильными, но и удобными. Поэтому важно учитывать Ergonomics при создании пространства. Выбирайте изделие, которое обеспечивает комфорт и уют, а не просто эстетичный вид.
В конечном счёте, рассматривать сочетания материалов и форм, а также правильно подобранный декор, позволит вам добиться желаемого эффекта. Ищите нестандартные решения, которые подчёркивают уникальность вашего стиля, а не заставляют его сливаться с типичными интерьерами.
Способы минимизации влияния на экологию
Энергетическая эффективность производства также играет важную роль. Выпускаемые изделия должны соответствовать современным стандартам, подразумевающим минимизацию потребления электроэнергии при изготовлении и эксплуатации. Установка солнечных панелей на производственных мощностях способна значительно уменьшить углеродный след.
На этапе эксплуатации стоит продумать возможность вторичного использования предметов. Например, можно организовать программы по возврату устаревших элементов для ремонта или переработки. Это стимулирует круговорот материалов и уменьшает количество отходов.
Регулярное обслуживание способствует продлению жизненного цикла изделий, что существенно снижает потребность в новых поставках и производстве. Использование высококачественных антикоррозийных покрытий также уменьшает необходимость в замене из-за повреждений.
Важно учитывать и транспортные расходы. Оптимизация логистических процессов, использование электромобилей для перевозки, способствует снижению выбросов. Учет расстояний при планировании доставки позволит минимизировать углеродный след.
Создание узловых центров для сборки и распределения позволяет снизить транспортные издержки и время доставки. Это также положительно скажется на окружающей среде, уменьшая уровень загрязнения.
https://joycom.ru/mebel-dlya-terrasy/mebel-iz-iskusstvennogo-rotanga/
DonDonfaita
| #
dark web market darknet markets links
mostbet_ifKa
| #
mostbet chrono mostbet chrono .
Willieavamy
| #
The table games online gleam—top notch! bet on red
Willieavamy
| #
I won $1100 on a slot—still hyped! bac bo
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена требуемыми документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – наш стандарт – мы на связи, чтобы ответить на ваши вопросы по мере того как предоставлять решения под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Лента титановая Рў20 – ГОСТ 2171-90 РљСЂСѓРі титановый Рў20 – ГОСТ 2171-90 является идеальным выбором для РјРЅРѕРіРёС… промышленных применений. Отличительной чертой этого изделия является высокая прочность Рё стойкость Рє РєРѕСЂСЂРѕР·РёРё, что делает его незаменимым РІ условиях агрессивной среды. Благодаря своему легкому весу Рё отличным механическим свойствам, РєСЂСѓРі используется РІ авиации, машиностроении Рё РґСЂСѓРіРёС… высоких технологиях. Если РІС‹ хотите использовать надежный Рё качественный материал, вам стоит купить РљСЂСѓРі титановый Рў20 – ГОСТ 2171-90. РћРЅ обеспечит необходимую долговечность Рё надежность РІ вашей работе.
Posihipmeali
| #
Visit https://toptenscasinos.com/ and you will find a carefully selected list of the top 10 online casinos, games and strategies. Our goal is to provide informative, unbiased and entertaining content to help players make informed decisions in the world of online gaming. Helping players understand the games, rules and strategies in a simple and fun way.
уличные зонты
| #
Уютная терраса с металлической мебелью для комфортного отдыха
Уютная терраса с металлической мебелью для отдыха
Создание комфортного пространства на открытом воздухе требует не только вкуса, но и понимания особенностей различных материалов. Выбор мебели из металла – это разумный шаг, учитывая её долговечность и простоту в уходе. Сочетая стильный дизайн и практичность, она обеспечивает отличные условия для вечерних встреч с друзьями или семейных посиделок под солнцем.
Перед тем как выбрать предметы интерьера, стоит обратить внимание на несколько факторов: учитывайте климатические условия вашего региона. Например, алюминий не подвержен коррозии и идеально подходит для влажных мест. Отличная альтернатива – нержавеющая сталь, особенно если вы хотите добавить элементы современного стиля. Какую бы марку не выбрать, предлагаемые варианты обеспечат необходимую устойчивость и надежность.
Выбор текстур и дополнительных элементов тоже играет немалую роль. К примеру, отделка с порошковым покрытием делает поверхность устойчивой к повреждениям, а также обеспечивает возможность выбирать различные цвета. Не забывайте и о комфорте: добавление мягких подушек создаст атмосферу расслабленности, а использование теплых светильников позволит продлить приятные вечера на свежем воздухе.
Как выбрать металлические конструкции для площадки в зависимости от стиля и функционала
При выборе конструкций из металла важно учитывать их гармоничное сочетание с окружающей обстановкой. Для современного стиля подойдут минималистичные изделия с простыми формами и холодными оттенками. Такие модели, как правило, отличаются лаконичностью и функциональностью, что делает их универсальными.
Если вы предпочитаете классический подход, выбирайте разнообразные модели с изысканными деталями и декоративными элементами. Ковка, например, добавит нотку элегантности и станет акцентом во дворе.
Для стиля прованс или кантри хороши изделия с эффектом старения, которые визуально создают атмосферу старины и уюта. Крашенные в пастельные тона конструкции гармонично впишутся в такие ландшафты, подчеркнув природную красоту.
Что касается функционала, необходимо определиться, как именно вы планируете использовать пространство. Если вы мечтаете о месте для семейных сборищ, выбирайте долговечные столы и стулья с достаточным количеством посадочных мест. Для создания зоны отдыха лучше подходят диваны и кресла с мягкими подушками.
Не забудьте о защите от неблагоприятных погодных условий. Выбирайте модели с антикоррозийным покрытием или те, которые легко очищаются. Это позволит сохранить внешний вид на долгое время.
Также стоит подумать о дополнениях: подушки, пледы или столики сделают атмосферу более теплой и приветливой. Каждый элемент должен подчеркивать общую идею и сочетаться между собой.
Идеи по оформлению открытого пространства с металлической мебелью
Чтобы создать привлекательное и стильное место для времяпрепровождения, начните с выбора качественной конструкции. Выберите модели из алюминия или нержавеющей стали, которые устойч mat к атмосферным условиям. Их элегантные линии и разнообразие форм могут стать центральным элементом вашего дизайна.
Оформляя обстановку, используйте яркие текстильные акценты. Пуховые подушки, пледы и накидки добавят мягкости и сделают атмосферу более расслабляющей. Выбор текстиля с водоотталкивающей пропиткой позволит сохранить свежесть даже после дождя.
Забудьте о холодных и однообразных цветах. Экспериментируйте с палитрой: глубоко синие, оливковые и бордовые оттенки в сочетании с металлическими поверхностями создадут гармоничное пространство. Также можно рассмотреть вариант неоново-желтых или ярко-розовых подушек для молодежного стиля.
Добавьте живые растения, которые будут контрастировать с современными формами. Деревья в кадках, цветочные композиции или вертикальные садики оживят пространство и создадут ощущение близости к природе. К тому же, поддержка зелени улучшит микроклимат.
Освещение имеет большое значение. Установите декоративные светильники, гирлянды или фонари, чтобы обеспечить мягкий свет в вечернее время. Встраиваемые светильники в пол или на стенах создадут уютную атмосферу и выделят ключевые зоны вашего дизайна.
Для обеспечения удобства и практичности стоит разместить несколько каркасов для хранения: специальные контейнеры под сиденьями или открытые полки. Они помогут организовать пространство, сохраняя порядок и функциональность.
https://joycom.ru/ulichniy-zont/
Sazrizk
| #
Заказать диплом под заказ возможно используя официальный сайт компании. 1mog.listbb.ru/viewtopic.phpf=6&t=10604
Donaldlaf
| #
bitcoin dark web darknet market links
купить садовые качели
| #
Обзор террасной мебели материалы для комфортного отдыха
Террасная мебель обзор популярных материалов для отдыха
При выборе уюта на открытом воздухе важно учитывать, как качественные компоненты могут определить атмосферу вашего пространства. Существует множество вариантов, каждый из которых имеет свои плюсы и минусы. Рассмотрим, какие сочетания материала придают не только эстетическую привлекательность, но и долговечность.
Деревянные конструкции давно зарекомендовали себя в создании естественной гармонии. Разнообразие пород, таких как акация, тик и лиственница, может обеспечить не только стильный внешний вид, но и стойкость к различным погодным условиям. Обработка защитными средствами значительно продлевает срок службы такой инфраструктуры, сохраняя ее первоначальные качества.
Металлические элементы, в частности, алюминий и нержавеющая сталь, становятся все популярнее благодаря своей лёгкости и прочности. Они устойчивы к коррозии и не требуют особого ухода, что делает их идеальными для состава разнообразных конструкций. Современные технологии позволяют придавать металлическим деталям разнообразные отделки, что открывает горизонты для креативных решений.
Не забудем и о пластике. Этот материал представляет собой идеальное решение для тех, кто ищет лёгкость и разнофункциональность. Суррогатные полимеры могут имитировать текстуру дерева и камня, при этом оставаясь устойчивыми к выцветанию и повреждениям. Таким образом, использование высококачественного пластика добавляет не только современный стиль, но и практичность.
Как выбрать подходящие компоненты для уличной мебели
При покупке предметов для открытых пространств важно учесть не только внешний вид, но и способность материалов противостоять внешним воздействиям. Неправильный выбор может привести к быстрому ухудшению состояния изделий. Рассмотрим ключевые элементы, на которые следует обратить внимание.
Первое, что стоит проверить, это устойчивость к влаге. Некоторые виды древесины, такие как тик или кедр, обладают естественной влагостойкостью, что позволяет им сохранять свои качества под открытым небом. Альтернативой могут стать изделия из пластика или композитов, которые не боятся дождя и снега.
Второй аспект – это прочность. Металлические конструкции, например, из алюминия, легко справляются с нагрузками и не боятся деформации при активном использовании. Однако важно выбирать модели, обработанные от коррозии, чтобы продлить их срок службы.
Не менее значимой характеристикой является комфорт. Обивка сидений из специального водоотталкивающего текстиля обеспечит удобство даже в условиях повышенной влажности. Также стоит обратить внимание на наполнители, которые не сохраняют влагу и быстро высыхают.
Цвет и текстура тоже играют роль. Яркие оттенки могут быстро выгореть под солнцем, в то время как темные тона менее подвержены этому процессу. Важно подобрать решения, гармонирующие с окружающим пространством.
Долговечность – критический фактор. Для этого стоит обратить внимание на сертификаты и гарантии от производителей. Модели с хорошей репутацией, прошедшие строгие тесты, обеспечат надежность на долгие годы.
Наблюдая за этими аспектами, можно создать пространство, которое будет служить не только как место для отдыха, но и станет эстетическим дополнением к вашему участку.
Преимущества и недостатки популярных материалов для мебели на свежем воздухе
Дерево – традиционный выбор. Оно привносит тепло и уют. Устойчивое к механическим повреждениям, хорошо пропускает воздух. Однако требует регулярного ухода. Лак или масло помогут защитить от влаги и насекомых, но с течением времени могут потребоваться замены частей.
Металл обладает прочностью и долгим сроком службы. Алюминий и сталь хорошо противостоят погодным условиям. Легкий и поддается переработке, алюминий не ржавеет. Но холодные поверхности могут быть нелицеприятными летом, а сталь требует дополнительного антикоррозийного покрытия.
Пластик выгоден благодаря своей легкости и разнообразии форм. Он легко чистится и устойчив к ультрафиолету. Однако низкое качество может привести к быстрому износу. Важно выбирать изделия из переработанного или качественного первичного пластика.
Текстиль, используемый для мягкой мебели, обеспечивает комфорт. Водонепроницаемые и ультрафиолетовые свойства позволяют использовать его на улице. Тем не менее, со временем могут появляться загрязнения, а некоторые ткани не выдерживают низких температур.
Каждый из этих материалов имеет свои нюансы. Выбор стоит делать, исходя из климата, частоты использования и желаемого уровня ухода. Правильный подход к выбору позволит создать уютное пространство на открытом воздухе.
https://joycom.ru/sadovie-kacheli/
generic rizatriptan pills
| #
Medicament information. Brand names.
generic rizatriptan pills
Actual what you want to know about drug. Get information now.
Pingrutty
| #
dark market 2025 darkmarket list
Cazrysu
| #
Приобрести диплом института по выгодной стоимости можно, обратившись к проверенной специализированной фирме. Мы можем предложить документы ВУЗов на Ваш выбор, которые расположены на территории всей России. 4x4zubry.by/forum/messages/forum1/topic53820/message350845/result=new#message350845
Willieavamy
| #
The live games online rock—fan love! mines
Toliknaf
| #
dark markets darknet market
@seomax_nakrutka_pf
| #
Доброго!
Накрутка ПФ — эффективный способ улучшить позиции сайта в Яндексе. Поддерживаем положительную динамику в Яндекс.Вебмастере. Индивидуальный подход к каждому проекту и точная аналитика. Telegram: Contact @seomax_nakrutka_pf Поддерживаем положительную динамику в Яндекс.Вебмастере.|Обеспечиваем рост видимости за счёт прокачки ПФ через Яндекс. Поддерживаем положительную динамику в Яндекс.Вебмастере. Индивидуальный подход к каждому проекту и точная аналитика. Telegram: Contact @seomax_nakrutka_pf Поддерживаем положительную динамику в Яндекс.Вебмастере.|Увеличиваем поведенческий трафик и снижаем отказ от сайта. Анализируем нишу, моделируем естественное поведение, добиваемся результата. Настраиваем стратегию под цели клиента и анализируем результат. Telegram: Contact @seomax_nakrutka_pf Гибкая настройка сценариев поведения и работа по ключевым фразам.|
накрутка пф быстротоп, софт по накрутке пф, купить робот для накрутки пф
накрутка пф 5173454, яндекс пф, накрутка пф сервисы
Удачи!
Jamesdusty
| #
Цветы пережили трёхчасовую пробку и всё равно выглядели свежо!
заказ цветов томск с доставкой
Trefrso
| #
Приобрести диплом университета. Приобретение документа о высшем образовании через надежную компанию дарит ряд плюсов. Данное решение позволяет сэкономить время и серьезные средства. pro-eltern.de/read-blog/609
Планета как дом
| #
Планета как дом
A Brief History of Elemental Analysis » Artemis Analytical
Willieavamy
| #
I wish online paid swift—drag off! plinko
1win_gkKt
| #
1win pariuri https://1win5027.ru/ .
Rabyitabe
| #
dark web sites darkmarket url
Willieavamy
| #
The online thrill bites—stay firm! plinko
Willieavamy
| #
Online bingo chills—cool gang! dragon tiger
DonDonfaita
| #
darknet market darknet drug links
binance kodu
| #
Your point of view caught my eye and was very interesting. Thanks. I have a question for you.
Pingrutty
| #
dark websites dark market 2025
does promethazine and codeine go bad
| #
Medication information for patients. What side effects?
does promethazine and codeine go bad
Everything news about medicines. Read here.
Donaldlaf
| #
darknet marketplace dark web market links
JupudtCow
| #
На сайте http://oknaksa.ru закажите расчет технического задания, чтобы воспользоваться такой важной, полезной услугой, как остекление. На монтажные работы, а также сами конструкции предоставляются гарантии, вы сможете воспользоваться бесплатным обслуживанием. А все работы выполняются точно по ГОСТу, в соответствии с требованиями. Предоставляются исчерпывающие и содержательные консультации. Вам будет предложено несколько вариантов решения проблемы. Компания располагает огромным количеством готовых проектов.
melbet_fupa
| #
мелбет кж melbet1001.ru .
metamask chrome extension
| #
MetaMask Chrome works flawlessly! I use it daily for DeFi transactions and NFT trading. Smooth and secure experience every time.
HomerFoday
| #
ремонт телевизоров в Красноярске Ремонт телевизоров любых моделей в Красноярске: ЖК, LCD, LED, плазменных панелей. Выезд на дом. Диагностика – БЕСПЛАТНО! Доступные цены. Срочный ремонт от 2-х часов.
Toliknaf
| #
dark web market dark web marketplaces
Cazrowt
| #
Заказать диплом о высшем образовании поспособствуем. Купить диплом в Ижевске – diplomybox.com/kupit-diplom-izhevsk
melbet_rkpa
| #
мелбет kg http://melbet1001.ru .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
поставляемая продукция:
Штамповая поковка 85 мм 3Х2Н2МВФ ГОСТ 1133-71 Купить штамповые поковки высокого качества от производителя. Гарантия прочности и долговечности. Широкий выбор форм и размеров. Доставка по России.
Thomaslob
| #
plinko deutschland
Thomaslob
| #
plinko играть
1win_iaMr
| #
1win партнерка вход 1win партнерка вход .
Sazrgfs
| #
Покупка документа о высшем образовании через надежную компанию дарит множество плюсов для покупателя. Данное решение помогает сберечь время и существенные финансовые средства. Однако, преимуществ значительно больше.Мы готовы предложить дипломы любых профессий. Дипломы изготавливаются на подлинных бланках. Доступная стоимость в сравнении с крупными тратами на обучение и проживание в чужом городе. Заказ диплома ВУЗа будет выгодным шагом.
Заказать диплом о высшем образовании: rusd-diplomj.ru/poluchite-diplom-s-zaneseniem-v-reestr-bistro-i-legko/
Rabyitabe
| #
darknet sites darknet sites
1win_vjpl
| #
игра ракета на деньги 1win https://1win7006.ru .
Pingrutty
| #
darknet drug market darknet websites
HuvizoTon
| #
Предприятие «Энерго Техстрой 2000» поставляет производственные материалы, технику российского производства. Ими смогут воспользоваться любые производственные предприятия страны. На сайте https://best-vendor.ru изучите полный ассортимент товаров, которые вы сможете приобрести в любое время, а доставка происходит наиболее комфортным для вас способом
DonDonfaita
| #
dark websites dark web market links
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их происхождение. Превосходное обслуживание – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы а также находить ответы под особенности вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
Порошок магниевый HK31A – ASTM B90 Рзделия РёР· магния HK31A – ASTM B90 – это высококачественная продукция, отличающаяся своей легкостью Рё прочностью. Рзготовленные РїРѕ стандартам ASTM B90, РѕРЅРё обеспечивают отличные механические свойства Рё устойчивость Рє РєРѕСЂСЂРѕР·РёРё. Рти изделия идеально РїРѕРґС…РѕРґСЏС‚ для использования РІ различных отраслях, включая автомобильную Рё аэрокосмическую. Если РІС‹ хотите купить Рзделия РёР· магния HK31A – ASTM B90, то можете быть уверены РІ РёС… надежности Рё долговечности. Заказывая Сѓ нас, РІС‹ получаете отличный баланс между ценой Рё качеством, что делает эти изделия лучшим выбором для вашего бизнеса.
Thomaslob
| #
plinko app recensioni
XaFiUNecope
| #
На сайте https://officepro54.ru представлено огромное количество мебели, которая идеально подходит для организации офисного пространства. Вся она выполнена из инновационных, уникальных материалов, а потому прослужит очень долго, не синтезирует в воздух опасных веществ. В разделе вы найдете кабинет руководителя, функциональные и вместительные столы для переговоров, шкафы-купе, сейфы, акустические системы, ученическую мебель и многое другое. Постоянно действуют акции. На продукцию установлены привлекательные цены.
Thomaslob
| #
mines app download
can i purchase fosamax tablets
| #
Meds information for patients. Brand names.
can i purchase fosamax tablets
Actual about meds. Get here.
Thomaslob
| #
plinko application
Thomaslob
| #
monopoly big baller result
Thomaslob
| #
betonred
Donaldlaf
| #
darknet drugs darknet markets onion address
Cazrrdj
| #
Мы можем предложить дипломы любых профессий по приятным тарифам. ЗАДАТЬ ВОПРОС — kyc-diplom.com/discussions.html
Thomaslob
| #
fishin frenzy slots
Thomaslob
| #
fishin frenzy free spins
Mazreki
| #
Где приобрести диплом специалиста?
Готовый диплом со всеми печатями и подписями 100% отвечает стандартам Министерства образования и науки, неотличим от оригинала. Не стоит откладывать личные мечты на долгие годы, реализуйте их с нами – отправьте заявку на изготовление документа уже сегодня! Диплом о среднем образовании – не проблема! nashforum.listbb.ru/viewtopic.phpf=6&t=26182
melbet_gjen
| #
mel bet сайт https://www.melbet1002.ru .
Toliknaf
| #
bitcoin dark web darknet websites
GarryZoona
| #
Дорого игрок, идешь где можно купить скины КС 2 или КСГО? Отлично,твой поиск официально завершен! В этой подборке сайтов вы найдете самые лучшие магазины для покупки дешевых скинов КС 2 купить скины кс2 . Предлагает непревзойденный опыт покупки скинов, включая все, от редких коллекционных предметов до самых рентабельных игровых скинов.
1win_vxKt
| #
1win молдова http://www.1win5027.ru .
Rabyitabe
| #
darknet marketplace dark market link
Pingrutty
| #
tor drug market darknet sites
melbet_coen
| #
melbet https://melbet1002.ru .
mostbet_bjKa
| #
motbet http://www.mostbet5006.ru .
Najarsvug
| #
Visita el sitio https://lucky-lady-charm.online/ donde podras leer una resena completa de la maquina tragamonedas Lucky Lady Charm Deluxe. Aprendera consejos y recomendaciones utiles sobre como jugar a la tragamonedas, lineas de pago y apuestas, aprendera todo sobre los bonos, asi como consejos sobre como ganar en Lucky Lady Charm.
DonDonfaita
| #
darkmarket url dark market
melbet_rgen
| #
мелбет http://www.melbet1002.ru .
Pixunlipglice
| #
Система менеджмента управления качеством содержит критерии менеджмента и стандарты, по которым проводится последующая сертификация предприятия. Чтобы получить сертификат СМК, нужно обратиться в аккредитованный сертификационный центр. Сертификация систем менеджмента качества является документальным подтверждением того, что система менеджмента сертифицированной компании соответствует требованиям того или иного стандарта. Подробнее: https://ok.ru/group/70000034956977
Thomaslob
| #
betclic cote d’ivoire
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы
switching from lexapro to duloxetine
| #
Medicines information sheet. Generic Name.
switching from lexapro to duloxetine
Everything trends of medicines. Read here.
Thomaspoext
| #
Доставили ночью, как и просили!
букет пионов
Thomaslob
| #
betonred casino
Donaldlaf
| #
dark web sites dark markets
Sazrdzz
| #
Приобрести диплом под заказ вы сможете используя сайт компании. animesocial.su/blogs/64421/Услуги-по-изготовлению-дипломов
mostbet_nqmr
| #
1вин rossvya http://mostbet5007.ru .
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности
Toliknaf
| #
darknet markets onion address dark web market list
Pingrutty
| #
tor drug market darknet market links
Mazrtfo
| #
Мы готовы предложить дипломы любой профессии по невысоким тарифам. Всегда стараемся поддерживать для клиентов адекватную ценовую политику. Важно, чтобы документы были доступны для подавляющей массы наших граждан.
Приобретение диплома, подтверждающего окончание института, – это выгодное решение. Заказать диплом о высшем образовании: diplom-bez-problem.com/kupit-diplom-texnikuma-ob-okonchanii/
Haroldemugs
| #
Looking for it AI Editing? Xnudes.app is the best NSFW generator with AI pictures. The Desire chat engine never forgets information about you, it’s incredibly immersive and realistic, and it’s the most immersive chat experience you’ve ever seen in an artificial intelligence service. Using deep knowledge models, these generators can produce visual illustrations in any variations.
site
| #
Nicely put, Thanks a lot!
skladi_huSi
| #
склады хранение для вещей склады хранение для вещей .
Rabyitabe
| #
darknet drug links dark market 2025
mostbet_srmr
| #
1 win.kg http://mostbet5007.ru .
Thomaslob
| #
betonred opinie
Thomaslob
| #
red on bet
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности
Cazrhlk
| #
Приобрести диплом ВУЗа по выгодной стоимости вы сможете, обращаясь к надежной специализированной фирме. Мы можем предложить документы университетов, которые находятся в любом регионе России. onelife.forumex.ru/viewtopic.phpf=3&t=731
DonDonfaita
| #
darknet websites darkmarket 2025
mostbet_xbmr
| #
1win.online mostbet5007.ru .
Thomaslob
| #
betonred casino
Thomaslob
| #
aviator bet
Richarddix
| #
Портал для женщин https://gracefulwoman.kyiv.ua которые ценят красоту жизни. Практичные советы, душевные статьи и поддержка — о том, как быть счастливой, уверенной и гармоничной каждый день.
HerbertBEf
| #
Современный женский https://happylady.kyiv.ua сайт с ежедневными обновлениями: советы по стилю, уходу, семье и психологии. Всё, что волнует и вдохновляет женщин сегодня — в одном месте.
PhilipARted
| #
Портал про авто https://impactspreadsms.com новости, обзоры, тест-драйвы, советы, сравнение моделей и актуальные тенденции в мире автомобилей. Всё для автолюбителей и профессионалов.
Virgilnains
| #
Онлайн-портал про автомобили https://impactspreadsms.com с каталогом моделей, тестами, аналитикой, ценами и отзывами. Удобный поиск и полезные материалы — для тех, кто выбирает с умом.
Thomaslob
| #
dragon tiger download
site
| #
Very good stuff. Appreciate it.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Плоская мишень из фтористого магния
cost generic hydrea pills
| #
where can i get generic hydrea price
can i order generic levaquin without a prescription
| #
Drugs information leaflet. What side effects can this medication cause?
can i order generic levaquin without a prescription
Everything information about medicines. Read now.
https://accordbrokers.co.nz/operation-free-assange-engaged/
| #
I do noot even understand how I ended up here, but I assumed this publish was
once good. I do not understand who you’re but certainly you’re going to a
well-known blogger when yoou aren’t already. Cheers!
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации —
Donaldlaf
| #
darknet markets onion address darknet markets onion
melbet_uzpa
| #
мелбет http://melbet1001.ru/ .
Pingrutty
| #
darkmarket link darknet market links
Thomaslob
| #
plinko game download pakistan
Toliknaf
| #
dark web market urls dark market onion
wenizMiz
| #
Tent3302.ru предлагает необходимые автодетали. Мы продаем запчасти на Газель Некст. Под заказ двигатель Газель Камминз 2.8 поставляем. Стараемся поддерживать цены на самом низком уровне. Продажи выполняем со склада за безналичный и наличный расчет. Ищете раздатка газ-66 гур камаз гур зил 130 кпп газ 53 редуктор газон некст? Tent3302.ru – тут можете детальную о нас информацию отыскать. Продаем качественную продукцию. Шины на Газель особенным спросом у клиентов пользуются. Стараемся накладные расходы и транспортные максимально сократить. Вы можете быть уверены в вежливом и грамотном обслуживании.
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы —
Sazrilm
| #
Приобрести документ ВУЗа можно в нашей компании в Москве. Мы оказываем услуги по производству и продаже документов об окончании любых университетов РФ. Вы сможете получить необходимый диплом по любым специальностям, включая документы Советского Союза. Даем гарантию, что в случае проверки документа работодателями, подозрений не возникнет. diplomskiy.com/visshee-obrazovanie-kupit-diplom-s-zaneseniem-4/
Sazroiz
| #
Приобрести диплом ВУЗа!
Приобрести диплом университета по доступной стоимости вы можете, обращаясь к надежной специализированной компании. Заказать диплом о высшем образовании: kupitediplom0027.ru/nastoyashij-diplom-s-reestrom-garantiya-podlinnosti
Rabyitabe
| #
dark web market darknet websites
crypto casinos australia
| #
crypto casinos australia
A Brief History of Elemental Analysis » Artemis Analytical
plinko argent reel avis
| #
plinko argent reel avis
A Brief History of Elemental Analysis » Artemis Analytical
Самоидентичность сегодня
| #
Very nice post. I just stumbled upon your blog and wished to say that I have really enjoyed browsing your blog posts.
In any case I’ll be subscribing to your rss feed and I hope you write again very soon!
Jeffreyset
| #
Ищете курсы по программированию со скидкой? Рекомендую заглянуть сюда https://say.la/Skladchik
Thomaslob
| #
fishin frenzy free play
plinko gambling
| #
Guide to Integrated Online Games with Contextual Insights
Guide to Playing Integrated Online Games with Context
The modern interactive entertainment experience transcends mere amusement, evolving into a complex fusion of strategy, storytelling, and community engagement. As participants seek immersive experiences that challenge their creativity and analytical skills, understanding the nuances of these platforms becomes increasingly significant. This exploration dissects various methodologies that enhance user experience through intricate systems of interaction.
One pivotal aspect to consider is how design elements interplay with player psychology. Incorporating elements such as adaptive difficulty levels or narrative branching can significantly influence user retention and satisfaction. By analyzing user behavior patterns, developers can tailor experiences that resonate deeply with individual preferences, thus creating a more fulfilling atmosphere.
Furthermore, examining the social dynamics at play reveals how collaboration and competition shape player engagement. Creating environments that encourage teamwork or healthy rivalry fosters a unique sense of community, which can elevate the overall experience. Leveraging these social factors not only enhances enjoyment but also promotes longevity within platforms, making it critical for developers to innovate continually.
How to Analyze Player Behavior for Enhanced Engagement
Understanding player actions requires a thorough approach, integrating various data sources to build a comprehensive profile. Begin by collecting quantitative metrics such as session length, frequency of play, and in-game purchases. These figures provide a solid foundation for identifying patterns and trends in user engagement.
Qualitative data is equally important; implement surveys or in-game feedback mechanisms to capture player sentiments. This can reveal motivations behind specific behaviors, such as why a player might abandon a task or seek assistance. Use tools like heatmaps to visualize areas where players commonly interact or disengage, facilitating targeted improvements.
Segment your audience based on behavior types. For instance, differentiate between casual players who enjoy exploration and competitive players focused on achievements. Tailoring experiences to these segments enhances personal connection and satisfaction. Consider creating adaptive challenges that respond to player skill levels, ensuring that each user feels both challenged and entertained.
Employ A/B testing to experiment with various game mechanics or narrative paths. Track how these changes impact engagement metrics. By analyzing the data, discern which elements resonate most with players and iterate based on those insights.
Analyze player interactions in real-time through analytics platforms. This allows for immediate responses to fluctuations in engagement, whether it be increased interest in certain features or a sudden drop in participation. Adjust gameplay dynamics or introduce timely events to re-engage users effectively.
Lastly, develop a community around your offering. Encourage players to share experiences and suggestions, fostering a sense of belonging. Engagement often flourishes in environments where players feel heard and valued, leading to enhanced retention and satisfaction.
Implementing Real-Time Feedback Mechanisms in Online Gaming Environments
The integration of real-time feedback systems is crucial for shaping player experiences and enhancing engagement levels. Leveraging data analytics can provide meaningful insights into player behavior, preferences, and patterns. Implementing these mechanisms allows developers to adapt gameplay dynamically.
One effective approach is to utilize telemetry data. This data can track player actions, decision-making processes, and pacing throughout a session. For instance, mapping out areas where players frequently fail can lead to targeted adjustments in difficulty or guidance, which helps in maintaining player interest without causing frustration.
Another strategy involves incorporating instant feedback loops. By using notifications or in-game prompts that offer suggestions based on performance can guide players to make better decisions. For example, if a player frequently misses key objectives, tailoring hints or visual cues can aid in their understanding, enhancing overall satisfaction.
Additionally, employing machine learning algorithms can optimize feedback. These models can analyze patterns and predict player needs, offering personalized advice or altering challenges in real-time. This level of customization can make a significant impact on player retention and overall experience.
Creating community-driven feedback channels is also beneficial. Enabling players to share experiences through forums or social media can foster a sense of belonging and collaboration. Real-time polls or surveys can gather reactions on specific features or gameplay changes, providing developers with timely user input.
Implementing these mechanisms requires robust infrastructure. Ensuring low-latency connections and reliable data processing is essential for maintaining a smooth user experience. The technical architecture should be designed to handle spikes in activity, particularly during peak times or events.
Finally, continuous iteration is key. Regularly revisiting feedback mechanisms to assess their performance can uncover new opportunities for improvement. By monitoring player satisfaction and engagement metrics, developers can refine their approaches and adjust strategies accordingly.
Here is my blog :: https://plinko-reviews.co.uk
GunefUsano
| #
На сайте http://www.lilibum.ru вы найдете много анкет реальных людей для того, чтобы завести знакомства, найти свою вторую половинку и просто интересно провести время, если пригласите кого-то на свидание. Для того чтобы составить мнение о человеке, можно обменяться фотографиями и пообщаться на определенные темы, которые вас интересуют. Самые привлекательные и интересные люди вашего города находятся на этом портале. Знакомьтесь, влюбляйтесь и создавайте семьи. Для получения доступа ко всем функциям, пройдите регистрацию.
DonDonfaita
| #
darknet market lists dark web market list
JamesNob
| #
Свежие новости Украины https://fraza.kyiv.ua и мира — политика, экономика, общество, культура, технологии. Главные события дня, оперативные обновления и аналитика от экспертов.
JohnnieInoft
| #
Модный журнал онлайн https://icz.com.ua одежда, аксессуары, макияж, прически, уличный стиль и haute couture. Следите за последними тенденциями и читайте советы экспертов индустрии.
DennisRap
| #
Современный портал https://lady.kyiv.ua для женщин: мода, уход, любовь, дети, стиль жизни и вдохновение. Полезный контент, тренды и темы, близкие каждой.
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности
Thomaslob
| #
demo fortune tiger
Davidfet
| #
Актуальные новости Украины https://lenta.kyiv.ua и мира на одном сайте. Подборка ключевых событий, факты, интервью, мнения и видео. Честно, быстро и без фейков.
Pingrutty
| #
darkmarket link darknet site
Cazrbjf
| #
Где приобрести диплом по необходимой специальности?
Мы изготавливаем дипломы любой профессии по разумным ценам. Важно, чтобы дипломы были доступными для большого количества граждан. Заказать диплом об образовании diplomgorkiy.com/kupit-diplom-vuza-s-zaneseniem-v-reestr-21/
can i order motrin
| #
Pills information for patients. Effects of Drug Abuse.
can i order motrin
Everything what you want to know about drugs. Get information now.
Donaldlaf
| #
dark web market urls darknet market
LesterLarty
| #
Сервисный центр Мобиопт – Ремонт телефонов Киров
Markcen
| #
Новостройки или вторичка? Узнайте подробности
Toliknaf
| #
darknet site dark market
can i purchase nemasole without rx
| #
get nemasole without insurance
Mazrlox
| #
Мы готовы предложить дипломы психологов, юристов, экономистов и любых других профессий по приятным ценам. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Важно, чтобы дипломы были доступны для большинства наших граждан.
Покупка документа, подтверждающего обучение в университете, – это грамотное решение. Заказать диплом любого ВУЗа: diplom5.com/kupit-diplom-ekb-2/
1win_geKt
| #
1 win md http://1win5027.ru/ .
Sazrylk
| #
Заказать диплом института по невысокой стоимости возможно, обратившись к надежной специализированной компании. Мы предлагаем документы об окончании любых ВУЗов РФ. Приобрести диплом университета– kupitediplom.ru/kupit-diplom-v-reestre-bistro-i-legko/
melbet_pren
| #
melbet http://www.melbet1002.ru .
Sazrcrz
| #
Мы изготавливаем дипломы любой профессии по приятным ценам.
Вы приобретаете документ через надежную и проверенную фирму. Заказать диплом о высшем образовании– http://povzroslomu.kabb.ru/viewtopic.phpf=3&t=971/ – povzroslomu.kabb.ru/viewtopic.phpf=3&t=971
Rabyitabe
| #
darknet market lists best darknet markets
site
| #
This is nicely expressed! .
plinko
| #
plinko
A Brief History of Elemental Analysis » Artemis Analytical
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы
dragon money_dbKl
| #
dragon казино dragon казино .
Thomaslob
| #
jogo do tigrinho fortune tiger
kalkylyator OSAGO_yson
| #
страховка на машину стоимость калькулятор seodict.ru .
Thomaslob
| #
bet on red free spins
1win_enPn
| #
1win футбол https://1win7019.ru/ .
ArthurBoN
| #
https://telegra.ph/Kak-vybrat-kolpaki-na-stolby-dlya-zabora-starinnogo-doma-04-18
Treflaa
| #
Приобрести диплом любого университета. Покупка документа о высшем образовании через проверенную и надежную компанию дарит ряд достоинств для покупателя. Такое решение позволяет сэкономить как длительное время, так и значительные денежные средства. rodina.listbb.ru/viewtopic.php?f=3&t=2495
Pingrutty
| #
darkmarket 2025 tor drug market
DonDonfaita
| #
darknet drug store dark market link
Iariorddv
| #
Приобрести диплом университета по доступной цене возможно, обратившись к надежной специализированной фирме. Приобрести документ о получении высшего образования вы можете в нашей компании. dip-lom-rus.ru/kupit-diplom-v-moskve-s-zaneseniem-v-reestr-14
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
поставляемая продукция:
Латунное литое кольцо 400С…600 РјРј ЛЦ40РЎРґ Купите латунные литые кольца РѕС‚ производителя. Великолепное сочетание прочности Рё эстетики. Высокое качество РїРѕ доступной цене. РЁРёСЂРѕРєРёР№ выбор форм Рё диаметров. Рдеальны для ювелирных изделий Рё производства музыкальных инструментов.
1win_vfpl
| #
1вин rossvya 1вин rossvya .
1win_avmi
| #
1win партнерка вход 1win7007.ru .
Sazruap
| #
Всегда думал что купить диплом о высшем образовании это миф и нереально, но все оказалось не так, изначально искал информацию про: купить диплом бакалавра в ростове на дону, диплом купить с занесением в ростове, диплом электромеханика морского судна купить ростов, купить диплом в омске о среднем образовании, купить диплом специалиста в омске, потом про дипломы вузов, подробнее здесь diplomybox.com/kupit-diplom-tekhnikuma-kolledzha-v-sankt-peterburge
Thomaslob
| #
fortune tiger
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы
Matthewhot
| #
https://mymoscow.forum24.ru/?1-6-0-00026564-000-0-0-1741947496
RickyMum
| #
Главные новости https://lentanews.kyiv.ua из Украины и со всего мира — ежедневно и без искажений. Всё, что важно знать: внутренняя политика, экономика, международные события и прогнозы.
WilliamAgown
| #
Онлайн-портал для современных https://madrasa.com.ua женщин. Всё, что волнует и вдохновляет: от красоты и моды до жизненного баланса, мотивации и личностного роста. Будь собой — с нами.
DanielCheag
| #
Портал для женщин https://maleportal.kyiv.ua мода, красота, здоровье, отношения, карьера, семья и вдохновение. Актуальные темы, полезные советы и поддержка для каждой женщины — всё в одном месте.
where to buy requip without a prescription
| #
Medication prescribing information. Short-Term Effects.
where to buy requip without a prescription
Actual trends of drugs. Get here.
Bernardflelf
| #
Клуб для беременных https://mam.ck.ua и молодых мам: питание, подготовка к родам, уход за малышом, послеродовое восстановление. Полезная информация, консультации и поддержка в одном месте.
WilliamDix
| #
Мы готовы предложить дипломы любой профессии по выгодным тарифам. Дипломы производят на фирменных бланках Купить диплом о высшем образовании fastdiploms.com
site
| #
Thanks! I value this.
Raymondhah
| #
http://forum.toonboom.ru/index.php?/topic/5111-тонизирующий-напиток-eazy-energy/
Donaldlaf
| #
darknet market lists darkmarkets
mostbet_ojmr
| #
1vin https://www.mostbet5007.ru .
dragon money_then
| #
драгон мани кэшбэк https://www.dragon-money42.com .
dragon money_jqSi
| #
драган мани dragon-money36.com .
Toliknaf
| #
best darknet markets dark web market
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – наш стандарт – мы на связи, чтобы улаживать ваши вопросы а также предоставлять решения под специфику вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
РўСЂСѓР±Р° висмутовая РџРћРЎР’Рё 42-10,5 РўСЂСѓР±Р° висмутовая РџРћРЎР’Рё 42-10,5 предназначена для использования РІ специализированных областях, таких как медицина Рё химическая промышленность. Благодаря высокой стойкости Рє РєРѕСЂСЂРѕР·РёРё Рё отличным теплопроводным свойствам, данная труба обеспечивает надежную эксплуатацию РІ условиях, требующих постоянного контроля Р·Р° температурными режимами. Если РІС‹ ищете качественное решение, которое соответствует современным требованиям, РІС‹ можете купить РўСЂСѓР±Р° висмутовая РџРћРЎР’Рё 42-10,5. Ртот РїСЂРѕРґСѓРєС‚ будет отличным выбором для ваших проектов, обеспечивая эффективность Рё долговечность.
Thomaslob
| #
dragon tiger game download 51 bonus
Thomaslob
| #
dragon tiger predictor
Jeffreyset
| #
Ищете курсы по программированию со скидкой? Рекомендую заглянуть сюда https://say.la/Skladchik
Raymondhah
| #
https://www.floristic.ru/forum/groups/moskva-d895-toniziruyuschii-napitok-eazy-energy.html#gmessage1382
Rabyitabe
| #
darkmarket dark market 2025
Pingrutty
| #
dark web drug marketplace darknet market lists
1win_nqmi
| #
1 вин 1 вин .
top online casinos
| #
top online casinos
A Brief History of Elemental Analysis » Artemis Analytical
Dnrtcou
| #
Мы можем предложить дипломы любой профессии по выгодным тарифам. Мы можем предложить документы ВУЗов, которые расположены в любом регионе России. Дипломы и аттестаты печатаются на “правильной” бумаге самого высокого качества. Это позволяет делать государственные дипломы, не отличимые от оригинала. diafan.rusheat.ru/forum/legalnoe-oformlenie-diplomov
1win_jlKr
| #
мотбет http://1win5028.ru/ .
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации —
Mazrcfo
| #
Мы предлагаем дипломы любой профессии по приятным ценам. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Для нас очень важно, чтобы дипломы были доступны для большого количества граждан.
Покупка диплома, который подтверждает окончание института, – это грамотное решение. Купить диплом любого ВУЗа: 1magistr.ru/kupit-vuz-diplom/
DonDonfaita
| #
darkmarket dark market 2025
how to buy generic bactrim without a prescription
| #
cost of generic bactrim without prescription
Thomaslob
| #
betonred
Charlesonexy
| #
Сайт для деловых людей https://manorsgroup.com.ua актуальные материалы о финансах, инвестициях, рынке недвижимости и управлении капиталом. Аналитика, обзоры, экспертные мнения и полезные инструменты.
Robertjer
| #
Женский онлайн-журнал https://mcms-bags.com о красоте, моде, психологии, отношениях и стиле жизни. Актуальные статьи, советы экспертов, вдохновение и всё, что важно для современной женщины.
dragon money_tgKl
| #
dragonmoney сайт https://www.dragon-money37.com .
Lazrgan
| #
Где заказать диплом специалиста?
Купить диплом университета по выгодной стоимости вы можете, обращаясь к надежной специализированной компании.: kupit-diplomyz24.com
Harryquaby
| #
бонусы казино за депозиты в криптовалюте
BrandonCreex
| #
Новостной портал https://mediashare.com.ua с актуальной информацией из Украины, мира, политики, экономики, технологий, культуры и общества. Только проверенные источники и объективные материалы.
should i take potassium with hydrochlorothiazide
| #
Medicament information sheet. Cautions.
should i take potassium with hydrochlorothiazide
Some news about medicines. Get information now.
1win_fmKr
| #
мостбет казино войти http://1win5028.ru .
dragon money_efen
| #
dragon money официальный сайт играть онлайн http://dragon-money42.com/ .
DennisStush
| #
cd alarm clock radio alarm clock
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Мишень из ниобия для распыления
Donaldlaf
| #
dark web market urls dark market link
1win_qrSi
| #
1win зайти https://www.1win7018.ru .
kalkylyator OSAGO_uhon
| #
рассчитать страховку на машину онлайн калькулятор http://seodict.ru/ .
1win_tdpl
| #
1win kg https://1win7006.ru/ .
Eanrola
| #
Для удачного продвижения вверх по карьере понадобится наличие диплома о высшем образовании. Приобрести диплом о высшем образовании у сильной компании: dip-lom-rus.ru/kupit-kachestvennij-texnicheskij-diplom-bistro-i-vigodno/
dragon money_qyEr
| #
казино dragon мани http://dragon-money40.com .
Harryquaby
| #
cryptoboss casino официальный зеркало
Toliknaf
| #
darknet drug links darknet links
1win_nwSi
| #
1win kg https://www.1win7018.ru .
Pingrutty
| #
darknet markets url onion dark website
Thomaslob
| #
plinko jogo
Markcen
| #
Новостройки или вторичка? Полезные рекомендации —
RakijamPreap
| #
Посетите B2BOX https://b2box.ru/ – это уникальное комплексное решение для создания упаковки всех видов и направлений. Мы делаем упаковку, которая продает! Упаковочные решения для ритейла, создание индивидуальной и SRP упаковки, услуги дизайнерского бюро, производство транспортной упаковки, собственное плоттерное производство, все виды печати – это и многое другое вы найдете, посетив наш сайт.
Rabyitabe
| #
dark market onion darknet links
mostbet_hxmt
| #
мостбет зеркало http://mostbet6033.ru .
Hijamnetly
| #
На сайте https://pilmi.ru вы найдете огромное количество фильмов самых разных жанров, включая комедии, мелодрамы, драмы, романтические. Для того чтобы подыскать наиболее подходящий вариант, нужно воспользоваться специальным каталогом. Вашему вниманию представлены и ожидаемые любопытные новинки этого года, которые произведут эффект. Все это можно просматривать в любое время и на различных устройствах. Прямо сейчас воспользуйтесь возможностью посмотреть азиатское кино. Используйте поиск для облегчения выбора.
iribet
| #
Ao se cadastrar no iribet, você ganha um bônus de
100$ para começar sua jornada no cassino com o
pé direito! Não importa se você é um novato ou um apostador experiente, o bônus de boas-vindas é a
oportunidade perfeita para explorar todas as opções que o site tem a oferecer.
Jogue seus jogos favoritos, descubra novas opções de apostas
e aproveite para testar estratégias sem risco, já
que o bônus ajuda a aumentar suas chances de ganhar.
Cadastre-se hoje e comece com 100$!
dragon money_eoen
| #
драгон мани ставки драгон мани ставки .
ArthurBoN
| #
https://telegra.ph/Kolpak-na-stolb-zabora-cena-i-sposoby-ehkonomii-04-18
internetomskCah
| #
лучший интернет провайдер омск
domashij-internet-omsk001.ru
провайдеры домашнего интернета омск
Sazrugg
| #
Купить диплом под заказ вы сможете используя сайт компании. jobs.ria-kj.com/employer/frees-diplom
DonDonfaita
| #
dark market link dark market 2025
Dnrtbva
| #
Мы изготавливаем дипломы любых профессий по приятным ценам. Мы готовы предложить документы ВУЗов, расположенных на территории всей России. Дипломы и аттестаты выпускаются на бумаге высшего качества. Это дает возможности делать настоящие дипломы, не отличимые от оригинала. 1001vieclam.forumvi.com/login
Iariorxdd
| #
Заказать диплом ВУЗа по доступной цене вы можете, обращаясь к надежной специализированной фирме. Заказать документ о получении высшего образования вы имеете возможность в нашей компании. diplomc-v-ufe.ru/kupit-diplom-s-zaneseniem-v-reestr-rossii-prosto-i-bistro
dragon money_qqEr
| #
драгон мани войти драгон мани войти .
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации —
Jariorbra
| #
Мы предлагаем быстро и выгодно купить диплом, который выполнен на бланке ГОЗНАКа и заверен печатями, штампами, подписями. Диплом пройдет лубую проверку, даже при использовании специального оборудования. w77515cs.beget.tech/2025/04/11/kupit-diplom-dlya-trudoustroystva.html
Thomaslob
| #
plinko game online real money
Thomaslob
| #
fortune tigrinho
Mazriht
| #
Мы готовы предложить дипломы психологов, юристов, экономистов и прочих профессий по приятным тарифам. Всегда стараемся поддерживать для заказчиков адекватную политику цен. Важно, чтобы дипломы были доступны для большого количества наших граждан.
Заказ диплома, подтверждающего обучение в ВУЗе, – это разумное решение. Приобрести диплом о высшем образовании: diplomius-docs.com/kupit-diplom-texnikuma-v-moskve-2/
baby powder ice water hack
| #
Woah! I’m really digging the template/theme of this site.
It’s simple, yet effective. A lot of times it’s very hard to
get that “perfect balance” between user friendliness and visual appearance.
I must say that you’ve done a superb job with this.
Additionally, the blog loads very quick for me on Safari.
Outstanding Blog!
my page – baby powder ice water hack
1win_rgKr
| #
мостбет скачать казино http://1win5028.ru .
Donaldlaf
| #
darknet markets 2025 darknet market list
amlodipine blurry vision
| #
Drug prescribing information. Generic Name.
amlodipine blurry vision
Everything trends of meds. Read information here.
Pingrutty
| #
darknet markets darknet site
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы
dragon money_sySi
| #
драгон мани войти драгон мани войти .
Rozewrdblisy
| #
Ищете, где заказать надежную кухню на заказ по вашим размерам за адекватные деньги? Посмотрите портфолио кухонной фабрики GLORIA – https://gloriakuhni.ru/ . Все проекты выполнены в Санкт-Петербурге и области. На каждую кухню гарантия 36 месяцев, более 800 цветовых решений. Большое разнообразие фурнитуры. Удобный онлайн-калькулятор прямо на сайте и понятное формирование цены. Много отзывов клиентов, видео-обзоры кухни с подробностями и деталями. Для всех клиентов – столешница и стеновая панель в подарок.
1win_khpl
| #
1win скачать kg http://www.1win7006.ru .
Diplomi_wmki
| #
можно ли купить диплом медсестры можно ли купить диплом медсестры .
Toliknaf
| #
darknet site darkmarket
can i purchase lozol without prescription
| #
buying cheap lozol tablets
greenbets
| #
Registre-se no greenbets – https://www.greenbets-br.com e Comece a Apostar com 100$ de Bônus!
Thomaslob
| #
betonred casino
Thomaslob
| #
aviator
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Керамическая мишень Licoo2 3N, плоская
Rabyitabe
| #
dark web markets darkmarket url
YuwiyEnuts
| #
На сайте https://xn—-7sbjhqn0bhjc0lk.xn--p1ai/ получите юридическую консультацию по различным вопросам. Компания Стратегия работает как с физическими, так и частными лицами. На все услуги установлены привлекательные расценки, чтобы воспользоваться ими смог каждый. Предприятие работает в этой сфере более 11 лет. В команде трудятся 11 специалистов, которые справятся с задачей независимо от сложности. К вашим услугам досудебный юрист, досудебное урегулирование, кредитный, семейный юрист, решение страховых споров.
mostbet_ohEn
| #
мастбет https://mostbet7003.ru/ .
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы
Mazrlrf
| #
Приобрести диплом о высшем образовании!
Мы предлагаем документы об окончании любых университетов Российской Федерации. Документы изготавливаются на подлинных бланках. jobs.recruithub.africa/profile/emily011340494
DavidSog
| #
Сервисный центр Мобиопт – Ремонт телефонов Киров
WilliamDix
| #
Мы готовы предложить дипломы любой профессии по приятным ценам. Дипломы производятся на настоящих бланках Приобрести диплом любого ВУЗа diplomnie.com
Lazraqg
| #
Где купить диплом по необходимой специальности?
Купить диплом института по доступной стоимости возможно, обратившись к проверенной специализированной компании.: institute-diplom.ru
Xazrune
| #
Приобрести диплом о высшем образовании!
Мы изготавливаем дипломы любой профессии по приятным тарифам. Вы приобретаете документ через надежную компанию. : gab.com/worksale/posts/114200173292852805
Lazrbmo
| #
Диплом любого университета России!
Без присутствия диплома трудно было продвинуться вверх по карьерной лестнице. Поэтому решение о покупке диплома стоит считать мудрым и рациональным. Приобрести диплом об образовании vieclamangiang.net/employer/gosznac-diplom-24
DonDonfaita
| #
dark websites darkmarket
Thomaslob
| #
red on bet
Sazrttk
| #
Заказать диплом о высшем образовании!
Приобрести диплом университета по выгодной стоимости вы сможете, обратившись к надежной специализированной фирме. Быстро заказать диплом: diplomaj-v-tule.ru/poluchite-ofitsialnij-diplom-s-vneseniem-v-gosreestr
Cazryex
| #
Заказать диплом о высшем образовании!
Мы готовы предложить документы ВУЗов, которые расположены в любом регионе Российской Федерации. Документы печатаются на “правильной” бумаге высшего качества: gurgaonplaygirl.copiny.com/question/details/id/1079665
Sazrbrf
| #
Мы готовы предложить дипломы любой профессии по доступным тарифам.– kupit-diplom24.com/kupit-diplom-s-registratsiej-v-reestre/
Pingrutty
| #
dark market link dark market url
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наши товары:
Кольцо РёР· драгоценных металлов палладиевое 200С…45С…1 РјРј РџРґРЎСЂ60-40 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
allwin
| #
Bônus de 100$ para Novos Jogadores no allwin – Cadastre-se Agora!
1win_zgKr
| #
мост бет https://1win5028.ru/ .
Sazrzew
| #
Задался вопросом: можно ли на самом деле купить диплом государственного образца в Москве? Был приятно удивлен — это реально и легально!
Сначала искал информацию в интернете на тему: купить диплом образования в кемерово, купить диплом швеи, купить диплом доктора наук, купить диплом в чебоксарах, купить диплом люди и получил базовые знания. В итоге остановился на материале: proffdiplomik.com/sterlitamak
Sazrjte
| #
Купить диплом о высшем образовании !
Приобретение диплома университета России в нашей компании – надежный процесс, поскольку документ заносится в реестр. Приобрести диплом института diplom-kaluga.ru/kupit-diplom-s-zaneseniem-v-reestr-tsena-7
Donaldlaf
| #
dark web market urls dark market onion
https://russiancouncil.ru/petr-aven/?str=
| #
https://russiancouncil.ru/petr-aven/?str=%C5
Uazrxpq
| #
Приветствую!
Для многих людей, приобрести диплом университета – это острая потребность, возможность получить достойную работу. Однако для кого-то – это разумное желание не терять массу времени на учебу в универе. Что бы ни толкнуло вас на такой шаг, наша фирма готова помочь вам. Максимально быстро, качественно и недорого сделаем диплом любого ВУЗа и любого года выпуска на подлинных бланках с реальными печатями.
Основная причина, почему многие покупают документ, – желание занять определенную должность. К примеру, навыки и опыт позволяют специалисту устроиться на желаемую работу, а документального подтверждения квалификации не имеется. Если работодателю важно наличие “корочки”, риск потерять место работы очень высокий.
Заказать документ о получении высшего образования можно в нашем сервисе. Мы предлагаем документы об окончании любых университетов РФ. Вы сможете получить необходимый диплом по любым специальностям, любого года выпуска, в том числе документы образца СССР. Даем 100% гарантию, что при проверке документов работодателями, подозрений не появится.
Обстоятельств, которые вынуждают приобрести диплом ВУЗа много. Кому-то очень срочно нужна работа, а значит, нужно произвести хорошее впечатление на начальника при собеседовании. Некоторые желают попасть в серьезную компанию, для того, чтобы повысить собственный статус в социуме и в дальнейшем начать собственное дело. Чтобы не тратить массу времени, а сразу начать эффективную карьеру, используя имеющиеся знания, можно купить диплом через интернет. Вы сможете стать полезным в обществе, обретете денежную стабильность в кратчайшие сроки- купить аттестат за 9 класс
internetomskCah
| #
провайдеры домашнего интернета омск
domashij-internet-omsk002.ru
провайдеры омск
Sazrcvd
| #
Покупка документа о высшем образовании через проверенную и надежную компанию дарит ряд плюсов для покупателя. Это решение дает возможность сэкономить как дорогое время, так и существенные финансовые средства. Впрочем, преимуществ значительно больше.Мы можем предложить дипломы любой профессии. Дипломы производят на оригинальных бланках государственного образца. Доступная цена сравнительно с большими издержками на обучение и проживание в другом городе. Приобретение диплома об образовании из российского ВУЗа станет выгодным шагом.
Быстро купить диплом о высшем образовании: diplomgorkiy.com/kupit-diplom-visshego-obrazovaniya-s-zaneseniem-v-reestr-14/
teva venlafaxine xr 37 5mg and alcohol
| #
Medicines information. Drug Class.
teva venlafaxine xr 37 5mg and alcohol
Actual what you want to know about drug. Get information here.
Cazrgzi
| #
Где купить диплом по необходимой специальности?
Мы готовы предложить дипломы психологов, юристов, экономистов и других профессий по приятным ценам. Для нас очень важно, чтобы дипломы были доступными для большого количества граждан. Приобрести диплом об образовании diplom-ryssia.com/ofitsialnij-diplom-s-garantiej-zaneseniya-v-reestr/
Cazrisl
| #
Добрый день!
Мы изготавливаем дипломы любой профессии по приятным ценам. Цена может зависеть от той или иной специальности, года выпуска и образовательного учреждения: diplomanc.com/
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наша продукция:
Керамическая мишень Y2O3 – 4N, плоская форма
Cazrpqr
| #
Приобрести диплом института по доступной стоимости возможно, обращаясь к проверенной специализированной фирме. Мы можем предложить документы престижных ВУЗов, расположенных на территории всей России. ematejo.com/2025/04/09/kupit-diplom-bystro-i-prosto-160
Sazrauv
| #
Приобрести документ университета вы сможете у нас в столице. Мы оказываем услуги по производству и продаже документов об окончании любых университетов РФ. Вы получите необходимый диплом по любой специальности, включая документы старого образца. Даем гарантию, что при проверке документа работодателями, никаких подозрений не возникнет. diplomc-v-ufe.ru/kupit-diplom-s-reestrom-v-moskve-bistro-i-udobno-2/
Toliknaf
| #
darknet site darknet market lists
Thomaslob
| #
plinko game review
Thomaslob
| #
plinko ?????
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности
Sazrprb
| #
Заказать диплом на заказ в Москве возможно через сайт компании. s24.team/club/user/155/forum/message/1165/1167
mostbet_eoEn
| #
мостбет зеркало http://mostbet7003.ru .
Diplomi_pmKt
| #
Заказать диплом ВУЗа!
Мы предлагаем документы любых учебных заведений, которые расположены на территории всей Российской Федерации.
vacshidiplom.com/kupit-podlinnij-diplom-s-zaneseniem-v-reestr-5/
Jariorphy
| #
Приобрести диплом любого университета!
Наши специалисты предлагаютвыгодно заказать диплом, который выполнен на оригинальном бланке и заверен мокрыми печатями, штампами, подписями. Наш документ способен пройти любые проверки, даже при помощи специально предназначенного оборудования. Достигайте своих целей быстро и просто с нашей компанией- leadrp.listbb.ru/viewtopic.phpf=3&t=1182
Eanrroy
| #
Для эффективного продвижения по карьерной лестнице необходимо наличие диплома о высшем образовании. Приобрести диплом об образовании у надежной фирмы: diplomp-irkutsk.ru/kupit-diplom-sredne-texnicheskoe-2/
Jariorwcm
| #
Наша компания предлагает быстро и выгодно купить диплом, который выполняется на оригинальной бумаге и заверен печатями, штампами, подписями официальных лиц. Наш документ способен пройти лубую проверку, даже при помощи профессионального оборудования. sieuthidev.net/read-blog/32_kupit-diplom-s-zaneseniem-v-reestr-rossiya.html
Thomaslob
| #
vai de bet casino
Thomaslob
| #
jeu plinko
Rabyitabe
| #
dark web marketplaces darknet market
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Керамическая мишень для распыления бора – плоская
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их происхождение. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы и адаптировать решения под требования вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
Наша продукция:
Лист магниевый MAG 3 – BS 2970 Лента магниевая MAG 3 – BS 2970 представляет СЃРѕР±РѕР№ высококачественный материал, используемый РІ различных отраслях. Основная особенность этой ленты – высокая прочность Рё гибкость, что делает её идеальным выбором для применения РІ строительстве Рё производстве. Лента устойчива Рє воздействию влаги Рё различных химических соединений. Выбирая талантливое решение для ваших задач, РЅРµ упустите возможность купить Лента магниевая MAG 3 – BS 2970. Ртот РїСЂРѕРґСѓРєС‚ обеспечит надежность Рё долговечность РІ эксплуатации, что делает его незаменимым помощником РІ работе.
betafudew
| #
Центр «Доверие» предоставляет психологические услуги. Мы людям с трудностями, появляющихся у них в отношениях с окружающими и собой, помогаем справиться. Записаться к специалисту на личный прием вы можете по телефону на портале. Ищете психотерапия? Ack-group.ru/stati-po-psikhologii/psikhoterapiya – тут узнаете, что такое психотерапия, кому она показана и какие перед ней стоят цели. Также тут представлены расценки на психологические консультации. Всегда готовы поддержать вас на пути к самопознанию и эмоциональному равновесию.
Thomaslob
| #
spribe plinko free
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности
where buy macrobid pill
| #
can i purchase generic macrobid without prescription
1win_eaPn
| #
1win.pro 1win.pro .
Pingrutty
| #
dark web drug marketplace darknet drug links
DonDonfaita
| #
darknet websites darkmarket url
Sazrlqh
| #
Заказать диплом института по доступной цене вы сможете, обращаясь к надежной специализированной фирме. Мы оказываем услуги по продаже документов об окончании любых университетов РФ. Купить диплом о высшем образовании– poluchidiplom.com/kupite-diplom-s-zaneseniem-v-reestr-bistro-i-udobno-2/
Mazrsqz
| #
Мы предлагаем дипломы любой профессии по приятным ценам. Стараемся поддерживать для клиентов адекватную ценовую политику. Важно, чтобы дипломы были доступны для большого количества граждан.
Покупка документа, который подтверждает окончание института, – это грамотное решение. Приобрести диплом университета: diplommy.ru/diplom-stomatologa-kupit-3/
VefejnFueld
| #
Visit https://coinintel-hq.com/ and you will learn all about the world of cryptocurrency. Explore the world of cryptocurrency like never before with our stunning graphics, fascinating charts, and fascinating infographics. Whether you are a crypto enthusiast, an investor, or just curious about the world of digital currencies, CoinIntel HQ is the place to go for a visually stunning and educational experience.
bet nacional
| #
bet nacional Oferece 100$ de Bônus para Novos Jogadores – Registre-se Já!
Yaxayrtriz
| #
Посетите сайт https://artradol.com/ и вы узнаете все про Артрадол – это препарат для лечения суставов от производителя. Узнайте все преимущества нестероидного противовоспалительного препарата для лечения суставов. Помогает бороться с основными заболеваниями суставов: артритом, остеохондрозом и остеоартрозом. Подробности, инструкция применения и где купить можно узнать на сайте.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наша продукция:
“Мишень для распыления неодима 3N, плоская форма”
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы
Donaldlaf
| #
best darknet markets darknet markets 2025
Diplomi_elki
| #
диплом электромеханика морского судна купить ростов rusdiplomm-orig.ru .
where to buy cheap sumatriptan no prescription
| #
Meds prescribing information. Effects of Drug Abuse.
where to buy cheap sumatriptan no prescription
Everything trends of medicament. Read information now.
bet onred
| #
bet onred
A Brief History of Elemental Analysis » Artemis Analytical
Toliknaf
| #
darknet markets url darkmarket url
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
поставляемая продукция:
Однораструбный медный РѕР±РІРѕРґ РїРѕРґ пайку 42С…36С…1.1 РјРј 27С…29 РјРј твердая пайка Рњ1Р• ГОСТ 32590-2013 Выбирайте высококачественные медные однораструбные РѕР±РІРѕРґС‹ РїРѕРґ пайку для надежных соединений. Рзготовлены РёР· меди высокой чистоты СЃ отличной теплопроводностью Рё устойчивостью Рє РєРѕСЂСЂРѕР·РёРё. Рдеальны для систем водоснабжения Рё отопления.
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности
Thomaslob
| #
is plinko a con
Pingrutty
| #
best darknet markets best darknet markets
Rabyitabe
| #
dark market url dark market onion
Sazruhk
| #
Заказать диплом под заказ в Москве возможно используя сайт компании. skvortsy.listbb.ru/viewtopic.phpf=31&t=3274
internetomskCah
| #
подключить домашний интернет омск
domashij-internet-omsk003.ru
домашний интернет тарифы
Diplomi_ucKt
| #
Купить диплом университета!
Мы готовы предложить документы университетов, которые расположены на территории всей Российской Федерации.
diplomaj-v-tule.ru/kupit-diplom-s-zaneseniem-bistro-i-nadezhno-2/
1win_hcPn
| #
1win сайт вход http://1win7019.ru/ .
Sazrtpe
| #
Купить диплом университета по невысокой цене возможно, обращаясь к проверенной специализированной компании. Купить документ о получении высшего образования вы сможете в нашей компании. netmitra.cyberei.com/read-blog/1853_kupit-diplom-s-registraciej-v-reestre.html
Jariorgaz
| #
Мы предлагаем максимально быстро приобрести диплом, который выполнен на бланке ГОЗНАКа и заверен мокрыми печатями, водяными знаками, подписями должностных лиц. Данный диплом способен пройти лубую проверку, даже при помощи специальных приборов. jenlabeschhen.phorum.pl/posting.php?mode=newtopic&f=1&sid=59ee9894d320873731740c32ff8f1f5d
Jariorpqq
| #
Приобрести диплом любого ВУЗа!
Наши специалисты предлагаютвыгодно и быстро заказать диплом, который выполнен на оригинальной бумаге и заверен мокрыми печатями, водяными знаками, подписями. Наш документ способен пройти любые проверки, даже при использовании профессионального оборудования. Решите свои задачи максимально быстро с нашей компанией- mosdom.flybb.ru/viewtopic.phpf=2&t=555
Iariorlbc
| #
Приобрести диплом института по выгодной цене возможно, обращаясь к надежной специализированной фирме. Приобрести документ университета вы сможете в нашей компании. diplom4you.com/kupit-diplom-s-zaneseniem-12
Dnrtpse
| #
Мы изготавливаем дипломы любой профессии по разумным тарифам. Мы можем предложить документы техникумов, расположенных на территории всей РФ. Дипломы и аттестаты выпускаются на “правильной” бумаге самого высокого качества. Это дает возможность делать государственные дипломы, не отличимые от оригиналов. bodybuilding.net/members/worksale.html
Thomaslob
| #
plinko casino
Thomaslob
| #
jogo.do tigre
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы
DonDonfaita
| #
dark market url dark market list
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их соответствие стандартам. Превосходное обслуживание – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы и адаптировать решения под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
поставляемая продукция:
Фольга вольфрамовая W-Ce20 Фольга вольфрамовая W-Ce20 представляет СЃРѕР±РѕР№ высококачественный материал, предназначенный для различных промышленных применений. Обладая отличной термостойкостью Рё механической прочностью, РѕРЅР° идеально РїРѕРґС…РѕРґРёС‚ для работы РІ экстремальных условиях. Благодаря своей стабильности РїСЂРё высоких температурах, фольга РЅРµ теряет СЃРІРѕРёС… свойств даже РїСЂРё длительном нагреве. Если РІС‹ ищете надежное решение для СЃРІРѕРёС… производственных нужд, купить Фольга вольфрамовая W-Ce20 – это правильный выбор. Ртот РїСЂРѕРґСѓРєС‚ обеспечит долговечность Рё эффективность работы вашего оборудования.
mostbet_guEn
| #
mostbet kg отзывы http://mostbet7003.ru .
cost of generic diovan pill
| #
can i buy generic diovan price
Olivervoino
| #
https://www.a-sila.com/novosti/shlifuvannya-metalichnikh-poverkhon-vid.html
Donaldlaf
| #
darknet drugs dark markets
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации —
Mazrsmn
| #
Мы изготавливаем дипломы любых профессий по приятным тарифам. Всегда стараемся поддерживать для клиентов адекватную ценовую политику. Важно, чтобы документы были доступными для большого количества граждан.
Приобретение диплома, который подтверждает обучение в ВУЗе, – это грамотное решение. Купить диплом любого ВУЗа: diplom-bez-problem.com/kupit-diplom-bakalavra-8/
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
поставляемая продукция:
Циркониевый лист 0.4x1200x3000 РјРј Р110 РўРЈ 92-166-83 Выбирайте циркониевый лист ведущего поставщика РЅР° рынке. РќР° сайте Редметсплав.СЂС„ представлен широкий выбор циркониевых листов различных размеров, толщин Рё марок. Отличные свойства материала позволяют использовать его РІ самых требовательных отраслях промышленности. Гарантированное качество, выгодные цены Рё профессиональная поддержка!
Jamesbeism
| #
https://my-hepatit.ru/chto-takoe-seo-prostymi-slovami/
Thomaslob
| #
online plinko
can you get generic phenytoin for sale
| #
Medicine information sheet. Effects of Drug Abuse.
can you get generic phenytoin for sale
Everything information about medicine. Read information now.
Toliknaf
| #
darknet marketplace darknet markets links
Pingrutty
| #
dark web market urls dark web market links
Thomaslob
| #
aviator apk
Thomaslob
| #
betting on red
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наши товары:
Латуное внутреннее стопорное пружинное кольцо 280х3 мм ЛС ГОСТ 13943-86 Купить латунные внутренние стопорные кольца по ГОСТ от производителя. Надежное крепление и удержание элементов механизмов. Широкий выбор размеров и конфигураций. Высокое качество и надежность. Применение в машиностроении, автомобильной и других отраслях. Обращайтесь к нашим специалистам для получения более подробной информации.
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности
Rabyitabe
| #
darknet drug store dark market
Diplomi_fpki
| #
купить диплом о высшем москва купить диплом о высшем москва .
Thomaslob
| #
fishin frenzy even bigger catch demo
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы гарантируем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их происхождение. Опытная поддержка – наш стандарт – мы на связи, чтобы ответить на ваши вопросы и адаптировать решения под требования вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Лист молибденовый ЦСДМ Лист молибденовый ЦСДМ – это высококачественный металл, используемый РІ различных отраслях, включая авиацию, атомную энергетику Рё научные исследования. РћРЅ обладает уникальными свойствами, такими как высокая температура плавления Рё отличная коррозионная стойкость. Приобретая этот РїСЂРѕРґСѓРєС‚, РІС‹ гарантируете надежность СЃРІРѕРёС… изделий Рё продлеваете СЃСЂРѕРє РёС… службы. Лист молибденовый ЦСДМ идеально РїРѕРґС…РѕРґРёС‚ для создания компонентов, которые требуют выдерживания экстремальных условий. РќРµ упустите возможность купить Лист молибденовый ЦСДМ РїРѕ выгодной цене Рё обеспечьте СЃРІРѕРё проекты качественным материалом для надежных решений.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым сегодняшние вызовы превращаются в завтрашние возможности.
Наша продукция:
Плоская мишень Ho 3N
DonDonfaita
| #
dark web market darknet links
1win_dfon
| #
1win http://www.1win5029.ru .
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы
RukanAnort
| #
Посетите блог юридической компании Закон и Справедливость https://blog.femida-justice.com/ и вы сможете найти полезную информацию, советы и разборы по правовым вопросам. Выбирайте рубрику, которая вас интересует, а мы простыми словами расскажем о сложном. Информация, представленная в блоге, будет полезна как для физических, так и для юридических лиц. Мы постоянно добавляем различные статьи и обзоры законодательства. Подробнее в блоге.
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
Наша продукция:
РџРѕРєРѕРІРєР° прямоугольная РёР· конструкционной стали 250С…1040 РјРј 45РҐ Купить качественную конструкционную прямоугольную РїРѕРєРѕРІРєСѓ РёР· стали 250С…1040 РјРј СЃ маркировкой 45РҐ РЅР° Редметсплав.СЂС„. Прочная, износостойкая Рё надежная РїРѕРєРѕРІРєР° для различных областей промышленности Рё строительства. Рдеально РїРѕРґС…РѕРґРёС‚ для создания прочных конструкций.
LiwufueKal
| #
Our website https://managerscasino.com/ is the ultimate guide for players who want to take control of their casino experience. It is where I share my ideas, strategies and tips for players who want to be in control, not just another number on the casino profit margin. My goal is to help players navigate the casino world with confidence, knowledge and a winning attitude.
1win_siel
| #
1win. casino. http://1win1007.top .
MiltonFAw
| #
Авто сайт с обзорами https://microbus.net.ua новостями, тест-драйвами, каталогом моделей, советами по выбору и эксплуатации автомобилей. Всё, что нужно автолюбителю — в одном месте.
1win_ojea
| #
1win casino uganda 1win1004.top .
Heathzex
| #
Сайт для женщин https://miymalyuk.com.ua мода, красота, здоровье, отношения, карьера, семья и вдохновение. Актуальные статьи, советы и поддержка для современной женщины каждый день
Robertdaype
| #
Актуальные новости Украины https://newsportal.kyiv.ua и мира сегодня: главные события, мнения экспертов, интервью, прогнозы. Оставайтесь в курсе с лентой, которая обновляется 24/7.
ClydeCycle
| #
Главные новости Украины https://novosti24.kyiv.ua и мира — честно, быстро и понятно. События, которые формируют завтрашний день, в одной ленте.
mostbet_sbmt
| #
motsbet motsbet .
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их происхождение. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы а также находить ответы под особенности вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в гибкости нашего предложения
поставляемая продукция:
Рзделия РёР· титана РўРќ-1Рљ РџРѕРєРѕРІРєР° титановая РўРќ-1Рљ – это высококачественный материал, который прекрасно РїРѕРґС…РѕРґРёС‚ для использования РІ различных отраслях. Благодаря СЃРІРѕРёРј уникальным свойствам, таким как легкость Рё прочность, РѕРЅ находит применение РІ авиации, медицине Рё машиностроении. Точную обработку титановых РїРѕРєРѕРІРѕРє обеспечивают современные технологии, что гарантирует долгий СЃСЂРѕРє службы Рё надежность изделий. Если РІС‹ хотите купить РџРѕРєРѕРІРєР° титановая РўРќ-1Рљ, Сѓ нас РІС‹ найдете оптимальные цены Рё высокое качество. Закажите РїСЂСЏРјРѕ сейчас Рё оцените преимущества этого материала!
internetpermCah
| #
подключить интернет в перми в квартире
domashij-internet-perm001.ru
интернет домашний пермь
Donaldlaf
| #
darknet websites darknet market lists
Pingrutty
| #
dark web sites bitcoin dark web
доставка алкоголя ночью в москве на дом
| #
доставка алкоголя москва от alcodostavkashop88.ru — это стандарт качества. Мы доставляем быстро, точно и без компромиссов. У нас только лицензированный алкоголь, вежливые курьеры и сервис, за который нас рекомендуют друзьям. Будьте уверены — вы в надёжных руках.
Thomaslob
| #
plinko
can i order generic nolvadex without prescription
| #
Drugs prescribing information. Brand names.
can i order generic nolvadex without prescription
Everything about meds. Read information now.
CadecenPaund
| #
На сайте https://arenda24.by/ каждый желающий получает возможность взять в прокат спецтехнику, садовый инвентарь для выполнения всех необходимых хозяйственных работ. Если нашли подходящее объявление, то свяжитесь и договоритесь с продавцом для того, чтобы он на выгодных условиях передал технику. Напротив каждого варианта имеются его характеристики, особенности, детальное описание, чтобы потенциальный клиент сразу же сориентировался. Постоянно появляются новые объявления, что позволит найти все, что нужно.
mostbet_haEn
| #
мостбет кг https://mostbet7003.ru .
Cazrofo
| #
Заказать диплом о высшем образовании мы поможем. Купить аттестат в Тольятти – diplomybox.com/kupit-attestat-v-tolyatti
Toliknaf
| #
darknet sites darkmarket list
1win_hoel
| #
1win. casino. https://www.1win1007.top .
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности
1win_luea
| #
1win bet uganda https://www.1win1004.top .
Arbionis
| #
Arbionis
A Brief History of Elemental Analysis » Artemis Analytical
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица продукции подтверждена всеми необходимыми документами, подтверждающими их происхождение. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы а также находить ответы под специфику вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Магний M10413 – UNS Магний M10413 – UNS является РѕРґРЅРёРј РёР· наиболее востребованных материалов РІ современных промышленных приложениях. Обладая высокой прочностью Рё легкостью, РѕРЅ идеально РїРѕРґС…РѕРґРёС‚ для создания различных конструкций. Ртот сплав магния обеспечивает отличную РєРѕСЂСЂРѕР·РёРѕРЅРЅСѓСЋ стойкость Рё теплопроводность, что делает его идеальным выбором для производителей. Если РІС‹ ищете надежный материал для СЃРІРѕРёС… проектов, купите Магний M10413 – UNS Рё убедитесь РІ его качестве сами. РќРµ упустите возможность улучшить СЃРІРѕРё изделия СЃ помощью этого универсального сплава!
where can i get diflucan price
| #
buy generic diflucan for sale
1win_nsel
| #
1win casino http://1win1007.top/ .
Rabyitabe
| #
darknet market list darknet market links
1win_yzSi
| #
скачать 1win официальный сайт http://1win7018.ru .
LojaneAmous
| #
Потеря близкого человека — это тяжелое испытание, и в такой момент важно, чтобы всё было организовано достойно и без лишних хлопот. ГБУ «Ритуал» Москва https://ritualmsk.com/ предлагает полный спектр ритуальных услуг, беря на себя все заботы: от транспортировки и оформления документов до выбора места захоронения. Наши специалисты помогут вам организовать похороны в Москве по всем правилам, обеспечивая уважение к памяти усопшего и поддержку для родственников.
1win_zrea
| #
1win login ug http://www.1win1004.top .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Мишень из высокочистого бисмута (Bi) – плоская форма
Thomaslob
| #
monopoly big baller
kepirdrind
| #
В случае если желаете узнать самую ценную, полезную и важную информацию о трейдинге, вложениях, то нужно зайти на портал, где вы отыщете все необходимое на такую тему. На портале просто рассказывается то, как торговать на внебиржевом пространстве. https://fxidea.com/ рекомендует воспользоваться акциями каждому новичку. Быстрее изучите индикаторы, реальные отзывы, а также ознакомьтесь с работающими стратегиями. Вы сможете воспользоваться интересными материалами о криптовалюте, будете в курсе того, как можно получить деньги от форекса.
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы
DonDonfaita
| #
darknet site dark web market links
Pingrutty
| #
bitcoin dark web darkmarkets
WilliamDix
| #
Мы можем предложить дипломы любой профессии по приятным ценам. Дипломы производятся на фирменных бланках государственного образца Приобрести диплом о высшем образовании diplomnie.com
Thomaslob
| #
monopoly big baller
Thomaslob
| #
online plinko
Donaldlaf
| #
darknet links onion dark website
Lazrjja
| #
Где приобрести диплом специалиста?
Купить диплом ВУЗа по невысокой цене возможно, обращаясь к надежной специализированной компании.: institute-diplom.ru
Markcen
| #
Покупка квартиры без рисков? Полезные статьи и советы —
NetherexPro
| #
NetherexPro
A Brief History of Elemental Analysis » Artemis Analytical
deneme bonusu veren siteler
| #
deneme bonusu veren siteler
blog topic
can hydroxyzine cause low blood pressure
| #
Medication information leaflet. Effects of Drug Abuse.
can hydroxyzine cause low blood pressure
Best what you want to know about medication. Get information now.
1win_rwmi
| #
1win прямой эфир https://1win7007.ru .
1win_vumi
| #
1win казино https://1win7007.ru .
Toliknaf
| #
bitcoin dark web dark web market urls
ElijahCog
| #
Онлайн-медицинский портал https://novamed.com.ua справочник болезней, симптомы, анализы, лекарства, консультации специалистов и актуальные новости здравоохранения. Всё о здоровье — в одном месте.
AllanVot
| #
Портал о здоровье и медицине https://pravovakrayina.org.ua узнайте больше о своём организме, симптомах, лечении и профилактике. Удобный поиск, рекомендации врачей, база клиник и аптек.
GarrettBum
| #
читайте на автомобильном https://proauto.kyiv.ua портале: тест-драйвы, сравнения, автоаналитика, обзоры технологий и новинки автопрома. Только актуальные материалы и честные мнения.
Davidpak
| #
Современный авто портал https://quebradadelospozos.com свежие новости, каталог машин, рейтинг моделей, видеообзоры и полезные статьи. Помощь при покупке, советы по обслуживанию и анализ рынка.
1win_qhPn
| #
партнёрка 1win https://www.1win7019.ru .
Cryptify Flows
| #
Cryptify Flows
A Brief History of Elemental Analysis » Artemis Analytical
internetpermCah
| #
интернет домашний пермь
domashij-internet-perm002.ru
домашний интернет в перми
Alexiszex
| #
Забронировать квартиру на сутки онлайн – быстрый и удобный сервис, узнайте все детали https://logopond.com/Domovits/profile/743217/?filter=&page=
where to buy cheap provera without a prescription
| #
how to get cheap provera price
Rabyitabe
| #
dark web market urls darkmarket url
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы
AllanUtibe
| #
Сервисный центр Мобиопт – Ремонт телефонов в Кирове
ocenka professionalnih riskov_rfMa
| #
оценка профессиональных рисков в Москве оценка профессиональных рисков в Москве .
soglasovanie pereplanirovki kvartiri_mnkr
| #
перепланировка помещения перепланировка помещения .
Pingrutty
| #
darknet markets onion address darkmarket list
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
поставляемая продукция:
Молибденовая труба 0.4х14х1250 мм МЧ ТУ 48-19-251-85 Приобретите молибденовые трубы от компании Редметсплав и обеспечьте вашему проекту прочность, устойчивость к высоким температурам и коррозии. Наши товары соответствуют высочайшим стандартам качества, гарантируя долговечность и надежность. Широкий ассортимент и профессиональные консультации по выбору подходящего решения.
1s byhgalteriya soprovojdenie_izPi
| #
1с бухгалтерия сопровождение программного http://www.programmy-1s15.ru/ .
Jamesinduh
| #
Сайт cryptopp.ru помогает принимать обоснованные решения. Мы динамику криптовалюты с помощью нейронных сетей на основе исторических данных прогонозируем. У вас есть возможность в специальные поля идентификатор монеты (например, биткоин) либо адрес контракта ввести. Затем вам необходимо сеть выбрать. После чего на кнопку «Получение данных» нажать. Ищете надежный прогноз цен на монеты? Cryptopp.ru – тут опубликована детальная информация. Наша основная миссия – предоставить вам для лучшего понимания рынка криптовалют, аналитические данные и инструменты. Рады вам всегда!
Cryvon Wealth App
| #
Cryvon Wealth App
A Brief History of Elemental Analysis » Artemis Analytical
DonDonfaita
| #
dark web sites bitcoin dark web
Ganancia Airflux
| #
Ganancia Airflux
A Brief History of Elemental Analysis » Artemis Analytical
Thomaslob
| #
betonred bonus codes
Thomaslob
| #
plinko app review
can i buy cheap tadora tablets
| #
can i order tadora without a prescription
Thomaslob
| #
jackpot jackpot result
Space exploration
| #
Space exploration
blog topic
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы —
Mazrwqt
| #
Приобрести диплом ВУЗа!
Мы оказываем услуги по производству и продаже документов об окончании любых ВУЗов Российской Федерации. Документы производят на настоящих бланках. job.firm.in/employer/radiplomy
Mazriat
| #
Где приобрести диплом по актуальной специальности?
Полученный диплом со всеми печатями и подписями целиком и полностью отвечает запросам и стандартам, неотличим от оригинала – даже со специально предназначенным оборудованием. Не следует откладывать собственные мечты и цели на долгие годы, реализуйте их с нашей помощью – отправляйте быструю заявку на изготовление документа уже сегодня! Получить диплом о высшем образовании – не проблема! b98385gb.beget.tech/2025/03/28/kupit-diplom-v-lyubom-gorode-bystraya-dostavka.html
Donaldlaf
| #
dark web market dark market 2025
can i get inderal for sale
| #
Pills prescribing information. Effects of Drug Abuse.
can i get inderal for sale
Some what you want to know about medicament. Read here.
Sazrvcd
| #
Приобрести диплом университета!
Заказать диплом института по невысокой цене можно, обратившись к проверенной специализированной фирме. Купить диплом о высшем образовании: damdiplomisa.com/diplom-s-provodkoj-zakazat-ofitsialno-i-nadezhno
Sazrozx
| #
Мы изготавливаем дипломы любых профессий по приятным тарифам.– vuz-diplom.ru/dostupnie-tseni-na-diplom-s-zaneseniem-v-reestr/
Cazrwda
| #
Приобрести диплом любого ВУЗа!
Мы предлагаем документы институтов, которые расположены в любом регионе России. Документы выпускаются на “правильной” бумаге высшего качества: armgame.forumex.ru/viewtopic.phpf=78&t=571
mostbet_qnmt
| #
мостбет кг mostbet6033.ru .
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до крупных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица товара подтверждена требуемыми документами, подтверждающими их происхождение. Дружелюбная помощь – наш стандарт – мы на связи, чтобы улаживать ваши вопросы по мере того как предоставлять решения под особенности вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наша продукция:
Магний AZ63HP Магний AZ63HP – это высококачественный легированный металл, обладающий превосходной прочностью Рё легкостью. РћРЅ идеально РїРѕРґС…РѕРґРёС‚ для изготовления деталей, требующих устойчивости Рє РєРѕСЂСЂРѕР·РёРё Рё высоким температурам. Ртот сплав широко используется РІ авиационной Рё автомобильной промышленности. Благодаря СЃРІРѕРёРј уникальным свойствам, Магний AZ63HP обеспечивает долговечность Рё надежность изделий. РќРµ упустите возможность купить Магний AZ63HP Рё повысить эффективность вашего производства. Убедитесь РІ высоком качестве материала, выбирая его для СЃРІРѕРёС… проектов. Доверьтесь проверенному выбору!
Toliknaf
| #
dark market darknet links
1win_ahSi
| #
сайт 1win официальный сайт вход сайт 1win официальный сайт вход .
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации —
PimoloVoish
| #
Посетите сайт https://xn--j1abip.com// и вы сможете ознакомиться с ЖК Толк в Академическом районе города Екатеринбург. Узнайте все преимущества ЖК, его местоположение, инфраструктуру рядом. На сайте вы сможете выбрать квартиру или паркинг. Купить новую квартиру в ЖК от застройщика по выгодной цене рядом с Преображенским парком легко – мы предлагаем различные варианты оплаты и ипотеку по выгодной ставке. Подробнее на сайте.
skladi_dwSa
| #
личный склад в аренду http://www.hranim-veshi-msk24.ru .
Pingrutty
| #
darknet markets url dark web market urls
ocenka professionalnih riskov_sgMa
| #
оценка профессиональных рисков в Москве оценка профессиональных рисков в Москве .
Rabyitabe
| #
dark web market links dark market onion
soglasovanie pereplanirovki kvartiri_rekr
| #
перепланировки квартир перепланировки квартир .
Thomaslob
| #
vai de bet casino
1s byhgalteriya soprovojdenie_hzPi
| #
1с бухгалтерия сопровождение программного programmy-1s15.ru .
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наши товары:
Медный двухраструбный отвод под высокотемпературную пайку 90 градусов 64х55х1.4 мм 32.5х34.5 мм твердая пайка М1М ГОСТ Р52922-2008 Купите высококачественные медные отводы под высокотемпературную пайку для систем водоснабжения, отопления и кондиционирования воздуха. Превосходные технические характеристики, простота монтажа и долгий срок службы. Широкий выбор размеров и типов отводов. Доставка по России.
Thomaslob
| #
betonred casino login
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние возможности.
Наши товары:
Вращающаяся мишень хрома
DonDonfaita
| #
best darknet markets darknet market
RichardApold
| #
Современный женский журнал https://reyesmusicandevents.com стиль, уход, карьера, семья, рецепты и саморазвитие. Читайте свежие материалы каждый день — будь в тренде и в гармонии с собой.
Anthonybioxy
| #
Женский онлайн-журнал https://ruforums.net с разнообразными рубриками — от моды и макияжа до материнства и путешествий. Открывайте каждый день с новыми идеями и полезным контентом.
Jerrynax
| #
Сайт про авто https://rusigra.org обзоры машин, автоновости, тест-драйвы, сравнения моделей, советы водителям и полезные статьи. Всё, что нужно автолюбителям — на одном ресурсе.
Frankmek
| #
Журнал для женщин https://saralelakarat.com мода, красота, здоровье, психология, семья и вдохновение. Актуальные статьи, советы экспертов и интересные темы для женщин всех возрастов.
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности
Sazrndq
| #
Хорошо быть студентом, пока не придет пора писать диплом, что и произошло со мной, но не стоит отчаиваться, ведь есть хорошие компании что помогают с написанием и сдачей диплома на хорошие оценки!
Изначально искал информацию про купить диплом об окончании колледжа, купить диплом о среднем специальном образовании гознака, купить аттестат об окончании школы, купить диплом пгс, аттестат за 11 классов купить, потом попал на diplomybox.com/kupit-diplom-tekhnikuma-kolledzha-v-sochi
Sazraab
| #
Приобрести диплом о высшем образовании !
Приобретение диплома ВУЗа России в нашей компании является надежным процессом, поскольку документ заносится в реестр. Купить диплом ВУЗа dip-lom-rus.ru/visshee-obrazovanie-kupit-diplom-s-zaneseniem-10
1win_daon
| #
1 win md 1 win md .
Uazrrsk
| #
Здравствуйте!
Для многих людей, приобрести диплом ВУЗа – это острая потребность, удачный шанс получить достойную работу. Но для кого-то – это желание не терять время на учебу в ВУЗе. С какой бы целью вам это не потребовалось, мы готовы помочь вам. Оперативно, профессионально и по разумной стоимости сделаем диплом нового или старого образца на настоящих бланках со всеми печатями.
Главная причина, почему многие покупают документ, – желание занять хорошую работу. К примеру, знания дают возможность человеку устроиться на работу, а подтверждения квалификации не имеется. В случае если работодателю важно наличие “корочек”, риск потерять вакантное место очень высокий.
Заказать документ ВУЗа можно в нашем сервисе. Мы предлагаем документы об окончании любых университетов РФ. Вы сможете получить диплом по любой специальности, любого года выпуска, включая документы образца СССР. Даем 100% гарантию, что в случае проверки документов работодателями, никаких подозрений не возникнет.
Ситуаций, которые вынуждают купить диплом ВУЗа достаточно. Кому-то срочно нужна работа, в итоге нужно произвести впечатление на начальника во время собеседования. Некоторые задумали устроиться в большую компанию, чтобы повысить свой статус и в последующем начать свое дело. Чтобы не тратить драгоценное время, а сразу начать удачную карьеру, используя имеющиеся навыки, можно заказать диплом через интернет. Вы станете полезным в обществе, обретете финансовую стабильность быстро и просто- купить аттестат
1win_dwel
| #
1win descarga https://www.1win1007.top .
internetpermCah
| #
подключить домашний интернет пермь
domashij-internet-perm003.ru
домашний интернет тарифы
Cazrgbt
| #
Заказать диплом университета по доступной цене возможно, обратившись к надежной специализированной фирме. Мы готовы предложить документы ВУЗов на Ваш выбор, расположенных на территории всей РФ. roseninsel.kz/forum/topic/add/forum7
dilemeEngab
| #
Minivan.online предлагает услуги такси (8 мест) по доступным тарифам. Работают только опытные водители. Машины оборудованы детскими креслами, люлькой, а также бустером. Стараемся клиентам безопасные и комфортные поездки обеспечить. Ищете большое такси? Minivan.online – тут представлена о нашей компании подробная информация. Гарантируем своевременную подачу автомобиля. Готовы ответить на интересующие вас вопросы и помочь с оформлением заказа. Минивэн онлайн – надежность и безупречный сервис. Удостоверьтесь в этом лично!
Cazrzbv
| #
Где заказать диплом по актуальной специальности?
Мы можем предложить дипломы любой профессии по разумным ценам. Важно, чтобы дипломы были доступными для большого количества граждан. Быстро заказать диплом института diplomus-spb.ru/kupit-legalnij-diplom-s-reestrom-bistro-i-udobno/
1win_euon
| #
înregistrare 1win înregistrare 1win .
Cazrfzn
| #
Приветствую!
Мы изготавливаем дипломы любых профессий по приятным тарифам. Стоимость может зависеть от выбранной специальности, года получения и образовательного учреждения: diploman-russian.com/
Donaldlaf
| #
dark websites darknet markets 2025
1win_epea
| #
1win casino ug 1win1004.top .
can you get cheap bactrim without dr prescription
| #
Meds information sheet. Brand names.
can you get cheap bactrim without dr prescription
Best news about medicament. Read information now.
Sazrpgw
| #
Купить документ о получении высшего образования можно в нашей компании. Мы оказываем услуги по изготовлению и продаже документов об окончании любых ВУЗов Российской Федерации. Вы сможете получить необходимый диплом по любым специальностям, любого года выпуска, в том числе документы образца СССР. Даем 100% гарантию, что при проверке документа работодателями, подозрений не возникнет. rusd-diplomj.ru/kupit-diplom-texnikuma-s-reestrom-i-otzivami-3/
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от мелких партий до крупных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их соответствие стандартам. Превосходное обслуживание – наш стандарт – мы на связи, чтобы разрешать ваши вопросы а также предоставлять решения под требования вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в гибкости нашего предложения
поставляемая продукция:
Фольга висмутовая Sn96Ag2,5Bi1Cu0,5 – ISO 9453 Фольга висмутовая Sn96Ag2,5Bi1Cu0,5 – ISO 9453 является качественным материалом для пайки, обеспечивая отличные электрические Рё механические свойства. РћРЅР° РїРѕРґС…РѕРґРёС‚ для различных областей применения, включая электронику Рё энергетику. Уникальная формула восемьдесят шесть процентов содержания олова, серебра, висмута Рё меди гарантирует высокую устойчивость Рє РєРѕСЂСЂРѕР·РёРё Рё РёР·РЅРѕСЃСѓ. Купить Фольга висмутовая Sn96Ag2,5Bi1Cu0,5 – ISO 9453 можно РїСЂСЏРјРѕ сейчас, чтобы обеспечить надежность Рё долговечность ваших соединений. Обратите внимание РЅР° преимущества, которые предлагает данный РїСЂРѕРґСѓРєС‚.
ocenka professionalnih riskov_fvMa
| #
оценка профессиональных рисков в Москве оценка профессиональных рисков в Москве .
soglasovanie pereplanirovki kvartiri_arkr
| #
порядок согласования перепланировки квартиры порядок согласования перепланировки квартиры .
Trade Alora
| #
Trade Alora
A Brief History of Elemental Analysis » Artemis Analytical
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы —
Toliknaf
| #
darkmarket url dark web market
Pingrutty
| #
dark market link darkmarket 2025
melbet_unot
| #
melbet кыргызстан melbet1003.ru .
can i order cheap imdur pill
| #
how to buy imdur without a prescription
Jefferydut
| #
Наследие Магазин сувениров оптом и сувениры школьникам в Колпино. Подарочные кружки оптом и глобал опт в Новороссийске. Сувениры полиграфия и Казань сувениры где купить в Оренбурге. Как оригинально подарить подарок На день рождения мужчине и что подарить женщине На день рождения 40 лет идеи в Пензе. Сувениры оптом от производителя дешево и Екатеринбург сувениры: сумки оптом от производителя
Eanrsid
| #
Для удачного продвижения вверх по карьерной лестнице необходимо наличие официального диплома о высшем образовании. Приобрести диплом любого института у надежной компании: diplomaj-v-tule.ru/kupit-diplom-vuza-bistro-i-udobno-2/
protonix 40 mg oral tablet delayed release
| #
order cheap protonix
Rabyitabe
| #
darknet market list dark markets 2025
Jefferydut
| #
Магазин морских сувениров оптом и сувениры в Калининграде. Уф печать это и логотип Queen в Улан-Удэ. Уфа сувениры и венира сувениры в Таганроге. Кружка Начальнику На день рождения и что купить сестре На день рождения в Новосибирске. Кинетические сувениры оптом и сувениры гарри поттер купить: подарочные коробки картонные
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности
OscarReurb
| #
Дмв тест калифорния на русском
DonDonfaita
| #
dark market 2025 dark web market list
Thomaslob
| #
plinko nederland
dragon money_qben
| #
drgn зеркало drgn зеркало .
Thomaslob
| #
vipzino casino
RowezonPyday
| #
На сайте https://voronlaws.ru/ вы найдете Casino Dragon Money и его новое рабочее зеркало для того, чтобы открыть для себя удивительный мир огромного количества развлечений, щедрых бонусов, а также бесконечных выигрышей. На сайте вас ожидает увлекательный и эксклюзивный контент, который обязательно подарит много приятных и положительных эмоций. Клуб щедр на акции и бонусы, а потому здесь вы найдете все, что понравится и поможет заработать деньги. Рабочее зеркало ежедневно обновляется для вашей безопасности.
Jerryvip
| #
The convenient booking Zabljak service will help you find the perfect hotel for car travelers and active holiday lovers. A wide range of accommodation: from cozy guest houses to modern hotels with parking, Wi-Fi and breakfast. Book in advance and relax in comfort in the heart of Montenegro!
Pingrutty
| #
darknet markets url onion dark website
JamesMic
| #
Портал о машинах https://shpik.info от новостей и технологий до экспертных статей и автомобильной аналитики. Узнайте всё о текущих трендах на рынке и новинках автопрома.
RichardDab
| #
Онлайн-журнал для женщин https://zhenskiy.kyiv.ua которые ценят себя, любят жить ярко и стремятся к гармонии. Будь в курсе моды, заботься о себе и черпай вдохновение каждый день.
Donaldlaf
| #
dark market onion dark market onion
JosephSwilt
| #
Портал о машинах https://xiwet.com от новостей и технологий до экспертных статей и автомобильной аналитики. Узнайте всё о текущих трендах на рынке и новинках автопрома.
Markcen
| #
Новостройки или вторичка? Полезные рекомендации —
Thomaspoext
| #
Хризантемы стоят уже третью неделю – рекорд!
цветы
Willieavamy
| #
The sounds online hook—stay in! bet on red
Ronaldrhype
| #
Celine fake designer bags
Toliknaf
| #
darknet market list darkmarket 2025
internetrostovCah
| #
подключить проводной интернет ростов
domashij-internet-rostov001.ru
домашний интернет ростов
OscarReurb
| #
https://losangeles.zagranitsa.com/article/9420/kak-sdat-na-prava-v-kalifornii-top-oshibok-kotorye-ne-pozvoliat-vam-sdat-s-pervogo-raza
Ronaldrhype
| #
Celine bag replica
Ronaldrhype
| #
Louis Vuitton replica designer bags
Rabyitabe
| #
dark web market darknet drug store
Ronaldrhype
| #
Louis Vuitton replica designer bags
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности
MichaelHathe
| #
Наряд заказ на ритуальные услуги включают весь необходимый комплекс: перевозку тела, оформление документов, подбор гроба и венков, аренду катафалка, работу носильщиков и организацию прощания. Мы работаем круглосуточно, оказывая помощь в самый сложный момент. Консультации бесплатны, возможен выезд агента на дом или в больницу. Выезд ритуального агента на дом круглосуточно https://pohoronnoe-agentstvo.ru/
Gibopwget
| #
Ищете сельхозтехнику и оборудование по самым выгодным ценам от производителя? Посетите сайт Производителя ООО МИТЕХ https://mi-teh.ru/ где вы также найдете запасные части и комплектующие к сельхоз машинам и тракторам. Ознакомьтесь с нашим ассортиментом продукции на сайте.
Aktiva Sarnix
| #
Aktiva Sarnix
A Brief History of Elemental Analysis » Artemis Analytical
WilliamDix
| #
Мы готовы предложить дипломы любой профессии по невысоким ценам. Дипломы производят на подлинных бланках государственного образца Приобрести диплом о высшем образовании kupitediplom.ru
order sinemet
| #
Medicines information. What side effects can this medication cause?
order sinemet
Actual about drug. Read now.
DonDonfaita
| #
darknet drug market dark web link
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наши товары:
Никелевая капиллярная трубка 1.45х0.07х5000 мм НМг0.08в ГОСТ 13548-2016 Приобретите никелевые капиллярные трубки от Редметсплав.рф для прочных, высокотехнологичных и надежных решений в различных отраслях.
Lazriat
| #
Где приобрести диплом по актуальной специальности?
Приобрести диплом ВУЗа по выгодной стоимости вы можете, обращаясь к надежной специализированной компании.: kupite-diplom0024.ru
can you buy generic flagyl price
| #
can you get generic flagyl without prescription
Pingrutty
| #
darkmarket url darknet links
AllanUtibe
| #
Превосходит защитное стекло на телефон – Гидрогелевая пленка в Кирове
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы —
Thomaslob
| #
hothotfruit
Thomaslob
| #
casino bet on red
Willieavamy
| #
Online gambling feels light—risk on! sugar rush
Ronaldrhype
| #
fake Celine bag
Donaldlaf
| #
darkmarket 2025 dark markets
mostbet_apmt
| #
мостбет зеркало https://mostbet6033.ru .
BarryOrilk
| #
Журнал про животных https://zoobonus.com.ua интересные статьи о питомцах, дикой природе, уходе, воспитании и поведении. Всё для тех, кто любит животных и хочет узнать о них больше.
Wesleycaf
| #
Полный гид по недвижимости https://all2realt.com.ua как выбрать, купить, продать или арендовать квартиру, дом или коммерческий объект. Обзоры, цены, тенденции рынка и юридические тонкости.
Georgechoof
| #
РРєСЃРєСѓСЂСЃРёСЏ РёР· Калининграда https://kurshskaya-kosa-ekskursii.ru/ РЅР° Куршскую РєРѕСЃСѓ СЃ ранним выездом для встречи рассвета
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы по мере того как находить ответы под специфику вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
поставляемая продукция:
Проволока ниобиевая Ta-Nb40. 5Р’2РњР¦-1 Проволока ниобиевая Ta-Nb40. 5Р’2РњР¦-1 – это высококачественный металл, используемый РІ различных промышленных областях благодаря его уникальным свойствам. Ртот сплав отличается высокой прочностью Рё устойчивостью Рє РєРѕСЂСЂРѕР·РёРё. Проволока идеально РїРѕРґС…РѕРґРёС‚ для применения РІ аэрокосмической Рё электронной промышленности, Р° также РІ производстве деталей, работающих РІ агрессивных средах.Если вам нужна надежная Рё долговечная проволока, купите Проволока ниобиевая Ta-Nb40. 5Р’2РњР¦-1 РїСЂСЏРјРѕ сейчас. Отдайте предпочтение качеству Рё устойчивости, которые обеспечат успех ваших проектов.
ArthurRuics
| #
Включите в маршрут экскурсия из Пионерского на Куршскую косу посещение рыбного рынка.
Toliknaf
| #
darkmarket 2025 onion dark website
Thomaslob
| #
play fishin frenzy free
ArthurRuics
| #
Куршская коса экскурсии https://kurshskaya-kosa-ekskursii.ru/ предлагают уникальные маршруты по живописным природным ландшафтам
dragon money_fcen
| #
drgn1n casino drgn1n casino .
dragon money_lpEr
| #
dragon money официальный сайт играть http://www.dragon-money40.com .
Sazrzvx
| #
Купить диплом института по выгодной цене возможно, обратившись к проверенной специализированной фирме. Мы предлагаем документы об окончании любых ВУЗов Российской Федерации. Купить диплом о высшем образовании– diplomgorkiy.com/kupit-diplom-s-otzivami-bez-xlopot-legko-i-bistro/
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы
Ronaldrhype
| #
Gucci fake designer bags
Ronaldrhype
| #
Fendi replica designer bags
Zivutrttar
| #
Перевозка негабаритных грузов и доставка по всей территории России и СНГ на сайте https://negabarit-24.ru/ – это профессиональные услуги от нашей компании. Перевозка негабаритных грузов по России и странам СНГ, ЖД перевозки, перевозки манипулятором и международные перевозки. Гибкие варианты оплаты. Соблюдение сроков. Страхование грузов. Подробнее на сайте.
MichaelHathe
| #
Юго западное кладбище ритуальные услуги включают весь необходимый комплекс: перевозку тела, оформление документов, подбор гроба и венков, аренду катафалка, работу носильщиков и организацию прощания. Мы работаем круглосуточно, оказывая помощь в самый сложный момент. Консультации бесплатны, возможен выезд агента на дом или в больницу. Работаем официально, с договором и чёткой сметой: юго западное кладбище ритуальные услуги
where to buy generic epivir without insurance
| #
order generic epivir without rx
Rabyitabe
| #
dark web drug marketplace onion dark website
Sazrhly
| #
Приобрести диплом института!
Приобрести диплом университета по доступной цене можно, обратившись к надежной специализированной фирме. Купить диплом: diplomt-v-samare.ru/diplom-texnikuma-s-zaneseniem-v-reestr-otzivi-pokupatelej
Cazrwfl
| #
Купить диплом университета!
Мы можем предложить документы университетов, расположенных в любом регионе России. Дипломы и аттестаты печатаются на “правильной” бумаге высшего качества: msk.flybb.ru/viewtopic.phpf=2&t=1073
Mazrvlt
| #
Где приобрести диплом специалиста?
Полученный диплом с приложением отвечает стандартам Министерства образования и науки, никто не отличит его от оригинала – даже со специальным оборудованием. Не откладывайте собственные цели на потом, реализуйте их с нашей компанией – отправьте заявку на изготовление документа прямо сейчас! Заказать диплом о высшем образовании – быстро и просто! mamapoltava.listbb.ru/viewtopic.phpf=39&t=13407
Mazrhot
| #
Приобрести диплом об образовании!
Мы оказываем услуги по производству и продаже документов об окончании любых университетов Российской Федерации. Документы производятся на подлинных бланках. l67697qa.beget.tech/2025/04/08/diplomy-kolledzhey-vuzov-i-tehnikumov.html
Pingrutty
| #
dark web link dark websites
how to buy plan b without a prescription
| #
Meds prescribing information. Short-Term Effects.
how to buy plan b without a prescription
Actual information about meds. Get now.
DonDonfaita
| #
dark web market urls darknet markets onion
Markcen
| #
Как выбрать надёжного застройщика? Читайте проверенные советы
internetrostovCah
| #
подключить домашний интернет в ростове
domashij-internet-rostov002.ru
лучший интернет провайдер ростов
1win_cfon
| #
1win md 1win md .
Willieavamy
| #
Online casinos need gear—aid us! plinko
Ronaldrhype
| #
Bottega Veneta bag replica
Thomaslob
| #
avis jeu plinko
Thomaslob
| #
plinko big win
Sazregi
| #
Мы предлагаем дипломы любых профессий по разумным ценам.– diplomc-v-ufe.ru/kupit-nastoyashij-diplom-bistro-i-bezopasno/
Ronaldrhype
| #
fake Bottega Veneta bag
skladi_igSa
| #
камера хранения вещей в москве недорого http://hranim-veshi-msk24.ru .
Donaldlaf
| #
dark market url darknet markets url
Thomaslob
| #
beton red
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы —
where to get cheap valtrex without prescription
| #
where can i get cheap valtrex without prescription
Toliknaf
| #
darknet markets onion dark market 2025
plinko balls
| #
plinko balls
A Brief History of Elemental Analysis » Artemis Analytical
promethazine with codeine syrup is used for
| #
Medicament information leaflet. Generic Name.
promethazine with codeine syrup is used for
Best what you want to know about medicine. Get here.
Ronaldrhype
| #
Chanel replica designer bags
Ronaldrhype
| #
Fendi fake designer bags
Pingrutty
| #
darknet market list tor drug market
Rabyitabe
| #
dark web market links dark market url
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные рекомендации —
Sazrczx
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и любых других профессий по приятным тарифам.
Вы покупаете диплом в надежной и проверенной временем компании. Приобрести диплом о высшем образовании– http://orthograf.getbb.ru/viewtopic.phpf=2&t=1733/ – orthograf.getbb.ru/viewtopic.phpf=2&t=1733
Sazrtbp
| #
Купить диплом ВУЗа !
Покупка диплома университета России у нас – надежный процесс, так как документ будет заноситься в реестр. Приобрести диплом об образовании vuz-diplom.ru/poluchite-diplom-s-zaneseniem-v-reestr-bistro-i-legko-2
Sazrypx
| #
Будучи студентом, я наслаждался учебой до тех пор, пока не пришло время писать диплом. Но паниковать не стоило, ведь существуют компании, которые помогают с написанием и защитой диплома на отличные оценки!
Изначально я искал информацию по теме: куплю диплом месте, про купленный диплом, купить диплом в братске, купить диплом в саранске, купить диплом дополнительном образовании, затем наткнулся на diplomybox.com/kupit-attestat-v-kemerovo
Uazrdup
| #
Доброго времени суток!
Для некоторых людей, приобрести диплом ВУЗа – это острая необходимость, возможность получить выгодную работу. Но для кого-то – это разумное желание не терять огромное количество времени на учебу в ВУЗе. С какой бы целью вам это не потребовалось, мы готовы помочь. Максимально быстро, качественно и выгодно изготовим документ нового или старого образца на подлинных бланках со всеми подписями и печатями.
Главная причина, почему многие люди покупают документы, – получить хорошую работу. Например, способности и опыт дают возможность кандидату устроиться на привлекательную работу, однако подтверждения квалификации нет. Когда для работодателя важно наличие “корочки”, риск потерять место работы очень высокий.
Заказать документ университета вы сможете у нас. Мы оказываем услуги по продаже документов об окончании любых ВУЗов России. Вы сможете получить диплом по любой специальности, любого года выпуска, в том числе документы образца СССР. Даем гарантию, что в случае проверки документов работодателями, каких-либо подозрений не возникнет.
Ситуаций, которые вынуждают приобрести диплом о среднем образовании немало. Кому-то срочно нужна работа, и нужно произвести хорошее впечатление на начальство на протяжении собеседования. Другие мечтают устроиться в престижную компанию, для того, чтобы повысить свой статус и в дальнейшем начать свой бизнес. Чтобы не терять впустую годы жизни, а сразу начать эффективную карьеру, применяя имеющиеся знания, можно купить диплом через интернет. Вы сможете быть полезным для общества, получите денежную стабильность в кратчайшие сроки- диплом о среднем образовании купить
Thomaslob
| #
jogando tigrinho
Thomaslob
| #
czy gra plinko to oszustwo
Ronaldrhype
| #
Gucci bag replica
xenical without a prescription
| #
Medicine information. Long-Term Effects.
xenical without a prescription
Actual information about pills. Read information here.
DonDonfaita
| #
tor drug market dark markets 2025
Cazrswg
| #
Приобрести диплом института по доступной стоимости вы сможете, обращаясь к надежной специализированной компании. Мы можем предложить документы учебных заведений, которые находятся на территории всей РФ. oppozi.rx22.ru/viewtopic.phpf=37&t=2562
Ronaldrhype
| #
fake Bottega Veneta bag
Ronaldrhype
| #
Gucci replica designer bags
Willieavamy
| #
Are online casino reviews real or just ads? aviator juego
Cazrtyq
| #
Где приобрести диплом по необходимой специальности?
Мы можем предложить дипломы психологов, юристов, экономистов и прочих профессий по приятным тарифам. Важно, чтобы дипломы были доступными для большого количества наших граждан. Заказать диплом университета diplomk-v-krasnodare.ru/kupit-diplom-menedzhera-s-provodkoj-v-reestre/
Sazrttg
| #
Купить документ университета можно в нашей компании в столице. Мы оказываем услуги по изготовлению и продаже документов об окончании любых университетов РФ. Вы сможете получить диплом по любым специальностям, включая документы СССР. Даем 100% гарантию, что при проверке документа работодателем, подозрений не возникнет. diplomers.com/kupit-diplom-s-reestrom-v-rossii-bistro-i-nadezhno-7/
Cazrghh
| #
Привет!
Мы изготавливаем дипломы любой профессии по доступным тарифам. Стоимость может зависеть от определенной специальности, года получения и университета: п»їtutdiploms.com/
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности
marketplace-akkauntov-top.ru_nkmmvn
| #
маркетплейс аккаунтов соцсетей https://marketplace-akkauntov-top.ru
pemerintah
| #
pemerintah
A Brief History of Elemental Analysis » Artemis Analytical
Как учится молодёжь
| #
Как учится молодёжь
blog topic
Donaldlaf
| #
dark web marketplaces darkmarket link
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние возможности.
Наши товары:
Высокочистые гранулы титана
Bitcoin Apex
| #
Bitcoin Apex
A Brief History of Elemental Analysis » Artemis Analytical
internetrostovCah
| #
подключение интернета ростов
domashij-internet-rostov003.ru
провайдеры домашнего интернета ростов
Toliknaf
| #
darkmarket url dark web market urls
Ronaldrhype
| #
Bottega Veneta fake designer bags
Pingrutty
| #
dark markets 2025 darkmarket 2025
Ronaldrhype
| #
fake Bottega Veneta bag
Eanrzpw
| #
Для удачного продвижения вверх по карьерной лестнице требуется наличие диплома университета. Заказать диплом любого университета у проверенной фирмы: diplomg-kurerom.ru/kupit-attestati-za-11-klass-s-zaneseniem-v-reestr-8/
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации —
Ronaldrhype
| #
Celine replica designer bags
buying cheap asacol without a prescription
| #
cost asacol for sale
can you buy astelin without a prescription
| #
can you get astelin without a prescription
melbet_ujot
| #
melbet melbet .
Yatavtswops
| #
На сайте https://rozatoday.ru/ представлено огромное количество ярких, запоминающихся композиций, которые созданы из свежих цветов: ослепительных роз, волнующих пионов, трепетных тюльпанов, строгих гвоздик. В компании работают только знающие, компетентные флористы, которые создают изысканные композиции в соответствии с предпочтениями клиента. В обязательном порядке учитываются и актуальные мировые тенденции. Можно приобрести как небольшой букетик, который подходит для романтического свидания, так и внушительных размеров для грандиозного праздника.
Rabyitabe
| #
dark web link darknet links
plinko application
| #
plinko application
A Brief History of Elemental Analysis » Artemis Analytical
get cheap ampicillin for sale
| #
Medicines information for patients. Short-Term Effects.
get cheap ampicillin for sale
Best what you want to know about drugs. Get here.
can you buy generic norvasc price
| #
Drugs prescribing information. Brand names.
can you buy generic norvasc price
Actual what you want to know about drug. Get now.
Markcen
| #
Новостройки или вторичка? Узнайте подробности
XixigmPrign
| #
Ищете Нестероидный противовоспалительный обезболивающий препарат с фенилбутазоном и лидокаином? Посетите сайт https://ambenium.ru/ и узнайте о препарате Амбениум – это единственный нестероидный противовоспалительный препарат, зарегистрированный в России с усиленным обезболивающим эффектом – раствор для внутримышечного введения фенилбутазон и лидокаин. Подробности на сайте.
Ronaldrhype
| #
fake DIOR bag
aajogo
| #
Comece no aajogo
com 100$ de Bônus de Boas-Vindas!
Thomaslob
| #
jogo vai de bet
DonDonfaita
| #
dark market list darknet market
Thomaslob
| #
fake plinko ball game
Willieavamy
| #
Online casinos should cap—play safe! vincispin
vitaseal
| #
mitolyn is an advanced nutritional solution specifically created to help maintain balanced blood sugar levels and improve metabolic performance.
Ronaldrhype
| #
Fendi bag replica
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные статьи и советы —
Thomaslob
| #
online plinko
plinko official app
| #
plinko official app
A Brief History of Elemental Analysis » Artemis Analytical
Pingrutty
| #
dark market best darknet markets
Donaldlaf
| #
darknet drugs dark web market
Ronaldrhype
| #
Louis Vuitton fake designer bags
FohiyasDwert
| #
Visit https://block-snacks.com/ and stay up to date with all the latest developments in the blockchain and cryptocurrency space today and always. We provide complete and up-to-date opinions, reviews, guides, and introduce people from the cryptocurrency world to help the general public understand and successfully use these technologies. We search and publish only relevant and important news in the cryptocurrency world.
vuhayDwego
| #
Efizika.ru – сайт, где вы увидите виртуальные лабораторные работы по физике. Они помогают ученикам освоить физические законы и явления, а также вырабатывают навыки проведения опытов и анализа данных. Вы можете эксперимент провести, усвоить от параметров цепи работу электрического тока и зависимость мощности. Ищете виртуальная лабораторная работа по физике? Efizika.ru – тут имеется более детальная информация, посмотрите ее уже сегодня. При появлении вопросов выполнения работы, или интерпретации результатов, к учебнику по физике обратитесь. Желаем вам в эксперименте удачи!
1win_ulkl
| #
1win. com http://1win7009.ru .
Toliknaf
| #
dark websites darknet markets onion address
TugotWot
| #
Детский туроператор Флагман организует школьные экскурсии и туры для детей по Санкт-Петербургу и Северо-Западу. Поездки с Флагманом всегда превращаются в увлекательные приключения. Собственный автопарк, профессиональные водители и контроль безопасности в пути гарантируют комфортные путешествия для детей. Профессиональные экскурсоводы адаптируют маршруты под детскую аудиторию, делая их познавательными и интересными. Подробнее на официальном сайте https://flagmanspb.ru/
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности
VocasGot
| #
Боль в суставах может серьезно ухудшить качество жизни. Артроз, подагра и воспаление суставов — частые причины дискомфорта. Эффективное лечение возможно! На сайте https://lechsustavy.ru/ вы найдете натуральные средства для лечения суставов в домашних условиях. Забудьте про боль без операции — попробуйте проверенные препараты, которые помогут восстановить хрящи, снять воспаление и вернуть подвижность. Начните лечить суставы сегодня!
Rabyitabe
| #
dark markets darknet markets onion
1win_tnei
| #
1win méxico 1win1008.top .
internetvolgogradCah
| #
подключить интернет в квартиру омск
domashij-internet-volgograd001.ru
подключить домашний интернет омск
doxycycline sharp stomach pain
| #
Medicines information for patients. Effects of Drug Abuse.
doxycycline sharp stomach pain
All news about pills. Read now.
Ronaldrhype
| #
Celine bag replica
Ronaldrhype
| #
Celine replica designer bags
DonDonfaita
| #
dark market onion dark market
melbet_rxot
| #
melbet kg https://melbet1003.ru/ .
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы —
Willieavamy
| #
Online casinos should cut—fair play! plinko
mostbet_ygkr
| #
мостбет промокод мостбет промокод .
Thomaslob
| #
plinko reviews
Pingrutty
| #
darknet market lists darknet market list
minocycline no prescription
| #
Medicine information for patients. Cautions.
minocycline no prescription
Best news about drug. Get information here.
vitaseal
| #
mitolyn is an advanced nutritional solution specifically created to help maintain balanced blood sugar levels and improve metabolic performance.
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
Наши товары:
Кольцо РёР· драгоценных металлов золотое 50С…2С…0.5 РјРј Р—Р»99.9 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
Donaldlaf
| #
dark market 2025 darknet markets url
Markcen
| #
Хотите сдать квартиру в аренду? Читайте проверенные советы
Ronaldrhype
| #
Fendi bag replica
mostbet_spet
| #
mostbet kg скачать на андроид mostbet kg скачать на андроид .
Toliknaf
| #
darknet markets darknet websites
Thomaslob
| #
fortune mouse bet
JeffreyBip
| #
Экскурсии из Калининграда https://kurshskaya-kosa-ekskursii.ru/ на Куршскую косу с акцией «3 экскурсии — обзорная в подарок»
OscarReurb
| #
https://www.bazar.club/blog/post/take-a-dmv-test-in-the-usa
DavidSaree
| #
The best online slots big bamboo in one place: classics, new releases, jackpots and themed machines. Play without registration, test the demo or make real bets with bonuses.
1win_ezKl
| #
1win ставки официальный сайт 1win ставки официальный сайт .
VictorTus
| #
Профессиональное агентство рекламное агентство Витрувий: разработка рекламы, брендинг, digital-маркетинг, наружка и SMM. Комплексное продвижение для бизнеса любого масштаба.
mostbet_awkr
| #
mostbet официальный сайт https://www.mostbet5008.ru .
1win_lzPr
| #
1win официальный сайт вход https://1win7020.ru .
Rabyitabe
| #
darknet markets url dark web market urls
mostbet_simn
| #
мостбет скачать андроид http://mostbet7004.ru/ .
where can i buy verapamil without a prescription
| #
Medicament information. Short-Term Effects.
where can i buy verapamil without a prescription
Some trends of drugs. Read information now.
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до масштабных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица товара подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы разрешать ваши вопросы и адаптировать решения под специфику вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
РџРѕРєРѕРІРєР° магниевая MgAl3Zn – DIN 1729 Part 1 Лист магниевый MgAl3Zn – DIN 1729 Part 1 – это высококачественный материал, обладающий выдающимися механическими свойствами Рё устойчивостью Рє РєРѕСЂСЂРѕР·РёРё. Ртот магниевый сплав идеально РїРѕРґС…РѕРґРёС‚ для использования РІ различных отраслях, включая автомобилестроение Рё aerospace. Благодаря легкости Рё прочности, лист становится предпочтительным выбором для конструкционных элементов. Покупая Лист магниевый MgAl3Zn – DIN 1729 Part 1, РІС‹ обеспечиваете СЃРІРѕР№ проект надежным материалом. Заказывайте Сѓ нас Рё получите конкурентоспособные цены Рё профессиональную поддержку. РќРµ упустите возможность улучшить качество своей продукции!
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы
Ronaldrhype
| #
Fendi bag replica
Pingrutty
| #
darknet market list darknet market lists
1s byhgalteriya soprovojdenie_vbPi
| #
1с бухгалтерия сопровождение программного https://www.programmy-1s15.ru .
ocenka professionalnih riskov_nyMa
| #
оценка профессиональных рисков в организации https://www.ocenka-profriskov495.ru .
Ronaldrhype
| #
Bottega Veneta bag replica
DonDonfaita
| #
dark web market urls dark web market urls
Ronaldrhype
| #
Prada replica designer bags
melbet_wiot
| #
melbet kg http://www.melbet1003.ru .
Ronaldrhype
| #
Prada bag replica
Ronaldrhype
| #
Prada bag replica
1win_yvPr
| #
1.вин 1win7020.ru .
Willieavamy
| #
The music in online casino games is so catchy—keeps me in the zone! fortune tiger
mostbet_msmn
| #
mostbet скачать https://www.mostbet7004.ru .
profile
| #
Hi, i think that i saw you visited my website thus i came
to “return the favor”.I am trying to find things to improve
my site!I suppose its ok to use some of your ideas!!
OscarReurb
| #
https://www.forumdaily.com/voditelskie-prava-v-ssha-chto-nuzhno-znat-immigrantam/
Pixunlipglice
| #
Система менеджмента управления качеством содержит критерии менеджмента и стандарты, по которым проводится последующая сертификация предприятия. Чтобы получить сертификат СМК, нужно обратиться в аккредитованный сертификационный центр. Сертификация систем менеджмента качества является документальным подтверждением того, что система менеджмента сертифицированной компании соответствует требованиям того или иного стандарта. Подробнее: https://ok.ru/group/70000034956977
internetvolgogradCah
| #
провайдеры омск
domashij-internet-volgograd002.ru
подключение интернета омск
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные статьи и советы —
1win_smPr
| #
1 win казино http://www.1win7020.ru .
Donaldlaf
| #
darkmarket url tor drug market
mostbet_zrmn
| #
mostbet.kg mostbet7004.ru .
1win_nkoi
| #
1win uganda https://1win1005.top .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Высокочистая мишень для распыления олова
Thomaslob
| #
free plinko
Ronaldrhype
| #
Prada replica designer bags
Thomaslob
| #
online plinko
soglasovanie pereplanirovki kvartiri_sokr
| #
этапы согласования перепланировки этапы согласования перепланировки .
Ronaldrhype
| #
Prada fake designer bags
Toliknaf
| #
darknet market darknet market links
JohnnyFiede
| #
ОЧИСТКА АВТОТЕКИ Чистка автотеки – улучшаем историю вашего авто! Наш сервис помогает удалить записи о ДТП, скорректировать пробег, убрать данные о расчетах и очистить данные о такси и каршеринге. Профессионально, быстро, конфиденциально. Вернем вашему авто идеальную историю и повысим его рыночную стоимость.
synthroid buy online uk
| #
Medicine information for patients. Cautions.
synthroid buy online uk
Everything news about drugs. Get now.
1win_icoi
| #
1win uganda http://1win1005.top .
Sazrimo
| #
Купить диплом института по доступной стоимости можно, обратившись к надежной специализированной компании. Мы оказываем услуги по продаже документов об окончании любых ВУЗов России. Купить диплом о высшем образовании– poluchidiplom.com/kupit-diplom-s-zaneseniem-v-reestr-nadezhno-i-bistro/
Ronaldrhype
| #
Chanel replica designer bags
Ronaldrhype
| #
Chanel replica designer bags
Markcen
| #
Новостройки или вторичка? Полезные рекомендации —
Pingrutty
| #
darknet drugs dark markets 2025
mostbet_acOt
| #
mostbet apk скачать http://mostbet5009.ru/ .
mostbet_kxet
| #
скачать мостбет официальный сайт https://mostbet5010.ru/ .
how can i get diflucan for sale
| #
Medicines information for patients. Generic Name.
how can i get diflucan for sale
Some about meds. Read information here.
Rabyitabe
| #
dark web marketplaces dark market url
Ronaldrhype
| #
Louis Vuitton replica designer bags
DonDonfaita
| #
dark web link darknet drugs
прокарниз
| #
Закажите уникальные шторы на заказ, для офиса.
Качественные шторы на заказ, быстро.
Создание штор мечты, под ваш интерьер.
Эксклюзивные шторы на заказ, подчеркивающие ваш стиль.
Пошив штор на заказ для кухни, с индивидуальным подходом.
Индивидуальный дизайн штор, под любой бюджет.
Пошив штор для нестандартных окон, с учетом особенностей помещения.
Шторы на заказ с уникальным дизайном, по вашему желанию.
Классические шторы на заказ, по вашему проекту.
Пошив штор на заказ по индивидуальным меркам, от ведущих мастеров.
Премиальные ткани для штор на заказ, воплощая ваши идеи.
Изготовление штор на заказ быстро и недорого, по вашему желанию.
Модные шторы на заказ для вашего дома, под любой стиль интерьера.
Индивидуальный пошив штор на заказ, с использованием лучших тканей.
Пошив штор по индивидуальному дизайну, от профессиональных мастеров.
Высококачественные шторы на заказ, по желанию клиента.
сшить шторы на заказ сшить шторы на заказ . прокарниз
mostbet_pdkr
| #
мостбет кыргызстан мостбет кыргызстан .
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные статьи и советы —
mostbet_gpOt
| #
mostbest http://mostbet5009.ru .
Willieavamy
| #
The live games online shine—big fan! tom of madness
Sazrxdm
| #
Приобрести диплом института по выгодной цене вы можете, обратившись к проверенной специализированной компании. Купить документ института можно в нашей компании. tzuchimalaysia.ddns.net/kupit-diplom-gosudarstvennogo-obrazca-173-2
Ronaldrhype
| #
Gucci fake designer bags
Iariordiy
| #
Купить диплом института по доступной цене можно, обращаясь к надежной специализированной фирме. Приобрести документ ВУЗа можно у нас в столице. diplomservis.ru/kupit-diplom-bakalavra-s-zaneseniem-v-reestr-8
1s byhgalteriya soprovojdenie_fePi
| #
1с какие есть программы https://programmy-1s15.ru/ .
ocenka professionalnih riskov_gbMa
| #
проведение оценки профессиональных рисков в Москве https://ocenka-profriskov495.ru/ .
Donaldlaf
| #
darkmarket darknet market links
Trefsjy
| #
Заказать диплом об образовании. Заказ документа о высшем образовании через надежную компанию дарит ряд плюсов для покупателя. Это решение позволяет сберечь время и значительные денежные средства. vv.flybb.ru/viewtopic.php?f=23&t=2842
JupudtCow
| #
На сайте http://oknaksa.ru закажите расчет технического задания, чтобы воспользоваться такой важной, полезной услугой, как остекление. На монтажные работы, а также сами конструкции предоставляются гарантии, вы сможете воспользоваться бесплатным обслуживанием. А все работы выполняются точно по ГОСТу, в соответствии с требованиями. Предоставляются исчерпывающие и содержательные консультации. Вам будет предложено несколько вариантов решения проблемы. Компания располагает огромным количеством готовых проектов.
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности
patomquish
| #
Arenda24.by – интернет-платформа для опубликования по аренде строительной техники и оборудования объявлений. На нашем ресурсе вы сможете работать комфортно, ведь его создала истинная команда профессионалов. Ищете аренда техники москва строительной? Arenda24.by – портал, где вы найдете по аренде техники и любых инструментов объявления, и сможете организовать грузоперевозки. У нас есть объявления любых типов категорий. Опубликуйте объявление уже сегодня, и оно в кратчайшие сроки найдет своих клиентов. С нами вы доступ к качественной и популярной платформе получите.
Pingrutty
| #
dark web market links darknet drugs
Toliknaf
| #
darknet markets onion dark market url
Thomaslob
| #
aviator predictor
Ronaldrhype
| #
DIOR bag replica
Diplomi_pfKt
| #
Купить диплом любого университета!
Мы предлагаем документы ВУЗов, расположенных на территории всей РФ.
diplomg-kurerom.ru/diplom-s-zaneseniem-v-reestr-po-vigodnoj-tsene-2/
Dnrtapg
| #
Мы изготавливаем дипломы любых профессий по доступным ценам. Мы можем предложить документы ВУЗов, которые расположены на территории всей РФ. Документы делаются на “правильной” бумаге высшего качества. Это дает возможности делать государственные дипломы, которые не отличить от оригиналов. medrese1000-letie.ru/auth/register=yes
AlfonsoIters
| #
Косметолог в Санкт-Петербурге – Перманентный макияж
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние достижения.
Наши товары:
Плоская мишень из фтористого магния
internetvolgogradCah
| #
подключить домашний интернет в волгограде
domashij-internet-volgograd003.ru
подключение интернета омск
metoprolol bipacksedel
| #
Medicament information for patients. Generic Name.
metoprolol bipacksedel
Actual about medicine. Read here.
Jarioryhi
| #
Заказать диплом любого ВУЗа!
Наша компания предлагаетбыстро и выгодно заказать диплом, который выполняется на бланке ГОЗНАКа и заверен печатями, штампами, подписями. Диплом способен пройти любые проверки, даже с применением профессиональных приборов. Достигайте своих целей быстро и просто с нашей компанией- shuddha.co.za/2025/04/13/kupit-diplom-prostoe-reshenie-dlja-slozhnyh-zadach-28
Ronaldrhype
| #
fake Bottega Veneta bag
Ronaldrhype
| #
fake Bottega Veneta bag
Rabyitabe
| #
dark market 2025 onion dark website
Markcen
| #
Новостройки или вторичка? Полезные рекомендации —
Ronaldrhype
| #
Chanel bag replica
soglasovanie pereplanirovki kvartiri_sikr
| #
проектирование перепланировки проектирование перепланировки .
Irvingseeta
| #
Купить натуральный камень https://proff-kamen.ru/kamen-krasnodar в Краснодаре и Краснодарском крае по оптовой цене. Каталог с расценками, размеры и монтаж дагестанской плитки из ракушечника, песчаника, травертина, известняка для отделки фасада и цоколя в Краснодаре от производителя.
can you buy cheap oxcarbazepine without dr prescription
| #
Pills information for patients. Cautions.
can you buy cheap oxcarbazepine without dr prescription
All about meds. Read information here.
Stevenspods
| #
переезды услуги грузчиков услуги грузчиков грузоперевозки
Ronaldrhype
| #
Fendi fake designer bags
Donaldkiz
| #
перевозки квартирные с грузчиками помощь при переезде грузчики
Gregorystype
| #
переезд квартирный с грузчиками http://atkarsk.standart-express.ru
fixitcaree
| #
Сервисный центр ФикситЦентр – выполнит срочный ремонт жестких дисков в Москве
DonDonfaita
| #
darknet sites dark market list
mostbet_lrOt
| #
мостбет скачать https://mostbet5009.ru .
Willieavamy
| #
Are online jackpots real?—skeptic! Book of Ra
mostbet_dzkr
| #
motbet http://mostbet5008.ru .
1win_goei
| #
1win cassino http://1win1008.top/ .
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы —
Pingrutty
| #
darknet websites darknet market links
Donaldlaf
| #
dark markets 2025 darknet links
Ronaldrhype
| #
fake DIOR bag
Ronaldrhype
| #
DIOR fake designer bags
Toliknaf
| #
darknet market links darknet drug market
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наши товары:
Молибденовая труба 1х23х800 мм МЧ ТУ 48-19-251-85 Приобретите молибденовые трубы от компании Редметсплав и обеспечьте вашему проекту прочность, устойчивость к высоким температурам и коррозии. Наши товары соответствуют высочайшим стандартам качества, гарантируя долговечность и надежность. Широкий ассортимент и профессиональные консультации по выбору подходящего решения.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Керамическая мишень АZO для распыления
1win_sbPr
| #
1 win pro 1 win pro .
Markcen
| #
Инвестиции в жильё: с чего начать? Читайте проверенные советы
EverettFLock
| #
кайт хургада
can you get cheap levitra without dr prescription
| #
Medicine information for patients. Generic Name.
can you get cheap levitra without dr prescription
Best about medicament. Get now.
1win_skKl
| #
1win партнерка вход https://1win7008.ru .
Thomaslob
| #
plinko app review
1win_lpkl
| #
1.вин https://www.1win7009.ru .
Rabyitabe
| #
darkmarket link darknet websites
Thomaslob
| #
plinko casino
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наша продукция:
Керамическая мишень Ta2O5
Ronaldrhype
| #
Louis Vuitton bag replica
mostbet_xgmn
| #
мостбет войти http://mostbet7004.ru/ .
1win_yhKl
| #
1win ваучер 1win ваучер .
1win_oeei
| #
casino online 1win casino online 1win .
DonDonfaita
| #
dark markets darknet marketplace
mostbet_nrOt
| #
мостбет вход http://www.mostbet5009.ru .
Sheilaadurl
| #
РедМетСплав предлагает внушительный каталог качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их качество. Превосходное обслуживание – наш стандарт – мы на связи, чтобы разрешать ваши вопросы и предоставлять решения под особенности вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наша продукция:
РџРѕРєРѕРІРєР° кобальтовая HAYNES HR-160 РџРѕРєРѕРІРєР° кобальтовая HAYNES HR-160 – это высококачественный металлический сплав, обладающий выдающимися свойствами прочности Рё устойчивости Рє РєРѕСЂСЂРѕР·РёРё. Данная РїРѕРєРѕРІРєР° предназначена для применения РІ экстремальных условиях, таких как авиационная Рё энергетическая промышленности. Благодаря СЃРІРѕРёРј уникальным характеристикам, РѕРЅР° обеспечивает надежную работу оборудования Рё долгий СЃСЂРѕРє службы изделий. Если РІС‹ ищете надежное решение для вашего бизнеса, покупая РџРѕРєРѕРІРєР° кобальтовая HAYNES HR-160, РІС‹ выбираете качество Рё эффективность. Закажите сейчас Рё убедитесь РІ ее превосходстве!
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные рекомендации —
Pingrutty
| #
darkmarket link darknet marketplace
erfahrungen plinko
| #
erfahrungen plinko
A Brief History of Elemental Analysis » Artemis Analytical
Willieavamy
| #
Online casinos need tools—help us! fishin frenzy
Ronaldrhype
| #
DIOR bag replica
internetvoronezhCah
| #
интернет провайдер воронеж
domashij-internet-voronezh001.ru
подключить интернет в воронеже в квартире
Thomaspoext
| #
Доставили в отель к годовщине – романтика!
доставка цветов томск
RakijamPreap
| #
Посетите B2BOX https://b2box.ru/ – это уникальное комплексное решение для создания упаковки всех видов и направлений. Мы делаем упаковку, которая продает! Упаковочные решения для ритейла, создание индивидуальной и SRP упаковки, услуги дизайнерского бюро, производство транспортной упаковки, собственное плоттерное производство, все виды печати – это и многое другое вы найдете, посетив наш сайт.
Hiwajamacuri
| #
Посетите страницу https://www.drive2.ru/o/b/691804470933197117/ и вы сможете ознакомиться с тем как происходит оклейка плёнкой Porsche 911 от профессионалов. Посмотрите как стал выглядеть Porsche 911 Carrera GTS после оклейки. Оклейка Porsche 911 выполнялась пленкой Vega 230 ST – узнайте почему и все ее преимущества в статье. Porsche 911 удивительный автомобиль – вам будет интересно читать!
Ronaldrhype
| #
Prada fake designer bags
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных покупателей. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние успехи.
Наша продукция:
Плоская мишень Cofeb
seroquel generics
| #
Drug information for patients. What side effects?
seroquel generics
Everything about medicament. Read information now.
Donaldlaf
| #
darknet sites darknet markets links
Anthonyaston
| #
большой лед экран https://svetodiodnye-led-ekrany.ru/
Michaelrow
| #
видеостена купить https://videostena-1.ru/
Ronaldrhype
| #
Celine replica designer bags
Ronaldrhype
| #
Celine replica designer bags
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы
Toliknaf
| #
darknet markets onion dark website
Mazrxag
| #
Мы изготавливаем дипломы любых профессий по доступным тарифам. Всегда стараемся поддерживать для заказчиков адекватную ценовую политику. Важно, чтобы дипломы были доступными для подавляющей массы наших граждан.
Покупка диплома, который подтверждает обучение в ВУЗе, – это выгодное решение. Купить диплом о высшем образовании: nsk-diplom.com/diplom-meditsinskogo-kolledzha-kupit-2/
Sazrygk
| #
Заказать диплом ВУЗа по доступной стоимости возможно, обратившись к надежной специализированной компании. Заказать документ о получении высшего образования можно в нашей компании. impulserp.5nx.ru/viewtopic.phpf=3&t=947
mostbet_dpet
| #
мостбет казино войти mostbet5010.ru .
Rabyitabe
| #
darknet drug links darknet markets
mostbet_rset
| #
скачать мостбет http://mostbet5010.ru .
Ronaldrhype
| #
Louis Vuitton fake designer bags
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наши товары:
РўСЂСѓР±Р° РёР· драгоценных металлов палладиевая 400С…8С…0.1 РјРј РџРґР90-10 РўРЈ Гарантированное качество Рё изысканный дизайн – купите драгоценные трубы для ювелирных украшений, машиностроения Рё архитектуры. РЁРёСЂРѕРєРёР№ выбор Рё высокое качество! Доставка РїРѕ Р РѕСЃСЃРёРё.
fexaxWab
| #
Ellcon.ru создан для вашего комфорта, чтобы вы экономили время на поездки, и смогли, отыскать нужное не выходя, из собственного дома. у нас вы можете купить провода и аксессуары, электроустановочные изделия, климатические системы, кабели, высоковольтное оборудование по доступным ценам. Ищете кабель канал 20 20? Ellcon.ru – портал, выбрав его для покупки, вы нервы, деньги и время прилично сэкономите. Доставка товаров осуществляется только при 100% предоплате заказа. Стремимся к лучшему, чтобы ежедневно радовать клиентов. Удачных вам покупок!
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации —
Thomaslob
| #
plinko app
Pingrutty
| #
darkmarket list darknet websites
Thomaslob
| #
dragon tiger game download
Sazrmiy
| #
Приобрести диплом под заказ в Москве вы имеете возможность через официальный сайт компании. thunder-consulting.net/employer/frees-diplom
DonDonfaita
| #
dark web sites dark web markets
Willieavamy
| #
Online casinos should curb—care play! vai de bet
Ronaldrhype
| #
DIOR fake designer bags
Ronaldrhype
| #
fake Fendi bag
Ronaldrhype
| #
fake Fendi bag
site
| #
Point very well used!.
Diplomi_uvki
| #
купить настоящий диплом гознак купить настоящий диплом гознак .
Iariorcvy
| #
Купить диплом института по невысокой цене возможно, обратившись к надежной специализированной фирме. Купить документ университета вы можете у нас в столице. diplomt-nsk.ru/kupit-diplom-s-zaneseniem-v-reestr-otzivi-ot-klientov-2
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы
Diplomi_qfKt
| #
Приобрести диплом о высшем образовании!
Мы предлагаем документы учебных заведений, которые находятся в любом регионе России.
diplomt-nsk.ru/kupit-diplom-s-zaneseniem-v-reestr-bez-problem/
Thomaslob
| #
vai.de.bete
syhie transformatori kypit_hcsl
| #
трансформатор силовой трехфазный сухой трансформатор силовой трехфазный сухой .
pin up azerbaycan_zkkl
| #
pinup az?rbaycan pinup az?rbaycan .
Donaldlaf
| #
darknet markets url darknet markets url
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их качество. Опытная поддержка – то, чем мы гордимся – мы на связи, чтобы улаживать ваши вопросы и предоставлять решения под требования вашего бизнеса.
Доверьте ваш запрос профессионалам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
Наши товары:
Труба кобальтовая Stellite SF6 Труба кобальтовая Stellite SF6 – это высококачественный продукт, разработанный для применения в сложных условиях. Состав данной трубы включает кобальт и хром, что гарантирует отличную коррозионную устойчивость и долговечность. Основные области использования: нефтегазовая, химическая и металлургическая промышленность. Труба обладает высокой прочностью и сохраняет свои характеристики при высоких температурах. Если вы ищете надежное решение для ваших производственных нужд, вам следует купить Труба кобальтовая Stellite SF6. Оптимальный выбор для профессионалов, которым важна надежность и качество.
Dnrtpvm
| #
Мы предлагаем дипломы любой профессии по выгодным тарифам. Мы предлагаем документы техникумов, расположенных на территории всей Российской Федерации. Документы делаются на “правильной” бумаге самого высокого качества. Это дает возможности делать настоящие дипломы, которые невозможно отличить от оригиналов. chotaikhoan.me/employer/archive-diploma
site
| #
Regards. Quite a lot of info!
Toliknaf
| #
bitcoin dark web dark web market list
Jariorpgb
| #
Приобрести диплом ВУЗа!
Наши специалисты предлагаютвыгодно приобрести диплом, который выполнен на бланке ГОЗНАКа и заверен мокрыми печатями, штампами, подписями официальных лиц. Диплом пройдет лубую проверку, даже с использованием профессионального оборудования. Решите свои задачи быстро и просто с нашими дипломами- bk8plusco68.copiny.com/question/details/id/1087642
Ronaldrhype
| #
Prada fake designer bags
Ronaldrhype
| #
Prada fake designer bags
Ronaldrhype
| #
Prada fake designer bags
Cazrbon
| #
Мы готовы предложить дипломы любой профессии по приятным ценам. Купить диплом в городе Королев — kyc-diplom.com/geography/korolev.html
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности
internetvoronezhCah
| #
провайдеры интернета в воронеже
domashij-internet-voronezh002.ru
подключить интернет
titobdup
| #
Serialexpress приобрести увлекательные сериалы на dvd предлагает. Здесь можно найти уникальные диски. Вы от покупки будете в восторге, потому как отменное качество сериалов. Товары доступны по приемлемой стоимости и отлично упакованы. Ищете купить сериалы в Волгограде? Serialexpress.ru – ресурс, где представлена информация об оплате, рекомендуем вам с ней ознакомиться. Предлагаем накопительную систему скидок. Быть постоянным клиентом – выгодно. Быструю доставку гарантируем. При возникновении вопросов, на электронную почту нам пишите. Рады вам всегда!
otutejTON
| #
Компания Аллегро https://spb.tk-allegro.ru/ – это аренда автобуса в Санкт-Петербурге. У нас от 8 до 55 мест свой автопарк автобусов. Водители с большим опытом. У вас есть возможность заказать для любых мероприятий автобус: на экскурсию, выпускной, свадьбу и для школьников. Посетите сайт – узнайте больше о наших услугах и стоимости аренды или воспользуйтесь калькулятором на сайте, для расчёта стоимости.
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – ваш лучший выбор.
поставляемая продукция:
Медный тройник под высокотемпературную пайку 25х21х0.9 мм 16.4х18.4 мм твердая пайка М2РМ ГОСТ Р52922-2008 Купить медные тройники под высокотемпературную пайку от компании Редметсплав. Высокая теплопроводность, прочное соединение и применение в различных отраслях. Обращайтесь к нам для подробной информации и заказа.
losartan potassium 25 mg color
| #
Medication information. Effects of Drug Abuse.
losartan potassium 25 mg color
Some trends of medication. Get information here.
Pingrutty
| #
dark market link dark market link
Toliknaf
| #
dark market list darknet markets
DonDonfaita
| #
dark web market urls dark websites
Donaldlaf
| #
darknet drugs darknet drug links
AlfonsoIters
| #
Ремонт телефонов – Сервисный центр Мобиопт
Cazrwdw
| #
Получить диплом о высшем образовании поспособствуем. Купить диплом о высшем образовании в Перми – diplomybox.com/kupit-diplom-o-vysshem-obrazovanii-v-permi
Ronaldrhype
| #
fake DIOR bag
Thomaslob
| #
plinko free
Thomaslob
| #
bet on red
RandySeexy
| #
The popular service ai girlfriend generator offers to create an AI girl who understands you, can hold a conversation and even flirt. Ideal for those who are looking for emotions in a digital format.
Willieavamy
| #
Online casinos need flags—alert us! plinko
Hijamnetly
| #
На сайте https://pilmi.ru вы найдете огромное количество фильмов самых разных жанров, включая комедии, мелодрамы, драмы, романтические. Для того чтобы подыскать наиболее подходящий вариант, нужно воспользоваться специальным каталогом. Вашему вниманию представлены и ожидаемые любопытные новинки этого года, которые произведут эффект. Все это можно просматривать в любое время и на различных устройствах. Прямо сейчас воспользуйтесь возможностью посмотреть азиатское кино. Используйте поиск для облегчения выбора.
1win_ajkl
| #
баланс ван вин http://www.1win7009.ru .
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
поставляемая продукция:
Нержавеющая круглая РїРѕРєРѕРІРєР° 700 РјРј РР943 РўРЈ Узнайте больше Рѕ преимуществах нержавеющей круглой РїРѕРєРѕРІРєРё, ее прочности, устойчивости Рє РєРѕСЂСЂРѕР·РёРё Рё высокой теплопроводности. Нержавеющая круглая РїРѕРєРѕРІРєР° – идеальное решение для производства строительных материалов, трубопроводов, техники, медицинского Рё пищевого оборудования.
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы
Thomaslob
| #
fortune tiger bonus gratis sem deposito
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наши товары:
Циркониевая мишень для распыления
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент отборных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы гарантируем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена соответствующими документами, подтверждающими их происхождение. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы и находить ответы под специфику вашего бизнеса.
Доверьте вашу потребность в редких металлах профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Рзделия РёР· кобальта РРџ720 – РўРЈ 14-1-3457-82 Рзделия РёР· кобальта РРџ720 – РўРЈ 14-1-3457-82 предлагают высокую прочность Рё устойчивость Рє РєРѕСЂСЂРѕР·РёРё, что делает РёС… идеальными для применения РІ различных отраслях. Рти изделия обладают отличными механическими характеристиками Рё надежностью, что гарантирует долгий СЃСЂРѕРє службы РІ условиях эксплуатации. Если РІС‹ ищете качественные компоненты, то купить Рзделия РёР· кобальта РРџ720 – РўРЈ 14-1-3457-82 – это разумное решение. Рти изделия удовлетворяют всем современным стандартам Рё требованиям, Рё обеспечивают отличные результаты РїСЂРё использовании.
pin up azerbaycan_vekl
| #
pin up casino azerbaijan pin up casino azerbaijan .
Pingrutty
| #
darknet markets 2025 darkmarket list
syhie transformatori kypit_rgsl
| #
каталог сухих трансформаторов каталог сухих трансформаторов .
Ronaldrhype
| #
fake Chanel bag
Ronaldrhype
| #
Chanel fake designer bags
Rabyitabe
| #
dark web sites darknet markets url
Toliknaf
| #
darknet site darknet markets
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – идеальный вариант для вас.
Наши товары:
Кольцо РёР· драгоценных металлов золотое 4С…3С…0.5 РјРј Р—Р»99.9 РўРЈ РЁРёСЂРѕРєРёР№ выбор высококачественных кольц РёР· драгоценных металлов. Рлегантные украшения, созданные талантливыми мастерами. Уникальность Рё изысканность РІ каждой детали. Возможность заказа персонального кольца РїРѕ вашим пожеланиям. Подчеркните СЃРІРѕСЋ индивидуальность СЃ нашими кольцами.
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности
site
| #
You’ve made your stand very effectively!.
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент качественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Опытная поддержка – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы по мере того как находить ответы под особенности вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Пруток титановый 70РўРњ-Р’Р” – ГОСТ 10994-74 Проволока титановая 70РўРњ-Р’Р” – ГОСТ 10994-74 является высококачественным продуктом, который широко используется РІ различных отраслях, включая авиацию Рё машиностроение. РћРЅР° обладает отличной РєРѕСЂСЂРѕР·РёР№РЅРѕР№ стойкостью Рё высокой прочностью, что делает ее идеальным выбором для ответственных конструкций. Вам стоит купить Проволока титановая 70РўРњ-Р’Р” – ГОСТ 10994-74, если РІС‹ ищете надежный РёDurable материал для СЃРІРѕРёС… проектов. Ртот РїСЂРѕРґСѓРєС‚ поможет вам достичь максимальной эффективности Рё долговечности изделий.
site
| #
Nicely put. Thanks a lot.
1win_rjoi
| #
1win bet uganda https://www.1win1005.top .
Thomaslob
| #
plinko game app
Thomaslob
| #
is plinko a con
blogcaree
| #
Про гаджеты и электроннику, подробнее читайте – на сайте
Pingrutty
| #
darknet market links darknet links
GunefUsano
| #
На сайте http://www.lilibum.ru вы найдете много анкет реальных людей для того, чтобы завести знакомства, найти свою вторую половинку и просто интересно провести время, если пригласите кого-то на свидание. Для того чтобы составить мнение о человеке, можно обменяться фотографиями и пообщаться на определенные темы, которые вас интересуют. Самые привлекательные и интересные люди вашего города находятся на этом портале. Знакомьтесь, влюбляйтесь и создавайте семьи. Для получения доступа ко всем функциям, пройдите регистрацию.
1win_hqKl
| #
1 win.pro 1 win.pro .
can i buy cheap zoloft without prescription
| #
Medication information sheet. Generic Name.
can i buy cheap zoloft without prescription
Everything what you want to know about medicine. Read here.
Willieavamy
| #
I don’t get online shade—it’s fine! fishin frenzy
1win_ywei
| #
1win mx https://1win1008.top/ .
Markcen
| #
Хотите сдать квартиру в аренду? Читайте проверенные советы
Potolkilk
| #
Добрый день смелым!
Натяжные потолки позволяют реализовать любые световые сценарии. Интегрировать можно как традиционные люстры и светильники, так и современные светодиодные ленты и точечные источники света. Правильно подобранное освещение не только подчеркнёт достоинства помещения, но и позволит легко менять атмосферу от яркой и динамичной до мягкой и расслабляющей.
Вся информация на сайте – https://ru.pinterest.com/NovaPotolki/
натяжной потолок в детскую, натяжной потолок как, какой потолок дешевле
монтаж натяжных потолков, минусы сатинового натяжного потолка, в квартире натяжные потолки
Удачи!
pin up azerbaycan_itkl
| #
pinup az?rbaycan pinup az?rbaycan .
Ronaldrhype
| #
Fendi bag replica
syhie transformatori kypit_sysl
| #
трансформатор силовой трехфазный сухой трансформатор силовой трехфазный сухой .
Ronaldrhype
| #
Gucci bag replica
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица товара подтверждена соответствующими документами, подтверждающими их качество. Превосходное обслуживание – наш стандарт – мы на связи, чтобы улаживать ваши вопросы а также находить ответы под требования вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в гибкости нашего предложения
Наши товары:
Магний 705C – ASTM B752 Магний 705C – ASTM B752 является высококачественным сплавом, который отличается отличной прочностью Рё легкостью. Ртот материал идеально РїРѕРґС…РѕРґРёС‚ для применения РІ авиационной Рё автомобильной промышленности благодаря СЃРІРѕРёРј уникальным свойствам. Благодаря хорошей РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкости Рё отличной обрабатываемости, Магний 705C – ASTM B752 становится незаменимым выбором для производителей высокотехнологичных компонентов. Если РІС‹ ищете надежный Рё эффективный материал, РЅРµ упустите шанс купить Магний 705C – ASTM B752. Его применение повысит качество ваших изделий!
Donaldlaf
| #
tor drug market dark web sites
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Мишень из ниобия для распыления
can i purchase benicar price
| #
Medicine information for patients. Short-Term Effects.
can i purchase benicar price
Best trends of medicament. Read information now.
internetvoronezhCah
| #
подключение интернета воронеж
domashij-internet-voronezh003.ru
провайдеры домашнего интернета воронеж
Pingrutty
| #
dark web market urls darkmarket
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы —
site
| #
You actually expressed it really well.
MiriamcauSe
| #
Could the Invention of CHNS Analyzer in 1968 be Considered a Revolutionary Leap in Elemental Analysis, and if So, How Did It Impact the Analysis of Food, Soil, and Product Quality?
Slightly off the main thread here
A bit back I discovered a site called the Talabout platform.
It’s full of interviews with people from all walks of life — genuine, not your typical media stuff.
Sometimes the vibe there somehow aligns with the stuff being shared here.
Ever read anything there?
site
| #
Really lots of awesome facts.
Lazrgbe
| #
Диплом ВУЗа Российской Федерации!
Без наличия диплома очень непросто было продвигаться по карьере. Именно из-за этого решение о заказе диплома следует считать целесообразным. Приобрести диплом о высшем образовании veniaminv.flybb.ru/viewtopic.php?f=1&t=4278
Ronaldrhype
| #
Bottega Veneta replica designer bags
Xazrmwb
| #
Купить диплом академии!
Мы изготавливаем дипломы любых профессий по доступным тарифам. Вы заказываете диплом в надежной и проверенной временем компании. : pashasevkav.getbb.ru/posting.php?mode=post&f=31&sid=475838660d60673c73f88a5c4b5c71a7
DonDonfaita
| #
dark market link darkmarkets
Toliknaf
| #
darkmarket url darkmarket link
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы —
Thomaslob
| #
betonred casino no deposit bonus
Pingrutty
| #
dark web market darknet markets 2025
1win_jzoi
| #
1win bet uganda 1win1005.top .
Willieavamy
| #
The sounds online hook—stay in! plinko
Ronaldrhype
| #
Chanel bag replica
where buy fexofenadine pill
| #
Meds information sheet. Drug Class.
where buy fexofenadine pill
Best news about medication. Get information now.
Ronaldrhype
| #
Louis Vuitton bag replica
Mazrgxf
| #
Мы изготавливаем дипломы любой профессии по выгодным тарифам. Всегда стараемся поддерживать для клиентов адекватную политику цен. Важно, чтобы дипломы были доступны для большинства граждан.
Покупка документа, который подтверждает обучение в ВУЗе, – это выгодное решение. Купить диплом любого университета: kupite-diplom0024.ru/kupit-diplom-nedorogo-4/
Thomaslob
| #
betonred casino no deposit bonus
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы —
Pingrutty
| #
dark websites darknet drug store
Thomaslob
| #
plinko france
Ronaldrhype
| #
DIOR bag replica
Ronaldrhype
| #
DIOR bag replica
DonDonfaita
| #
darkmarket 2025 darknet markets
Rabyitabe
| #
dark market link darknet markets 2025
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем широкий ассортимент продукции, обеспечивая высочайшее качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с прекрасным качеством обслуживания.
Наша команда поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наши товары:
Лист свинцовый 29x1000x1500 С1 ГОСТ 9559-89 Выберите идеальный свинцовый лист для вашего проекта. Широкий выбор размеров и толщин для защиты от радиации, звукоизоляции, строительства, производства аккумуляторов и многих других целей. Мы предлагаем высококачественные продукты с гарантированной прочностью и устойчивостью. Подробности на сайте!
Sazrast
| #
Приобретение документа о высшем образовании через качественную и надежную фирму дарит ряд преимуществ для покупателя. Данное решение позволяет сберечь время и значительные финансовые средства. Тем не менее, плюсов намного больше.Мы изготавливаем дипломы любой профессии. Дипломы производят на настоящих бланках. Доступная цена в сравнении с огромными расходами на обучение и проживание. Заказ диплома института будет мудрым шагом.
Заказать диплом: freediplom.com/kupit-diplom-s-zaneseniem-v-reestr-bistro-i-prosto-7/
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности
Ronaldrhype
| #
Louis Vuitton fake designer bags
Yaxayrtriz
| #
Посетите сайт https://artradol.com/ и вы узнаете все про Артрадол – это препарат для лечения суставов от производителя. Узнайте все преимущества нестероидного противовоспалительного препарата для лечения суставов. Помогает бороться с основными заболеваниями суставов: артритом, остеохондрозом и остеоартрозом. Подробности, инструкция применения и где купить можно узнать на сайте.
Pingrutty
| #
darknet links darknet markets
get generic propecia online
| #
Medicine information sheet. Long-Term Effects.
get generic propecia online
Some information about pills. Read here.
best australian online casino
| #
best australian online casino
A Brief History of Elemental Analysis » Artemis Analytical
Ronaldrhype
| #
DIOR bag replica
Willieavamy
| #
The slot thrill online keeps me going! aviator game
Thomaslob
| #
plinko casino
internetomskCah
| #
тарифы интернет и телевидение омск
domashij-internet-omsk001.ru
лучший интернет провайдер омск
Rabyitabe
| #
dark web sites dark web market urls
Donaldlaf
| #
darknet markets onion darkmarket link
DonDonfaita
| #
darknet drug market dark web marketplaces
Ronaldrhype
| #
fake Louis Vuitton bag
Ronaldrhype
| #
fake Louis Vuitton bag
Markcen
| #
Как выбрать надёжного застройщика? Полезные статьи и советы —
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица товара подтверждена соответствующими документами, подтверждающими их происхождение. Дружелюбная помощь – наша визитная карточка – мы на связи, чтобы улаживать ваши вопросы и находить ответы под специфику вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
РўСЂСѓР±Р° висмутовая CAC911 – JIS H 5120-2016 РўСЂСѓР±Р° висмутовая CAC911 – JIS H 5120-2016 представляет СЃРѕР±РѕР№ высококачественный металлический РїСЂРѕРґСѓРєС‚, предназначенный для использования РІ различных отраслях. Рзготовленная РїРѕ международным стандартам, данная труба отличается отличной РєРѕСЂСЂРѕР·РёР№РЅРѕР№ стойкостью Рё механической прочностью. РћРЅР° идеально РїРѕРґС…РѕРґРёС‚ для систем, работающих РІ сложных условиях. Если РІС‹ ищете надежное решение, чтобы улучшить эффективность СЃРІРѕРёС… процессов, вам стоит купить РўСЂСѓР±Р° висмутовая CAC911 – JIS H 5120-2016. Ртот РїСЂРѕРґСѓРєС‚ обеспечит долговечность Рё надежность РІ эксплуатации. РќРµ упустите возможность воспользоваться преимуществами, которые дает современное оборудование.
Diplomi_jbki
| #
купить диплом москва купить диплом москва .
1win_khkl
| #
один вин официальный сайт http://1win7009.ru/ .
cost generic requip pill
| #
Pills information sheet. Cautions.
cost generic requip pill
Actual what you want to know about medication. Read here.
Pingrutty
| #
darknet markets url darkmarket
Thomaslob
| #
hot hot fruit demo
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы —
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
Имея широкий спектр редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние достижения.
Наша продукция:
Плоская мишень AlSc для распыления сплавов
Sheilaadurl
| #
В поисках надежного поставщика редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф гарантирует все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы выполнить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в определении подходящих продуктов и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – ваш лучший выбор.
Наша продукция:
Проволока РёР· палладия Рё его сплавов 0.28 РјРј РџРґРЎСЂ-40 ГОСТ 18390-73 Купить проволоку РёР· палладия Рё его сплавов 0.28 РјРј РџРґРЎСЂ-40 ГОСТ 18390-73 РЅР° сайте Редметсплав.СЂС„. Рдеальный выбор для точных Рё тонких работ, прочный Рё стойкий материал для производства электроники, медицинских устройств Рё ювелирных изделий.
Birogopisk
| #
Посетите https://igormylnikovchannel.ru/poezdki-v-taksi-s-det-mi/ и вы найдете полезную информацию для начинающих таксистов и опытных водителей на тему поездок в такси с детьми, начиная от безопасности поездок и ответственности и правилах перевозки детей, до разбора вопросов почему таксисты отказываются от поездок с детьми и многое другое. Подробнее на странице.
Donaldlaf
| #
dark web marketplaces dark web markets
DonDonfaita
| #
dark web marketplaces dark market url
Ronaldrhype
| #
Celine replica designer bags
Ronaldrhype
| #
Chanel replica designer bags
Pingrutty
| #
darknet markets 2025 darknet site
Willieavamy
| #
Online bingo flows—great crew! book of dead
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации —
1win_hzEi
| #
1win win 1win win .
Anonymous
| #
Can you be more specific about the content of your article? After reading it, I still have some doubts. Hope you can help me.
Ronaldrhype
| #
Chanel bag replica
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от мелких партий до обширных поставок, мы обеспечиваем оперативное исполнение вашего заказа.
Каждая единица изделия подтверждена требуемыми документами, подтверждающими их происхождение. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы разрешать ваши вопросы и предоставлять решения под специфику вашего бизнеса.
Доверьте вашу потребность в редких металлах специалистам РедМетСплав и убедитесь в множестве наших преимуществ
поставляемая продукция:
Проволока магниевая M12331 – UNS Полоса магниевая M12331 – UNS – это высококачественный материал, идеально подходящий для аэрокосмической Рё автомобильной промышленности. Обладая отличной РєРѕСЂСЂРѕР·РёРѕРЅРЅРѕР№ стойкостью Рё легкостью, эта полоса гарантирует надежность Рё долговечность РІ эксплуатации. Р’С‹ можете купить Полоса магниевая M12331 – UNS для ваших проектов, чтобы обеспечить оптимальные характеристики Рё производительность. Данная полоса также поддается механической обработке, что позволяет применять ее РІ самых различных продуктах. Оцените преимущества использования данного материала уже сегодня!
Pingrutty
| #
darknet market darknet market links
Thomaslob
| #
predictor aviator
Toliknaf
| #
darknet market lists dark web link
Thomaslob
| #
plinko casino
Rabyitabe
| #
dark market link darknet markets url
aviator game
| #
aviator game
A Brief History of Elemental Analysis » Artemis Analytical
Ronaldrhype
| #
Fendi fake designer bags
Markcen
| #
Как выбрать надёжного застройщика? Узнайте подробности
get tadacip without rx
| #
Meds information leaflet. Long-Term Effects.
get tadacip without rx
Best about medication. Get now.
1win_juPr
| #
1win casino uganda http://1win1006.top .
generic nortriptyline online
| #
Medicines information leaflet. Drug Class.
generic nortriptyline online
Everything what you want to know about drugs. Read information now.
1win_ivSn
| #
1 win официальный сайт 1win7026.ru .
mostbet_qyEt
| #
mosbet http://mostbet3021.ru/ .
Ronaldrhype
| #
Chanel fake designer bags
mostbet_heSr
| #
скачать mostbet на телефон https://www.mostbet7005.ru .
Pingrutty
| #
darknet sites dark market onion
internetomskCah
| #
дешевый интернет омск
domashij-internet-omsk002.ru
интернет домашний омск
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные рекомендации —
Willieavamy
| #
I wish online casinos had better tutorials for beginners! book of dead
DonDonfaita
| #
darknet drug market dark web sites
Rabyitabe
| #
darknet links darknet drug store
Ronaldrhype
| #
Prada replica designer bags
mostbet_ljEt
| #
скачать mostbet на телефон https://mostbet3021.ru/ .
Ronaldrhype
| #
Celine replica designer bags
mostbet_siSr
| #
mostber https://mostbet7005.ru/ .
Anya100mi
| #
Hello .!
I came across a 103 useful tool that I think you should dive into.
This platform is packed with a lot of useful information that you might find helpful.
It has everything you could possibly need, so be sure to give it a visit!
https://suradapoetica.es/estrategias-de-apuestas/apostando-con-ventajas-promociones-especiales-que-ofrecen-las-casas-de-apuestas-online/
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные рекомендации —
Pingrutty
| #
darknet markets url darknet links
mostbet_spEt
| #
служба поддержки мостбет номер телефона http://mostbet3021.ru/ .
blogcaree
| #
Про гаджеты и электроннику, подробнее читайте – в блоге
mostbet_ngSr
| #
мостбет вход http://www.mostbet7005.ru .
mostbet_sqmt
| #
mostbet игры http://www.mostbet6033.ru .
Donaldlaf
| #
dark websites dark markets 2025
DonDonfaita
| #
dark web market urls dark web market list
how to get cheap colchicine price
| #
Medicament information. Generic Name.
how to get cheap colchicine price
Best about drug. Read information here.
Rabyitabe
| #
onion dark website dark web link
skladi_qeSa
| #
где можно хранить вещи hranim-veshi-msk24.ru .
Ronaldrhype
| #
fake Chanel bag
Ronaldrhype
| #
Chanel replica designer bags
Emiletwext
| #
услуги сертификации
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы —
Pingrutty
| #
dark web link dark web marketplaces
Ronaldrhype
| #
fake Louis Vuitton bag
Willieavamy
| #
Online casinos need hold—too wild! jackpotraider
pliinko
| #
pliinko
A Brief History of Elemental Analysis » Artemis Analytical
Xazrteh
| #
Приобрести диплом о высшем образовании!
Мы можем предложить дипломы психологов, юристов, экономистов и других профессий по приятным ценам. Вы покупаете документ через надежную и проверенную временем фирму. : veniaminv.flybb.ru/viewtopic.php?f=1&t=4268
StevenCeste
| #
Punpfun.org is your one-stop shop for launching chaos into the chart galaxy—no barriers, just vibes
Try it now on Punpfun.net
can i purchase cheap erythromycin without insurance
| #
Pills information leaflet. Long-Term Effects.
can i purchase cheap erythromycin without insurance
Some news about medicines. Get here.
StevenCeste
| #
Punpfun.org is where memecoins go from 0 to viral in seconds—built for chaos, fun, and wild energy
Try it now on Punpfun.net
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для легализации товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы поставить любой запрос с прекрасным качеством обслуживания.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете надежность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что качество и уровень нашего сервиса – наилучшее решение для вашего бизнеса.
Наши товары:
Мишень РёР· драгоценного металла или его сплава кольцевая платина 10С…1.5 РјРј ПлПд95-5 РўРЈ Выберите уникальные мишени РёР· драгоценных металлов для напыления РѕС‚ Редметсплав.СЂС„. Обеспечивают высокую степень защиты Рё долговечность. Рдеальный выбор для промышленности, строительства Рё производства.
Gerardobut
| #
Ищите место для отдыха? туры в иорданию 2025 Мы предлагаем увлекательные экскурсии по Иордании, комфортный отдых и маршруты по главным достопримечательностям Иордании. Не знаете, что посмотреть в Иордании? Начните с Петры, Вади Рам и Мёртвого моря!
StevenCeste
| #
Punpfun.org makes memecoin culture faster, louder, and easier to join than ever before
Try it now on Punpfun.net
pin up azerbaycan_ypkt
| #
pin up az pin up az .
pin up azerbaycan_yzml
| #
pinup casino pinup casino .
Thomaslob
| #
plinko gratuit
Markcen
| #
Хотите сдать квартиру в аренду? Узнайте подробности
StevenCeste
| #
Punpfun.org makes it easy and fun to launch, discover, and follow the next big memecoin movement
Try it now on Punpfun.net
Donaldlaf
| #
tor drug market best darknet markets
Toliknaf
| #
darknet drug store dark web drug marketplace
Ronaldrhype
| #
Gucci fake designer bags
StevenCeste
| #
Punpfun.org gives you the tools to create, watch, and share your favorite new memecoins with ease
Try it now on Punpfun.net
DonDonfaita
| #
darknet markets onion address darkmarkets
Pingrutty
| #
darknet markets onion darknet sites
Ronaldrhype
| #
fake Gucci bag
Ronaldrhype
| #
Gucci replica designer bags
Ronaldrhype
| #
Prada fake designer bags
StevenCeste
| #
Punpfun.org delivers instant creation, smooth charts, and nonstop coin launches around the clock
Try it now on Punpfun.net
Ronaldrhype
| #
Gucci bag replica
site
| #
Terrific postings, Kudos.
Thomaslob
| #
fortune tiger bet
internetomskCah
| #
домашний интернет омск
domashij-internet-omsk003.ru
домашний интернет тарифы
Markcen
| #
Покупка квартиры без рисков? Полезные статьи и советы —
Shift Amrix
| #
Shift Amrix
A Brief History of Elemental Analysis » Artemis Analytical
Claudeweamp
| #
Всем привет! Если вы думаете, как провести незабываемый уикенд, загляните на раздел для коротких поездок. Мы с друзьями воспользовались рекомендацией и отправились в Харьков, чтобы посетить местный парк Горького и гастрономические заведения. Эти выходные были наполнены позитивом, впечатлениями и вкусной едой. Такой отдых я советую всем, кто ищет вдохновения для своих приключений.
Michaeldow
| #
Нужно купить бетон в Краснодаре с доставкой на объект? Предлагаем прочные и надежные строительные смеси для фундамента, заливки полов, дорожных работ и других задач. Производим по ГОСТу, соблюдаем все пропорции и сроки. Заказать можно по телефону или через сайт: доставка бетона краснодар. Доставим точно в срок и по выгодной цене
Sazrelf
| #
Покупка диплома через проверенную и надежную фирму дарит много плюсов. Это решение помогает сберечь как личное время, так и существенные финансовые средства. Тем не менее, только на этом выгоды не ограничиваются, достоинств намного больше.Мы предлагаем дипломы любых профессий. Дипломы производят на оригинальных бланках государственного образца. Доступная стоимость по сравнению с большими тратами на обучение и проживание. Покупка диплома о высшем образовании из российского университета является выгодным шагом.
Приобрести диплом о высшем образовании: diplomers.com/kupit-diplom-s-reestrom-i-otzivami-bistro-i-udobno-2/
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор отборных изделий из ценных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до обширных поставок, мы гарантируем быстрое выполнение вашего заказа.
Каждая единица изделия подтверждена всеми необходимыми документами, подтверждающими их соответствие стандартам. Дружелюбная помощь – то, чем мы гордимся – мы на связи, чтобы ответить на ваши вопросы а также находить ответы под требования вашего бизнеса.
Доверьте потребности вашего бизнеса специалистам РедМетСплав и убедитесь в множестве наших преимуществ
Наши товары:
Пруток титановый РџРў-7РњСЃРІ – ГОСТ 27265-87 Проволока титановая РџРў-7РњСЃРІ – ГОСТ 27265-87 представляет СЃРѕР±РѕР№ высококачественный материал, используемый РІ различных отраслях промышленности. Обладая отличными механическими свойствами Рё стойкостью Рє РєРѕСЂСЂРѕР·РёРё, эта проволока идеальна для производства компенсаторов, крепежа Рё РґСЂСѓРіРёС… конструкций. Если РІС‹ ищете надежный материал, проволока Титан РџРў-7РњСЃРІ является отличным выбором. РћРЅР° обеспечивает долговечность Рё стабильность РІ эксплуатации. РќРµ упустите возможность купить проволока титановая РџРў-7РњСЃРІ – ГОСТ 27265-87 Рё повысить качество своей продукции.
can i get fosamax pills
| #
Pills information sheet. Brand names.
can i get fosamax pills
Everything about medicament. Get here.
Pingrutty
| #
dark web markets darknet site
Donaldlaf
| #
dark market onion darknet sites
Toliknaf
| #
dark web sites onion dark website
DonDonfaita
| #
darkmarket list darknet site
Ronaldrhype
| #
Bottega Veneta replica designer bags
Willieavamy
| #
Online casino ads blast—quiet it! ganesha gold
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы
site
| #
Wow plenty of useful data!
Sazrnjv
| #
Заказать диплом института по выгодной цене можно, обратившись к проверенной специализированной компании. Купить документ университета можно у нас в Москве. veraciousrp.listbb.ru/viewtopic.phpf=82&t=2081
site
| #
Really all kinds of helpful information.
prodvijenie saita_pbsi
| #
раскрутка сайта в москве itechua.com/other/277983 .
pin up azerbaycan_gikt
| #
pinup az pinup az .
pin up azerbaycan_akml
| #
pin ap pin ap .
Pingrutty
| #
dark websites darkmarket link
blogcaree
| #
Про гаджеты и электроннику, подробнее читайте – в блоге
Toliknaf
| #
tor drug market dark web link
Markcen
| #
Хотите сдать квартиру в аренду? Полезные рекомендации —
Rabyitabe
| #
darknet market list dark web marketplaces
DonDonfaita
| #
tor drug market darknet market
Ronaldrhype
| #
fake Celine bag
Ronaldrhype
| #
fake DIOR bag
where can i get lisinopril pills
| #
Medicine information leaflet. Generic Name.
where can i get lisinopril pills
Best information about drugs. Read information now.
dobrowin
| #
Receba 100$ de Bônus no dobrowin e Comece a Apostar
Agora!
Thomaslob
| #
dragon tiger online india
Pingrutty
| #
dark web market urls dark web drug marketplace
1win_qePr
| #
1win uganda 1win uganda .
Willieavamy
| #
Online casinos should free—try it! betonred
Markcen
| #
Новостройки или вторичка? Полезные статьи и советы —
cost of coumadin no prescription
| #
Medicament information leaflet. Generic Name.
cost of coumadin no prescription
Actual about meds. Get information here.
pin up azerbaycan_anml
| #
pinup az pinup az .
mostbet_vpEt
| #
mostbet игры https://www.mostbet3021.ru .
prodvijenie saita_tusi
| #
продвижение сайтов в москве продвижение сайтов в москве .
Donaldlaf
| #
dark markets 2025 darknet marketplace
Toliknaf
| #
dark market 2025 best darknet markets
DonDonfaita
| #
dark web markets darknet sites
Rabyitabe
| #
darknet markets onion dark web market urls
1win_coPr
| #
1win bet uganda 1win bet uganda .
Ronaldrhype
| #
Louis Vuitton bag replica
site
| #
Kudos, Wonderful information.
Pingrutty
| #
dark market url darknet markets onion
Thomaslob
| #
aviator predictor online
blogcaree
| #
Про гаджеты и электроннику, подробнее читайте – тут
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы —
Trefoff
| #
Приобрести диплом о высшем образовании. Покупка диплома ВУЗа через качественную и надежную фирму дарит много плюсов для покупателя. Такое решение позволяет сэкономить время и значительные денежные средства. thegroomslist.co.uk/registered-employers/131249
1win_nzSn
| #
1вин сайт официальный 1вин сайт официальный .
Sazrpru
| #
Купить диплом под заказ в Москве можно через сайт компании. connectingsparks.com/read-blog/75477_mozhno-li-kupit-diplom-v-reestre.html
mostbet_dbSr
| #
мосбет казино http://mostbet7005.ru .
4657
| #
4657
A Brief History of Elemental Analysis » Artemis Analytical
1win_hzEi
| #
1win сайт 1win сайт .
site
| #
Helpful material, Appreciate it!
Diplomi_yiKt
| #
Заказать диплом университета!
Мы предлагаем документы любых учебных заведений, расположенных на территории всей РФ.
diplom-onlinex.com/kupit-diplom-s-reestrom-v-rossii-bistro-i-nadezhno-4/
EdwinGeopy
| #
GetGrass.io is a decentralized platform that lets users earn rewards by sharing their unused internet bandwidth. The collected traffic is used to gather public web data for training AI models, ensuring transparency and privacy. You can join the network through their browser extension or desktop app — getgrass io.
Ronaldrhype
| #
fake prada bag
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Работа с нами гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа требуемого количества.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым текущие трудности превращаются в завтрашние возможности.
Наша продукция:
Ниобиевый тигель высокой чистоты
Pingrutty
| #
dark market onion best darknet markets
Toliknaf
| #
dark web market urls darknet markets url
DonDonfaita
| #
darknet market lists darknet drug market
Rabyitabe
| #
dark market onion darkmarket
Anya100mi
| #
Hello guys!
I came across a 103 awesome tool that I think you should browse.
This resource is packed with a lot of useful information that you might find interesting.
It has everything you could possibly need, so be sure to give it a visit!
https://fass-heidelberg.de/tricks-zum-sieg/lotterie-gibt-es-reale-gewinnchancen-oder-nicht/
Markcen
| #
Хотите сдать квартиру в аренду? Читайте проверенные советы
Ronaldrhype
| #
Celine fake designer bags
site
| #
Appreciate it. A good amount of postings.
mostbet_kbkr
| #
mostbet скачать http://www.mostbet7006.ru .
Dnrttbx
| #
Мы готовы предложить дипломы психологов, юристов, экономистов и любых других профессий по приятным тарифам. Мы можем предложить документы ВУЗов, расположенных на территории всей РФ. Дипломы и аттестаты выпускаются на бумаге высшего качества. Это дает возможность делать государственные дипломы, не отличимые от оригиналов. talkotive.com/read-blog/1817_gde-kupit-diplom-s-provodkoj.html
Thomaslob
| #
plinko game review
Thomaslob
| #
jogo vai de bet
Willieavamy
| #
Online blackjack is my jam—simple yet strategic! sugar rush
Sazrfua
| #
Купить диплом института по выгодной цене вы сможете, обратившись к надежной специализированной фирме. Приобрести документ университета можно в нашем сервисе. jobboat.co.uk/employer/originals-diplomsi
buy motrin without prescription
| #
Medication information sheet. What side effects?
buy motrin without prescription
All news about drug. Get information here.
Cazrwcq
| #
Заказать диплом любого университета поможем. Купить аттестат в Ставрополе – diplomybox.com/kupit-attestat-v-stavropole
where buy generic ziprasidone without a prescription
| #
Meds information for patients. Long-Term Effects.
where buy generic ziprasidone without a prescription
Everything about medicament. Get information now.
Pingrutty
| #
dark web sites darknet market lists
blogcaree
| #
Про гаджеты и электроннику, подробнее читайте – в блоге
Markcen
| #
Инвестиции в жильё: с чего начать? Узнайте подробности
site
| #
Good write ups, Many thanks!
WilliamKab
| #
?Hola usuarios de apuestas
Las pГЎginas de apuestas EspaГ±a sin licencia estГЎn ganando popularidad rГЎpidamente.
ВїQuieres que siga generando mГЎs como estos para el resto de las palabras clave? TambiГ©n puedo organizarlos por categorГa o intenciГіn del usuario si prefieres.
Mas detalles en el enlace – http://casasdeapuestassinlicenciaespana.xyz/
?Que tengas excelentes ventajas!
1win_jsSn
| #
1win win https://www.1win7026.ru .
Ronaldrhype
| #
Chanel replica designer bags
Ronaldrhype
| #
Celine replica designer bags
Toliknaf
| #
dark web market urls dark web market
DonDonfaita
| #
darkmarket 2025 dark websites
Ronaldrhype
| #
Fendi bag replica
Ronaldrhype
| #
Fendi bag replica
Ronaldrhype
| #
Gucci bag replica
Pingrutty
| #
darknet drug store darknet markets
skladi_vcSa
| #
аренда склада под вещи москва http://www.hranim-veshi-msk24.ru/ .
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы —
Thomaspoext
| #
Букет с герберами – солнечно!
букет пионов
site
| #
Nicely put, Thanks!
1win_ciEi
| #
1 win регистрация 1win7010.ru .
Thomaslob
| #
plinko app
Thomaslob
| #
plinko online game
Thomaslob
| #
bet onred
Thomaslob
| #
red on bet casino
Toliknaf
| #
dark markets darknet drug store
Donaldlaf
| #
darknet markets darknet market
mostbet_zqkr
| #
mostbet промокод mostbet7006.ru .
Ronaldrhype
| #
Prada fake designer bags
DennisAnype
| #
Эскорт Москва – это о вкусе, статусе и удовольствии. Здесь ценят конфиденциальность, качество и высокий уровень сервиса. Каждая встреча – словно идеальный вечер, где ты главный герой, а рядом – безупречная спутница, знающая, чего ты хочешь https://byescorts.com/
Ronaldrhype
| #
Fendi bag replica
mostbet_ibmt
| #
мостюет http://www.mostbet6033.ru .
Markcen
| #
Хотите сдать квартиру в аренду? Полезные статьи и советы —
Willieavamy
| #
Online casinos feel odd on loss—check it! plinko
seroquel cost 300mg
| #
Medicine information sheet. Long-Term Effects.
seroquel cost 300mg
Some trends of medicine. Read information now.
Thomaslob
| #
is plinko a con
Pingrutty
| #
dark markets darknet market list
Diplomi_dxKt
| #
Купить диплом о высшем образовании!
Мы предлагаем документы институтов, расположенных в любом регионе Российской Федерации.
diplomaj-v-tule.ru/realno-li-kupit-diplom-s-reestrom-2/
sitecaree
| #
Обзоры, рейтинги и новости про новинки гаджетов, читайте – здесь
Stonehenge Blocktrade
| #
Stonehenge Blocktrade
A Brief History of Elemental Analysis » Artemis Analytical
1win_cxsa
| #
1win сайт вход https://1win7012.ru/ .
pin up azerbaycan_dkkl
| #
pinup az?rbaycan pinup az?rbaycan .
Dnrtyuz
| #
Мы готовы предложить дипломы психологов, юристов, экономистов и прочих профессий по приятным ценам. Мы готовы предложить документы техникумов, которые находятся на территории всей России. Документы выпускаются на “правильной” бумаге высшего качества. Это позволяет делать настоящие дипломы, которые не отличить от оригинала. bolsatrabajo.cusur.udg.mx/employer/archive-diploma
internetpermCah
| #
подключить интернет пермь
domashij-internet-perm001.ru
подключить интернет
front-end
| #
front-end
A Brief History of Elemental Analysis » Artemis Analytical
Ronaldrhype
| #
Bottega Veneta bag replica
Sazrvfh
| #
Приобрести диплом под заказ в столице возможно используя официальный портал компании. notebooks.ru/forum/user/145395
Markcen
| #
Покупка квартиры без рисков? Полезные рекомендации —
Richarddremo
| #
Без пересадок автобусная экскурсия из Калининграда на Куршскую косу за 1,5 часа.
Immediate LexiPro
| #
Immediate LexiPro
A Brief History of Elemental Analysis » Artemis Analytical
Toliknaf
| #
darkmarket 2025 dark web market
Rabyitabe
| #
dark web markets darknet markets onion
Donaldlaf
| #
onion dark website dark market link
Pingrutty
| #
darkmarket link dark web drug marketplace
syhie transformatori kypit_mtsl
| #
сухие трансформаторы цена сухие трансформаторы цена .
Ronaldrhype
| #
Chanel fake designer bags
1win_ghMl
| #
1win casino online 1win casino online .
1win_jfEi
| #
1вин официальный 1вин официальный .
can i buy cheap lopressor without rx
| #
Medicament prescribing information. What side effects can this medication cause?
can i buy cheap lopressor without rx
Everything information about medication. Get here.
prostafense
| #
prostafense is a premium, doctor-crafted supplement formulated to maintain optimal prostate function, enhance urinary performance, and support overall male wellness.
site
| #
site
A Brief History of Elemental Analysis » Artemis Analytical
1win_vrsa
| #
сайт 1win http://1win7012.ru/ .
Ernestcreri
| #
Преимущества работы рекламное агентство Витрувий — индивидуальный подход и креативные идеи. Помогаем бизнесу выделиться и привлечь нужную аудиторию.
Markcen
| #
Как выбрать надёжного застройщика? Полезные рекомендации —
DennisAnype
| #
Мы предлагаем выбор из лучших моделей города, чтобы ты мог получать максимум от каждого момента. Почувствуй эту роскошь. Почувствуй Эскорт Москва.
Pingrutty
| #
darknet drug store darkmarket list
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем обширный каталог продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф обеспечивает все стадии сделки, предоставляя полный пакет необходимых документов для оформления товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с высоким уровнем сервиса.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в подборе нужных изделий и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете достоверность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – наилучшее решение для вашего бизнеса.
Наши товары:
Полоса РёР· платины Рё ее сплавов 6 РјРј ПлРд-30 ГОСТ 24718-81 Рзучите разнообразие полос РёР· платины Рё ее сплавов РЅР° сайте Редметсплав.СЂС„. Наши высококачественные материалы обладают высокой прочностью, устойчивостью Рє РєРѕСЂСЂРѕР·РёРё Рё непревзойденными техническими характеристиками. Полосы РёР· платины Рё ее сплавов применяются РІ ювелирном деле, медицине Рё производстве специальных деталей, гарантируя надежность Рё качество продукции.
Ronaldrhype
| #
Celine fake designer bags
Thomaslob
| #
fortune.tiger
1win_egPr
| #
1win ug https://1win1006.top .
DonDonfaita
| #
darknet drug store darknet marketplace
Toliknaf
| #
dark market dark web market
Donaldlaf
| #
darknet marketplace dark markets 2025
Rabyitabe
| #
dark market onion darknet drug store
Willieavamy
| #
The live dealers online glow—real touch! aviator login
buying cheap zithromax without prescription
| #
Medicament prescribing information. Drug Class.
buying cheap zithromax without prescription
Some news about medicine. Get here.
Diplomi_skKt
| #
Приобрести диплом о высшем образовании!
Мы можем предложить документы любых учебных заведений, которые расположены в любом регионе Российской Федерации.
diploml-174.ru/skolko-stoit-diplom-s-zaneseniem-v-reestr-2/
Ronaldrhype
| #
fake Gucci bag
Ronaldrhype
| #
fake Gucci bag
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы
составление технического плана на объект недвижимости
| #
составление технического плана на объект недвижимости
A Brief History of Elemental Analysis » Artemis Analytical
1win_oiMl
| #
casino 1 win https://www.1win1009.top .
Pingrutty
| #
darknet marketplace bitcoin dark web
jazz clubs in montreal
| #
jazz clubs in montreal
A Brief History of Elemental Analysis » Artemis Analytical
Dnrtfzs
| #
Мы изготавливаем дипломы психологов, юристов, экономистов и других профессий по выгодным ценам. Мы можем предложить документы ВУЗов, расположенных на территории всей Российской Федерации. Дипломы и аттестаты печатаются на бумаге самого высокого качества. Это позволяет делать настоящие дипломы, которые не отличить от оригинала. cdltruckdrivingcareers.com/employer/archive-diploma
mostbet_wgMt
| #
мостбет кыргызстан https://mostbet7007.ru/ .
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы —
DonDonfaita
| #
darknet links darknet markets 2025
Trefcla
| #
Приобрести диплом о высшем образовании. Покупка официального диплома через надежную компанию дарит ряд плюсов. Это решение дает возможность сберечь как длительное время, так и существенные финансовые средства. ls.co-x.ru/2025/04/07/gde-nadezhno-kupit-diplom.html
Sheilaadurl
| #
РедМетСплав предлагает широкий ассортимент высококачественных изделий из нестандартных материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы обеспечиваем своевременную реализацию вашего заказа.
Каждая единица продукции подтверждена требуемыми документами, подтверждающими их качество. Опытная поддержка – наш стандарт – мы на связи, чтобы разрешать ваши вопросы и находить ответы под требования вашего бизнеса.
Доверьте потребности вашего бизнеса профессионалам РедМетСплав и убедитесь в гибкости нашего предложения
Наши товары:
Фольга магниевая ISO-MC21310 – BS ISO 16220 РўСЂСѓР±Р° магниевая ISO-MC21310 – BS ISO 16220 представляет СЃРѕР±РѕР№ высококачественный РїСЂРѕРґСѓРєС‚, который идеально РїРѕРґС…РѕРґРёС‚ для использования РІ различных отраслях. Рта магниевая труба отличается высокой прочностью Рё стойкостью Рє РєРѕСЂСЂРѕР·РёРё, что делает ее надежным выбором для тяжелых условий. РўСЂСѓР±Р° магниевая ISO-MC21310 – BS ISO 16220 легко поддается механической обработке Рё РїРѕРґС…РѕРґРёС‚ для сварки, что расширяет её применение РІ промышленных проектах. Если РІС‹ ищете надежный Рё долговечный РїСЂРѕРґСѓРєС‚, РЅРµ упустите возможность купить РўСЂСѓР±Р° магниевая ISO-MC21310 – BS ISO 16220 Рё получите отличный результат!
Toliknaf
| #
darknet marketplace dark web sites
Donaldlaf
| #
darknet market dark market onion
Sazrbzz
| #
Приобрести диплом под заказ возможно используя официальный сайт компании. mirstalkera.4admins.ru/viewtopic.phpf=50&t=3192
Rabyitabe
| #
bitcoin dark web darknet markets
pin up azerbaycan_qtkl
| #
pinup az?rbaycan pinup az?rbaycan .
prostafense
| #
prostafense is a premium, doctor-crafted supplement formulated to maintain optimal prostate function, enhance urinary performance, and support overall male wellness.
mostbet_dkMt
| #
поддержка мостбет mostbet7007.ru .
Ronaldrhype
| #
Bottega Veneta fake designer bags
Jariorszo
| #
Заказать диплом любого университета!
Мы предлагаемвыгодно купить диплом, который выполнен на бланке ГОЗНАКа и заверен мокрыми печатями, штампами, подписями должностных лиц. Наш диплом способен пройти любые проверки, даже при использовании специальных приборов. Достигайте свои цели быстро и просто с нашей компанией- mybuildhouse.ru/kupite-diplom-i-uverenno-vstupayte-v-novuyu-zhizn
Pingrutty
| #
best darknet markets dark markets
Jariorxgl
| #
Наша компания предлагает выгодно приобрести диплом, который выполняется на оригинальной бумаге и заверен мокрыми печатями, штампами, подписями. Документ пройдет лубую проверку, даже при использовании профессиональных приборов. tiktack.socialkhaleel.com/read-blog/2172_mozhno-li-kupit-diplom-zanesennyj-v-reestr.html
Thomaslob
| #
bet on red recenze
syhie transformatori kypit_mysl
| #
сухие трансформаторы купить сухие трансформаторы купить .
1win_rbsa
| #
1win официальный сайт скачать https://1win7012.ru/ .
JustinGrics
| #
Is it possible to rewind the clock on elemental analysis, using modern technology to recreate the first-ever CHNS analysis performed by Carlo Erba in 1968? If so, what challenges would we face in attempting to recreate this historic experiment, and what potential insights could such a project offer to our understanding of elemental analysis today?
Maybe not 100% in context, but felt like saying this
I just the other day discovered a team called sevenup.trance.mk.ua.
They’re a professional esports team focused on Battle Tactics. From what I saw — clean planning and tactical gameplay.
Anyone familiar with them?
Markcen
| #
Хотите разбираться в рынке недвижимости? Узнайте подробности
mostbet_kvmt
| #
скачать mostbet на телефон http://mostbet6033.ru/ .
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к материалам высочайшего качества, но и возможность заказа любого необходимого объема.
Имея широкий спектр продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие всем нормам.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Приступая к взаимодействию с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит надежного партнера, с которым сегодняшние вызовы превращаются в завтрашние успехи.
Наши товары:
Алюминиево-неодимовая плоская мишень
internetpermCah
| #
подключить интернет в перми в квартире
domashij-internet-perm002.ru
лучший интернет провайдер пермь
Ronaldrhype
| #
Prada bag replica
can i get cheap diflucan online
| #
Pills information sheet. Effects of Drug Abuse.
can i get cheap diflucan online
Some trends of medicines. Get here.
Thomaslob
| #
red on bet
how to get ceftin no prescription
| #
Drug information for patients. What side effects can this medication cause?
how to get ceftin no prescription
Everything news about medication. Read information now.
Willieavamy
| #
The excitement of online slots is electric! dragon hatch
Toliknaf
| #
best darknet markets dark market
Rabyitabe
| #
darknet market list dark web drug marketplace
Thomaslob
| #
plinko demo
1win_fmSn
| #
1 вин про 1win7026.ru .
Pingrutty
| #
dark web drug marketplace darknet site
Zuwontex App
| #
Zuwontex App
A Brief History of Elemental Analysis » Artemis Analytical
Markcen
| #
Новостройки или вторичка? Читайте проверенные советы
Ronaldrhype
| #
DIOR bag replica
Ronaldrhype
| #
DIOR bag replica
mostbet_cfkr
| #
мостбет мобильная версия скачать mostbet7006.ru .
Thomaslob
| #
mines website
mostbet_rikr
| #
мостбет кыргызстан скачать мостбет кыргызстан скачать .
Markcen
| #
Хотите разбираться в рынке недвижимости? Читайте проверенные советы
1win_wcMl
| #
1win. casino. https://1win1009.top .
Ronaldrhype
| #
Celine bag replica
plinko geld verdienen
| #
plinko geld verdienen
A Brief History of Elemental Analysis » Artemis Analytical
Pingrutty
| #
darkmarket url darknet site
Rabyitabe
| #
darkmarket url darknet market
Donaldlaf
| #
dark market onion darknet markets url
Ronaldrhype
| #
Fendi replica designer bags
1win_tnsa
| #
1вин. https://1win7012.ru .
Ronaldrhype
| #
fake Louis Vuitton bag
Cazrums
| #
Купить диплом о высшем образовании!
Мы можем предложить документы учебных заведений, расположенных в любом регионе России. Дипломы и аттестаты делаются на “правильной” бумаге самого высшего качества: odessaforum.getbb.ru/viewtopic.phpf=5&t=24483
Delta Ravex
| #
Delta Ravex
A Brief History of Elemental Analysis » Artemis Analytical
Sazrqib
| #
Приобрести диплом о высшем образовании!
Заказать диплом ВУЗа по невысокой стоимости возможно, обратившись к надежной специализированной фирме. Приобрести диплом о высшем образовании: good-diplom.ru/diplom-s-reestrom-podlinnost-garantirovana
vedaftak
| #
Хотите, чтобы ваш портал занимал лидирующие позиции в поисковых системах и привлекал тысячи пользователей? Вечные ссылки – ваш ключ к успеху! Наши достоинства: гибкие тарифы, оперативный результат, белые методы продвижения. Приступаем к выполнению заказа сразу, после оплаты. Ищете вечные ссылки для сайта купить? SeoBomba.ru – тут опубликована более подробная о нас информация, посмотрите ее в любое время. Также тут представлены ответы на популярные вопросы. Не упустите возможность вывести свой сайт на новый уровень. Позвоните нам, и мы проконсультируем вас.
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы
cost generic levaquin without prescription
| #
Medicines information sheet. Generic Name.
cost generic levaquin without prescription
All what you want to know about pills. Read information now.
Pingrutty
| #
darknet markets links darkmarket 2025
Ronaldrhype
| #
Gucci replica designer bags
+7 (499) 460-69-87
| #
Лучшие шторы для уютного загородного жилья
шторы в загородном доме шторы в загородном доме .+7 (499) 460-69-87
Willieavamy
| #
I wish online cut bets—low fun! plinko
Toliknaf
| #
darknet market list dark market link
DonDonfaita
| #
dark web marketplaces darknet markets 2025
Donaldlaf
| #
dark market dark web sites
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
С широким ассортиментом продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается необходимыми документами, удостоверяющими его происхождение и соответствие заданным критериям.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние успехи.
Наши товары:
Гранулы циркония
Ronaldrhype
| #
DIOR bag replica
Markcen
| #
Хотите разбираться в рынке недвижимости? Полезные статьи и советы —
Jarioroev
| #
Заказать диплом о высшем образовании!
Мы предлагаемвыгодно купить диплом, который выполнен на оригинальном бланке и заверен печатями, водяными знаками, подписями должностных лиц. Наш диплом пройдет лубую проверку, даже с применением специальных приборов. Достигайте своих целей быстро и просто с нашими дипломами- diveorthrive.com/read-blog/6815_diplom-vysshego-obrazovaniya-s-zaneseniem-v-reestr.html
Iariorple
| #
Заказать диплом ВУЗа по доступной стоимости можно, обратившись к надежной специализированной фирме. Купить документ института вы можете у нас в столице. diplomt-v-chelyabinske.ru/kupit-diplom-s-reestrom-v-moskve-7
Ronaldrhype
| #
Celine fake designer bags
how to get kamagra without a prescription
| #
Medicine prescribing information. Drug Class.
how to get kamagra without a prescription
Actual news about medicines. Read information now.
Ronaldrhype
| #
Gucci bag replica
Pingrutty
| #
dark market link darkmarket list
Thomaslob
| #
plinko come funziona
Thomaslob
| #
mines betting game
internetpermCah
| #
тарифы интернет и телевидение пермь
domashij-internet-perm003.ru
провайдеры интернета пермь
1win_dhMl
| #
1win cassino 1win cassino .
Jariorwzg
| #
Наши специалисты предлагают выгодно и быстро приобрести диплом, который выполнен на бланке ГОЗНАКа и заверен мокрыми печатями, штампами, подписями должностных лиц. Документ способен пройти любые проверки, даже с использованием специально предназначенного оборудования. romawki.flybb.ru/viewtopic.php?f=3&t=2385
Markcen
| #
Инвестиции в жильё: с чего начать? Полезные статьи и советы —
Rabyitabe
| #
bitcoin dark web dark web market
Toliknaf
| #
darknet site dark market link
DonDonfaita
| #
darkmarkets darknet drug links
WilliamDix
| #
Мы изготавливаем дипломы любых профессий по приятным тарифам. Дипломы изготавливаются на подлинных бланках государственного образца Заказать диплом о высшем образовании diplomf-v-irkutske.ru
BBLarry
| #
Could the invention of a time-traveling CHNS elemental analyzer revolutionize the study of historical artifacts’ elemental composition, potentially revealing long-lost secrets of ancient civilizations? How would this technology impact archaeology and our understanding of history?
Що робити, якщо на улюбленій футболці з’явилася пляма? Видаляємо плями правильно!
Trade Avita
| #
Trade Avita
A Brief History of Elemental Analysis » Artemis Analytical
Donaldlaf
| #
darknet markets url darknet drug market
Lazrjql
| #
Где заказать диплом специалиста?
Заказать диплом института по выгодной стоимости возможно, обратившись к надежной специализированной компании.: diplompro.ru
Ronaldrhype
| #
fake Fendi bag
Ronaldrhype
| #
DIOR replica designer bags
Pingrutty
| #
dark market 2025 dark market url
where to get generic xenical tablets
| #
Pills prescribing information. Short-Term Effects.
where to get generic xenical tablets
Everything information about medicament. Read information here.
Markcen
| #
Покупка квартиры без рисков? Читайте проверенные советы
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние возможности.
Наша продукция:
Высокочистые гранулы молибдена
Willieavamy
| #
Online casino ads hit hard—chill out! fortune tiger
Sheilaadurl
| #
В поисках достоверного источника редкоземельных металлов и сплавов? Обратите внимание на компанию Редметсплав.рф. Мы предлагаем внушительный выбор продукции, обеспечивая превосходное качество каждого изделия.
Редметсплав.рф защищает все стадии сделки, предоставляя полный пакет необходимых документов для законного использования товаров. Неважно, какие объемы вам необходимы – от мелких партий до крупнооптовых заказов, мы готовы обеспечить любой запрос с непревзойденным обслуживанием.
Наша команда службы поддержки всегда на связи, чтобы помочь вам в выборе товаров и ответить на любые вопросы, связанные с применением и характеристиками металлов. Выбирая нас, вы выбираете уверенность в каждой детали сотрудничества.
Заходите на наш сайт Редметсплав.рф и убедитесь, что наше качество и сервис – идеальный вариант для вас.
Наша продукция:
Медная капиллярная трубка 1.6х0.5 мм М1 ГОСТ 2624-2016 Покупайте медную капиллярную трубку у нас для идеального теплоотвода и надежности. Широкий выбор размеров и конфигураций для различных проектов. Отличная теплопроводность и гибкость монтажа, гарантирующие долговечность и экономию времени и денег.
Ronaldrhype
| #
Bottega Veneta replica designer bags
Call Girls in Anand Niketan
| #
Call Girls in Anand Niketan
A Brief History of Elemental Analysis » Artemis Analytical
DonDonfaita
| #
dark market onion best darknet markets
Toliknaf
| #
darkmarket 2025 darkmarket url
Rabyitabe
| #
darkmarket url dark web link
Pingrutty
| #
dark market 2025 darknet markets onion address
Donaldlaf
| #
darknet markets links darknet marketplace
Frankfemia
| #
Обзор на самые лучшие сайты для продажи скинов, которые имеют хорошую репутацию среди комьюнити.
Если вы ищете, продать вещи кс 2 где выгоднее продать скины CS2, CS:GO, DOTA 2 за реальные деньги или хотите узнать, как быстро и сразу продать скины с выводом на карту.
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир качественных инноваций. Партнерство с нашей компанией гарантирует не только доступ к премиальным продуктам, но и возможность заказа любого необходимого объема.
Обладая обширной линейкой продукции на основе редкоземельных материалов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых требовательных клиентов. Каждый продукт сопровождается всеми соответствующими документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе сотрудничества. В нашем лице вы найдете не просто продавца, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Выбирая сотрудничество с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение необходимыми напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит достойного партнера, с которым текущие трудности превращаются в завтрашние достижения.
Наша продукция:
Гранулы высокочистого ванадия
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности
Ronaldrhype
| #
Prada bag replica
Ronaldrhype
| #
Prada bag replica
Jariormdd
| #
Заказать диплом любого ВУЗа!
Мы предлагаемвыгодно приобрести диплом, который выполнен на оригинальной бумаге и заверен печатями, водяными знаками, подписями должностных лиц. Диплом способен пройти лубую проверку, даже при помощи специального оборудования. Достигайте своих целей быстро с нашим сервисом- dev.aimplace.io/employer/gosznac-diplom-24
Mazrsbu
| #
Где заказать диплом специалиста?
Готовый диплом с приложением отвечает условиям и стандартам Министерства образования и науки, никто не отличит его от оригинала – даже со специальным оборудованием. Не откладывайте свои цели на потом, реализуйте их с нами – отправьте простую заявку на диплом прямо сейчас! Диплом о высшем образовании – легко! zanasite.free.fr/laripostedumardi/doku.php
Cazrkcs
| #
Мы предлагаем дипломы любой профессии по невысоким ценам. Оставить отзыв — kyc-diplom.com/replies.html
mostbet_gmmt
| #
mostbet kg скачать https://mostbet6033.ru/ .
Pingrutty
| #
bitcoin dark web darknet markets url
Sheilaadurl
| #
РедМетСплав предлагает обширный выбор высококачественных изделий из редких материалов. Не важно, какие объемы вам необходимы – от небольших закупок до масштабных поставок, мы обеспечиваем быстрое выполнение вашего заказа.
Каждая единица продукции подтверждена соответствующими документами, подтверждающими их соответствие стандартам. Опытная поддержка – наш стандарт – мы на связи, чтобы улаживать ваши вопросы и адаптировать решения под особенности вашего бизнеса.
Доверьте ваш запрос специалистам РедМетСплав и убедитесь в широком спектре предлагаемых возможностей
поставляемая продукция:
Порошок ванадиевый Р’РР›-1 Порошок ванадиевый Regular – Stratcor представляет СЃРѕР±РѕР№ высококачественный материал, широко используемый РІ различных отраслях. РћРЅ применяется РІ металлургии, как добавка для улучшения прочности стали Рё РґСЂСѓРіРёС… сплавов. Благодаря своей высокой чистоте, этот порошок обеспечивает отличные механические свойства Рё стойкость Рє РєРѕСЂСЂРѕР·РёРё. Приобретая Порошок ванадиевый Regular – Stratcor, РІС‹ получаете надежный РїСЂРѕРґСѓРєС‚, который отвечает всем современным стандартам. РќРµ упустите возможность купить Порошок ванадиевый Regular – Stratcor Рё повысить качество СЃРІРѕРёС… производственных процессов. Позаботьтесь Рѕ вашем бизнесе уже сегодня!
Ronaldrhype
| #
fake Bottega Veneta bag
Markcen
| #
Покупка квартиры без рисков? Узнайте подробности
Sheilaadurl
| #
Сотрудничество с компанией НАПЫЛЕНИЕ РФ, специализирующейся на поставке редкоземельных материалов для различных методов напыления, открывает перед вашим предприятием двери в мир высококлассных инноваций. Закупка у нас гарантирует не только доступ к премиальным продуктам, но и возможность заказа требуемого количества.
С широким ассортиментом редкоземельных элементов, от мышьяка до циркония, компания НАПЫЛЕНИЕ РФ готова удовлетворить потребности самых разных заказчиков. Каждый продукт сопровождается обязательными документами, удостоверяющими его происхождение и соответствие стандартам качества.
Кроме того, НАПЫЛЕНИЕ РФ понимает важность профессионального взаимодействия и поддержки на каждом этапе партнерских отношений. В нашем лице вы найдете не просто поставщика, но и партнера, готового предложить квалифицированную помощь и поддержку, опираться на ваши конкретные потребности и адаптировать условия поставки под особенности вашего производства.
Заключая партнерство с НАПЫЛЕНИЕ РФ, вы гарантируете себе плавное и бесперебойное снабжение ценными напыляемыми материалами, позволяющее вам без остановки реализовывать самые передовые и технологичные проекты. Ваш бизнес получит крепкого союзника, с которым сегодняшние вызовы превращаются в завтрашние достижения.
Наша продукция:
Титановая плоская мишень
Jamesedunc
| #
Discover Montenegro Zabljak Montenegro crystal clear sea, picturesque bays, mountain landscapes and ancient fortresses. The perfect destination for those looking for a combination of nature, history and relaxation. Detailed guides, recommendations, photos and route ideas.
Toliknaf
| #
darknet drug links darknet websites
Crecedizo Solidez
| #
Crecedizo Solidez
A Brief History of Elemental Analysis » Artemis Analytical
Terrymooma
| #
SPA-салон в Москве https://uslugi.yandex.ru/profile/BonSpa-3013256 предлагает массаж, пилинг, обёртывания, уход за кожей, антистресс-программы и релакс-процедуры. Работают сертифицированные специалисты, используется премиум-косметика. Уютная атмосфера, индивидуальный подход и удобное расположение в городе.
Ronaldrhype
| #
fake Chanel bag
Donaldlaf
| #
darkmarket 2025 darknet websites
Ronaldrhype
| #
Chanel replica designer bags
Willieavamy
| #
The mobile casino dips—lift it! tome of madness
Jarioromn
| #
Мы предлагаем быстро приобрести диплом, который выполняется на оригинальном бланке и заверен печатями, штампами, подписями. Документ пройдет лубую проверку, даже с использованием профессиональных приборов. silton.ru/forum/user/8036
mostbet_frKa
| #
официальный сайт мостбет http://www.mostbet3023.ru .